
Evolutionarily Developed Alternatively Spliced Exons Containing Translation Initiation Sites


Abstract

1. Introduction
2. Materials and Methods
2.1. Phylogenetic Classification of Human Exons Using In Silico Analyses
2.1.1. Classification of Human Exons Based on Coding Information
2.1.2. Estimation of the Tissue-Specific PSIs of Human 5UC-ASEs
2.1.3. Detection of 5UC-ASEs and Genes Orthologous to the Human Genes That Contain 5UC-ASEs in Model Organisms
2.1.4. Gene Ontology (GO) Analysis of the Genes That Contain 5UC-ASEs
2.2. In Silico Analysis of Functional RBPs Binding Around 5UC-ASEs
2.3. Antibodies
2.4. Cell Cultures and Transfection
2.5. RNA Extraction and RT-PCR
2.6. Immunoblotting
2.7. Polysome Profiling Analysis
2.8. Construction of Minigenes and Expression Vectors for Splicing Analysis
2.9. Tethered Function Assay of MATR3
3. Results
3.1. In Silico Analysis Identified a Unique Class of Exons Containing 5′ Untranslated Region (UTR) and Coding Sequence (CDS)
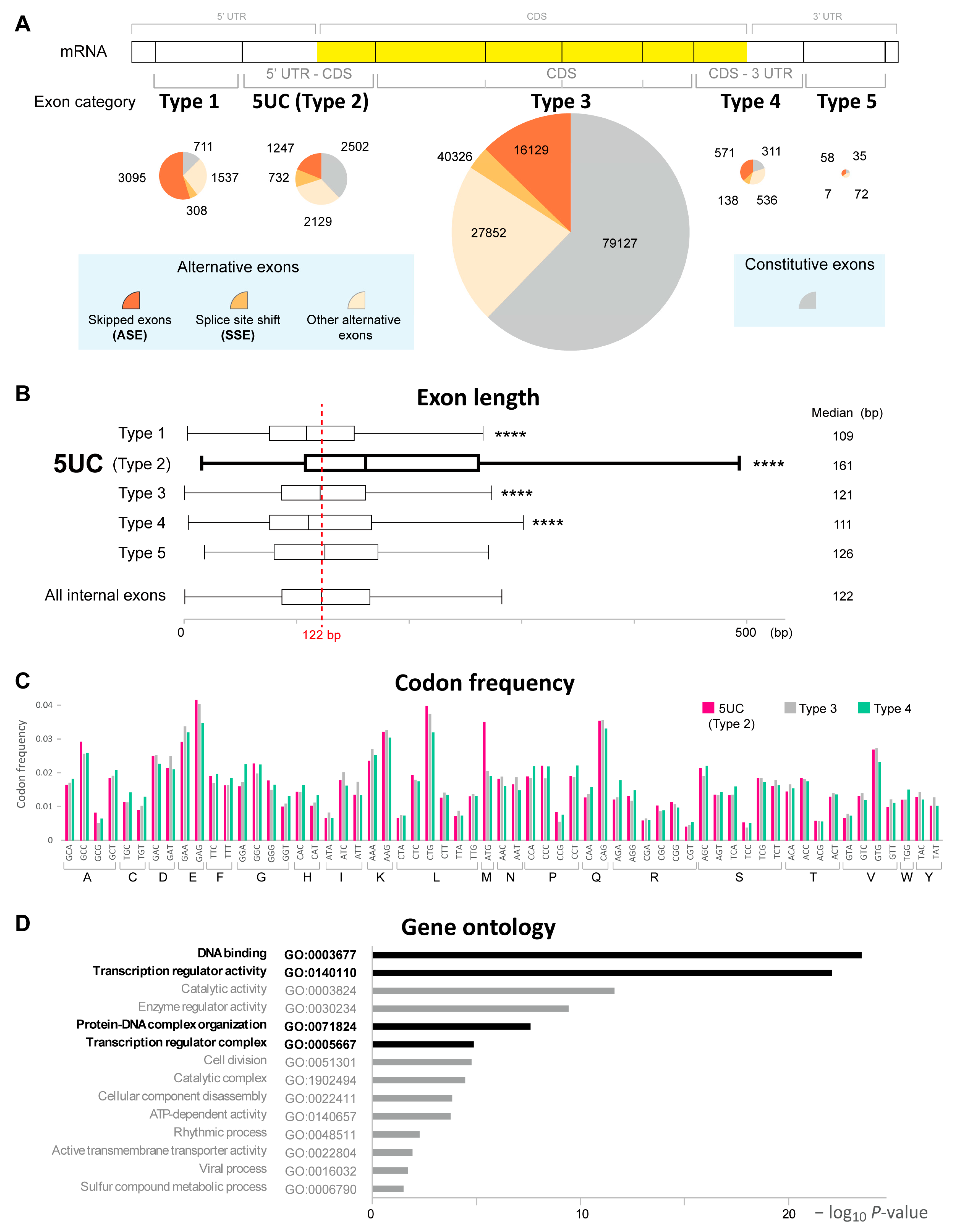
3.1.1. The Classification of Human Internal Exons Based on the Coding Information
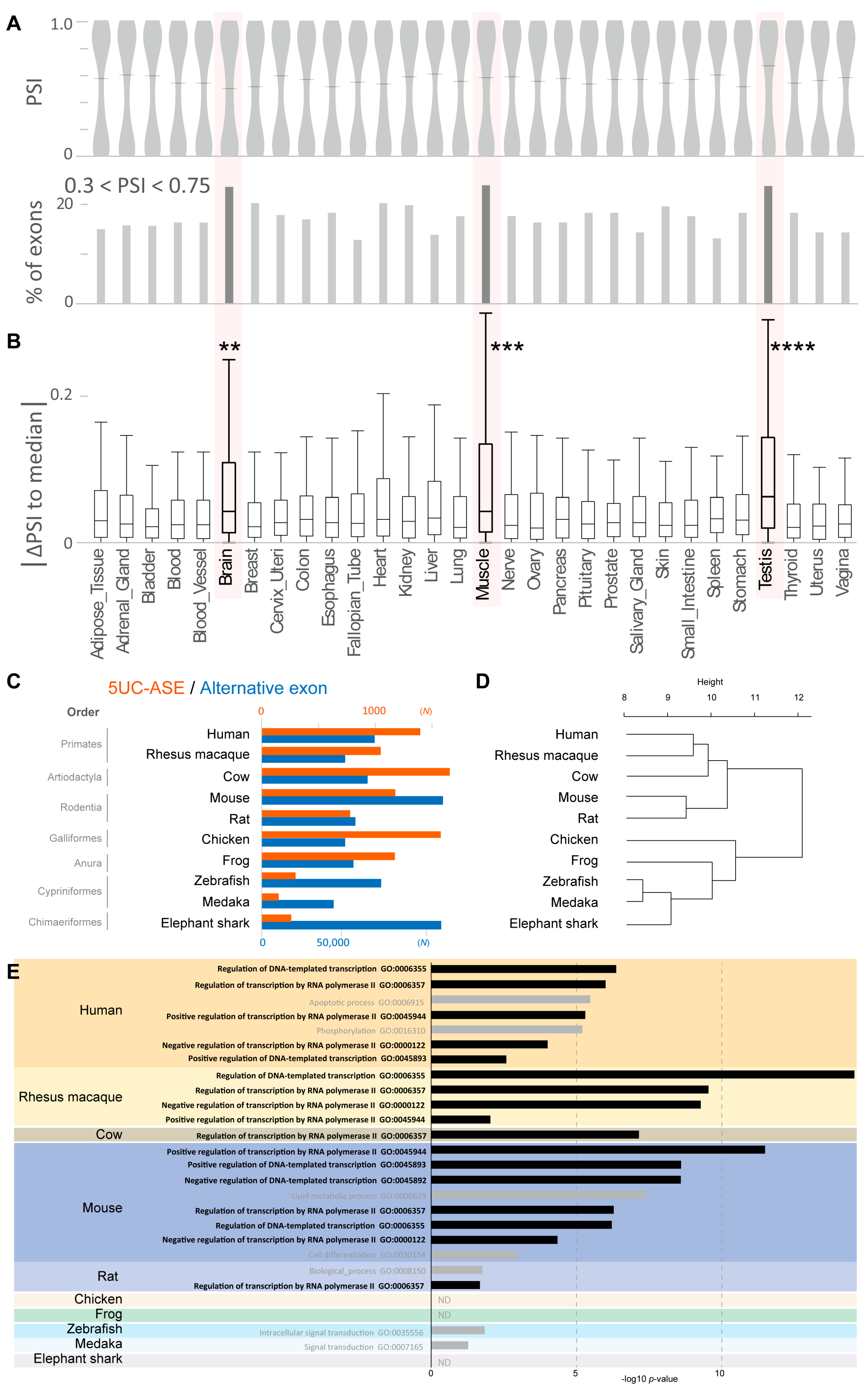
3.1.2. Alternative Splicing of 5UC Exons in Human Tissues
3.1.3. Development of 5UC-ASEs During Vertebrate Evolution
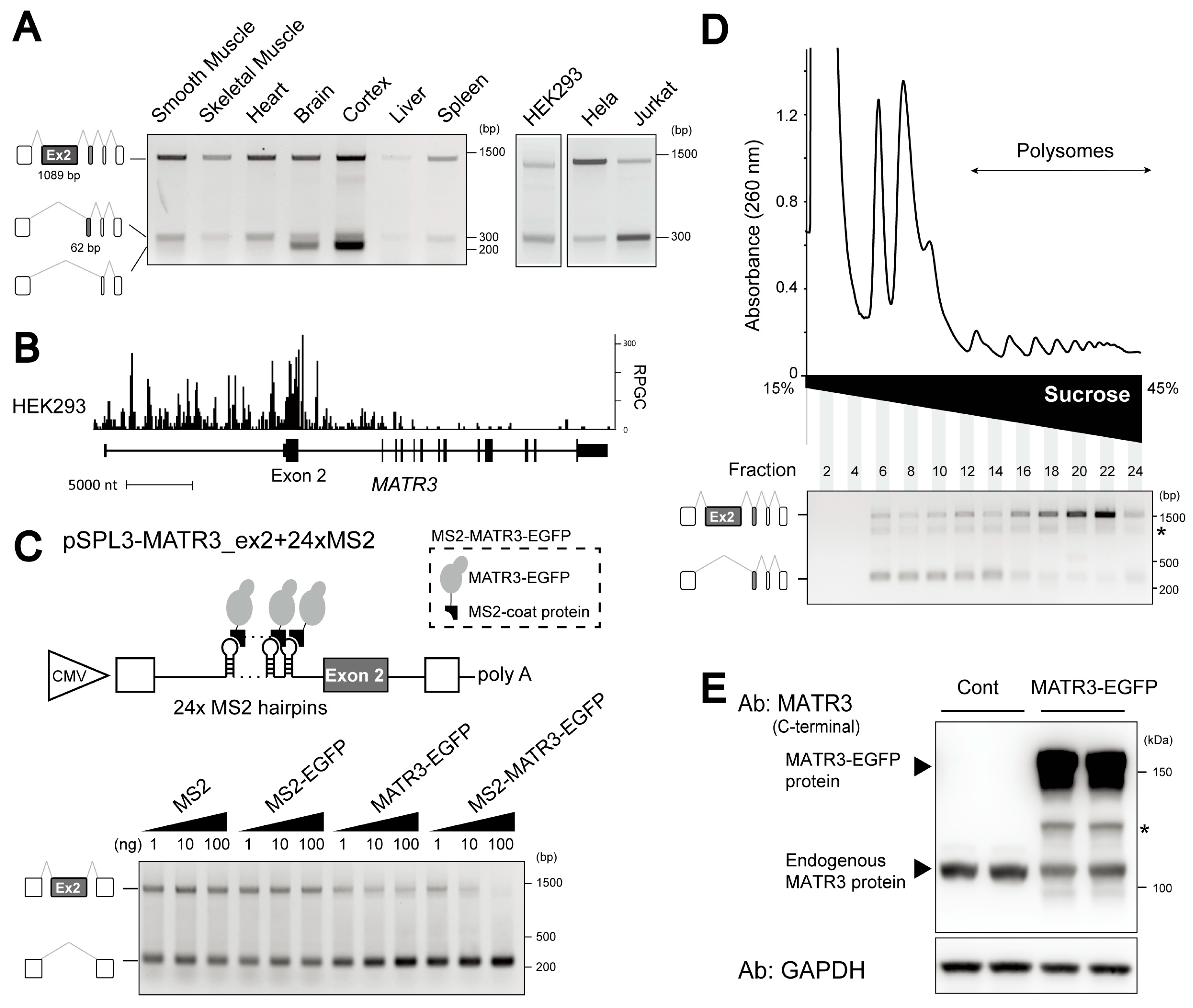
3.2. Identification of MATR3 as a Splicing Repressor of 5UC-ASEs
3.3. Abundant Expression of the MATR3 mRNA Isoform Lacking a 5UC-ASE, Exon 2
3.4. The MATR3 mRNA Isoform Lacking Exon 2 Is Not Translated
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Amaral, P.; Carbonell-Sala, S.; De La Vega, F.M.; Faial, T.; Frankish, A.; Gingeras, T.; Guigo, R.; Harrow, J.L.; Hatzigeorgiou, A.G.; Johnson, R.; et al. The status of the human gene catalogue. Nature 2023, 622, 41–47. [Google Scholar] [CrossRef]
- Mazin, P.V.; Khaitovich, P.; Cardoso-Moreira, M.; Kaessmann, H. Alternative splicing during mammalian organ development. Nat. Genet. 2021, 53, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Merkin, J.; Russell, C.; Chen, P.; Burge, C.B. Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Science 2012, 338, 1593–1599. [Google Scholar] [CrossRef]
- Garg, K.; Green, P. Differing patterns of selection in alternative and constitutive splice sites. Genome Res. 2007, 17, 1015–1022. [Google Scholar] [CrossRef]
- Ule, J.; Blencowe, B.J. Alternative Splicing Regulatory Networks: Functions, Mechanisms, and Evolution. Mol. Cell 2019, 76, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Kuroyanagi, H. Fox-1 family of RNA-binding proteins. Cell. Mol. Life Sci. 2009, 66, 3895–3907. [Google Scholar] [CrossRef] [PubMed]
- Conboy, J.G. Developmental regulation of RNA processing by Rbfox proteins. Wiley Interdiscip. Rev. RNA 2017, 8, e1398. [Google Scholar] [CrossRef] [PubMed]
- Ule, J.; Ule, A.; Spencer, J.; Williams, A.; Hu, J.S.; Cline, M.; Wang, H.; Clark, T.; Fraser, C.; Ruggiu, M.; et al. Nova regulates brain-specific splicing to shape the synapse. Nat. Genet. 2005, 37, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Movassat, M.; Forouzmand, E.; Reese, F.; Hertel, K.J. Exon size and sequence conservation improves identification of splice-altering nucleotides. RNA 2019, 25, 1793–1805. [Google Scholar] [CrossRef]
- Xing, Y.; Lee, C. Assessing the application of Ka/Ks ratio test to alternatively spliced exons. Bioinformatics 2005, 21, 3701–3703. [Google Scholar] [CrossRef]
- Irimia, M.; Weatheritt, R.J.; Ellis, J.D.; Parikshak, N.N.; Gonatopoulos-Pournatzis, T.; Babor, M.; Quesnel-Vallieres, M.; Tapial, J.; Raj, B.; O’Hanlon, D.; et al. A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell 2014, 159, 1511–1523. [Google Scholar] [CrossRef]
- Kawachi, T.; Masuda, A.; Yamashita, Y.; Takeda, J.I.; Ohkawara, B.; Ito, M.; Ohno, K. Regulated splicing of large exons is linked to phase-separation of vertebrate transcription factors. EMBO J. 2021, 40, e107485. [Google Scholar] [CrossRef]
- Lev-Maor, G.; Goren, A.; Sela, N.; Kim, E.; Keren, H.; Doron-Faigenboim, A.; Leibman-Barak, S.; Pupko, T.; Ast, G. The "alternative" choice of constitutive exons throughout evolution. PLoS Genet. 2007, 3, e203. [Google Scholar] [CrossRef]
- Johnson, J.O.; Pioro, E.P.; Boehringer, A.; Chia, R.; Feit, H.; Renton, A.E.; Pliner, H.A.; Abramzon, Y.; Marangi, G.; Winborn, B.J.; et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat. Neurosci. 2014, 17, 664–666. [Google Scholar] [CrossRef]
- Senderek, J.; Garvey, S.M.; Krieger, M.; Guergueltcheva, V.; Urtizberea, A.; Roos, A.; Elbracht, M.; Stendel, C.; Tournev, I.; Mihailova, V.; et al. Autosomal-dominant distal myopathy associated with a recurrent missense mutation in the gene encoding the nuclear matrix protein, matrin 3. Am. J. Hum. Genet. 2009, 84, 511–518. [Google Scholar] [CrossRef]
- Coelho, M.B.; Attig, J.; Bellora, N.; Konig, J.; Hallegger, M.; Kayikci, M.; Eyras, E.; Ule, J.; Smith, C.W. Nuclear matrix protein Matrin3 regulates alternative splicing and forms overlapping regulatory networks with PTB. EMBO J. 2015, 34, 653–668. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.M.; Barmada, S.J. Matrin 3 in neuromuscular disease: Physiology and pathophysiology. JCI Insight 2021, 6, e143948. [Google Scholar] [CrossRef] [PubMed]
- Rayaprolu, S.; D’Alton, S.; Crosby, K.; Moloney, C.; Howard, J.; Duffy, C.; Cabrera, M.; Siemienski, Z.; Hernandez, A.R.; Gallego-Iradi, C.; et al. Heterogeneity of Matrin 3 in the developing and aging murine central nervous system. J. Comp. Neurol. 2016, 524, 2740–2752. [Google Scholar] [CrossRef] [PubMed]
- Coelho, M.B.; Attig, J.; Ule, J.; Smith, C.W. Matrin3: Connecting gene expression with the nuclear matrix. Wiley Interdiscip. Rev. RNA 2016, 7, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Q. Statistical features of human exons and their flanking regions. Hum. Mol. Genet. 1998, 7, 919–932. [Google Scholar] [CrossRef]
- Cui, Y.; Cai, M.; Stanley, H.E. Comparative Analysis and Classification of Cassette Exons and Constitutive Exons. BioMed. Res. Int. 2017, 2017, 7323508. [Google Scholar] [CrossRef]
- Frankish, A.; Carbonell-Sala, S.; Diekhans, M.; Jungreis, I.; Loveland, J.E.; Mudge, J.M.; Sisu, C.; Wright, J.C.; Arnan, C.; Barnes, I.; et al. GENCODE: Reference annotation for the human and mouse genomes in 2023. Nucleic Acids Res. 2023, 51, D942–D949. [Google Scholar] [CrossRef]
- Trincado, J.L.; Entizne, J.C.; Hysenaj, G.; Singh, B.; Skalic, M.; Elliott, D.J.; Eyras, E. SUPPA2: Fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol. 2018, 19, 40. [Google Scholar] [CrossRef]
- Katz, Y.; Wang, E.T.; Airoldi, E.M.; Burge, C.B. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods 2010, 7, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.W.; Amode, M.R.; Austine-Orimoloye, O.; Azov, A.G.; Barba, M.; Barnes, I.; Becker, A.; Bennett, R.; Berry, A.; Bhai, J.; et al. Ensembl 2024. Nucleic Acids Res. 2024, 52, D891–D899. [Google Scholar] [CrossRef]
- Yates, A.; Beal, K.; Keenan, S.; McLaren, W.; Pignatelli, M.; Ritchie, G.R.; Ruffier, M.; Taylor, K.; Vullo, A.; Flicek, P. The Ensembl REST API: Ensembl Data for Any Language. Bioinformatics 2015, 31, 143–145. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef] [PubMed]
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Vilo, J.; Peterson, H. gprofiler2—An R package for gene list functional enrichment analysis and namespace conversion toolset g:Profiler. F1000Res 2020, 15, 9. [Google Scholar] [CrossRef]
- Luo, Y.; Hitz, B.C.; Gabdank, I.; Hilton, J.A.; Kagda, M.S.; Lam, B.; Myers, Z.; Sud, P.; Jou, J.; Lin, K.; et al. New developments on the Encyclopedia of DNA Elements (ENCODE) data portal. Nucleic Acids Res. 2020, 48, D882–D889. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Carbon, S.; Ireland, A.; Mungall, C.J.; Shu, S.; Marshall, B.; Lewis, S.; Ami, G.O.H.; Web Presence Working Group. AmiGO: Online access to ontology and annotation data. Bioinformatics 2009, 25, 288–289. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology Consortium; Aleksander, S.A.; Balhoff, J.; Carbon, S.; Cherry, J.M.; Drabkin, H.J.; Ebert, D.; Feuermann, M.; Gaudet, P.; Harris, N.L.; et al. The Gene Ontology knowledgebase in 2023. Genetics 2023, 224, iyad031. [Google Scholar] [CrossRef]
- Ramirez, F.; Ryan, D.P.; Gruning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dundar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef] [PubMed]
- Sahashi, K.; Masuda, A.; Matsuura, T.; Shinmi, J.; Zhang, Z.; Takeshima, Y.; Matsuo, M.; Sobue, G.; Ohno, K. In vitro and in silico analysis reveals an efficient algorithm to predict the splicing consequences of mutations at the 5′ splice sites. Nucleic Acids Res. 2007, 35, 5995–6003. [Google Scholar] [CrossRef] [PubMed]
- Coulon, A.; Ferguson, M.L.; de Turris, V.; Palangat, M.; Chow, C.C.; Larson, D.R. Kinetic competition during the transcription cycle results in stochastic RNA processing. Elife 2014, 3, e03939. [Google Scholar] [CrossRef]
- Masuda, A.; Shen, X.M.; Ito, M.; Matsuura, T.; Engel, A.G.; Ohno, K. hnRNP H enhances skipping of a nonfunctional exon P3A in CHRNA1 and a mutation disrupting its binding causes congenital myasthenic syndrome. Hum. Mol. Genet. 2008, 17, 4022–4035. [Google Scholar] [CrossRef] [PubMed]
- Masuda, A.; Okamoto, T.; Kawachi, T.; Takeda, J.I.; Hamaguchi, T.; Ohno, K. Blending and separating dynamics of RNA-binding proteins develop architectural splicing networks spreading throughout the nucleus. Mol. Cell 2024, 84, 2949–2965.e2910. [Google Scholar] [CrossRef] [PubMed]
- GTEx Consortium. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef]
- Kumar, S.; Suleski, M.; Craig, J.M.; Kasprowicz, A.E.; Sanderford, M.; Li, M.; Stecher, G.; Hedges, S.B. TimeTree 5: An Expanded Resource for Species Divergence Times. Mol. Biol. Evol. 2022, 39, msac174. [Google Scholar] [CrossRef]
- Kalsotra, A.; Cooper, T.A. Functional consequences of developmentally regulated alternative splicing. Nat. Rev. Genet. 2011, 12, 715–729. [Google Scholar] [CrossRef] [PubMed]
- Bolisetty, M.T.; Beemon, K.L. Splicing of internal large exons is defined by novel cis-acting sequence elements. Nucleic Acids Res. 2012, 40, 9244–9254. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, J.D. A survey on intron and exon lengths. Nucleic Acids Res. 1988, 16, 9893–9908. [Google Scholar] [CrossRef]
- Bieberstein, N.I.; Carrillo Oesterreich, F.; Straube, K.; Neugebauer, K.M. First exon length controls active chromatin signatures and transcription. Cell Rep. 2012, 2, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Polydorides, A.D.; Okano, H.J.; Yang, Y.Y.; Stefani, G.; Darnell, R.B. A brain-enriched polypyrimidine tract-binding protein antagonizes the ability of Nova to regulate neuron-specific alternative splicing. Proc. Natl. Acad. Sci. USA 2000, 97, 6350–6355. [Google Scholar] [CrossRef]
- Damianov, A.; Ying, Y.; Lin, C.H.; Lee, J.A.; Tran, D.; Vashisht, A.A.; Bahrami-Samani, E.; Xing, Y.; Martin, K.C.; Wohlschlegel, J.A.; et al. Rbfox Proteins Regulate Splicing as Part of a Large Multiprotein Complex LASR. Cell 2016, 165, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Muller-McNicoll, M.; Rossbach, O.; Hui, J.; Medenbach, J. Auto-regulatory feedback by RNA-binding proteins. J. Mol. Cell Biol. 2019, 11, 930–939. [Google Scholar] [CrossRef]
- Boelens, W.C.; Jansen, E.J.; van Venrooij, W.J.; Stripecke, R.; Mattaj, I.W.; Gunderson, S.I. The human U1 snRNP-specific U1A protein inhibits polyadenylation of its own pre-mRNA. Cell 1993, 72, 881–892. [Google Scholar] [CrossRef]
- Ayala, Y.M.; De Conti, L.; Avendano-Vazquez, S.E.; Dhir, A.; Romano, M.; D’Ambrogio, A.; Tollervey, J.; Ule, J.; Baralle, M.; Buratti, E.; et al. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2011, 30, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Rajgor, D.; Hanley, J.G.; Shanahan, C.M. Identification of novel nesprin-1 binding partners and cytoplasmic matrin-3 in processing bodies. Mol. Biol. Cell 2016, 27, 3894–3902. [Google Scholar] [CrossRef]
- Giordano, G.; Sanchez-Perez, A.M.; Montoliu, C.; Berezney, R.; Malyavantham, K.; Costa, L.G.; Calvete, J.J.; Felipo, V. Activation of NMDA receptors induces protein kinase A-mediated phosphorylation and degradation of matrin 3. Blocking these effects prevents NMDA-induced neuronal death. J. Neurochem. 2005, 94, 808–818. [Google Scholar] [CrossRef]
- De Marco, G.; Lomartire, A.; Manera, U.; Canosa, A.; Grassano, M.; Casale, F.; Fuda, G.; Salamone, P.; Rinaudo, M.T.; Colombatto, S.; et al. Effects of intracellular calcium accumulation on proteins encoded by the major genes underlying amyotrophic lateral sclerosis. Sci. Rep. 2022, 12, 395. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exon Class | Total | Alternatively Skipped Exon (ASE) | Splice-Site-Shifted Exons (SSE) | Other Alternative Exon | Constitutive Exon |
|---|---|---|---|---|---|
| Type 1 | 5651 | 3095 | 308 | 1537 | 711 |
| 5UC (Type 2) | 6610 | 1247 | 732 | 2129 | 2502 |
| Type 3 | 127,144 | 16,129 | 4036 | 27,852 | 79,127 |
| Type 4 | 1556 | 571 | 138 | 536 | 311 |
| Type 5 | 172 | 58 | 7 | 72 | 35 |
| Protein-Coding Genes | 5UC-ASE Genes | ||||||
|---|---|---|---|---|---|---|---|
| Species | Genes | Transcript Isoforms | Exons | Alternative Exons | Genes | Transcript Isoforms | ASEs |
| Human | 20,042 | 64,128 | 289,850 | 66,286 | 1130 | 3153 | 1398 |
| Rhesus macaque | 21,369 | 48,237 | 256,946 | 48,971 | 878 | 1581 | 1048 |
| Cow | 23,492 | 59,313 | 268,947 | 62,243 | 1388 | 2065 | 1657 |
| Mouse | 21,833 | 102,034 | 451,167 | 106,475 | 907 | 2480 | 1178 |
| Rat | 23,049 | 45,829 | 264,477 | 54,998 | 701 | 899 | 778 |
| Chicken | 16,711 | 44,314 | 241,811 | 49,013 | 1293 | 1865 | 1578 |
| Frog | 21,759 | 48,976 | 268,133 | 53,862 | 944 | 1701 | 1173 |
| Zebrafish | 25,107 | 50,807 | 313,568 | 70,100 | 268 | 407 | 298 |
| Medaka | 23,587 | 37,433 | 253,363 | 42,102 | 168 | 238 | 181 |
| Elephant shark | 19,415 | 49,321 | 260,786 | 105,543 | 243 | 359 | 259 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takeda, J.-i.; Okamoto, T.; Masuda, A. Evolutionarily Developed Alternatively Spliced Exons Containing Translation Initiation Sites. Cells 2025, 14, 11. https://doi.org/10.3390/cells14010011
Takeda J-i, Okamoto T, Masuda A. Evolutionarily Developed Alternatively Spliced Exons Containing Translation Initiation Sites. Cells. 2025; 14(1):11. https://doi.org/10.3390/cells14010011
Chicago/Turabian StyleTakeda, Jun-ichi, Takaaki Okamoto, and Akio Masuda. 2025. "Evolutionarily Developed Alternatively Spliced Exons Containing Translation Initiation Sites" Cells 14, no. 1: 11. https://doi.org/10.3390/cells14010011
APA StyleTakeda, J.-i., Okamoto, T., & Masuda, A. (2025). Evolutionarily Developed Alternatively Spliced Exons Containing Translation Initiation Sites. Cells, 14(1), 11. https://doi.org/10.3390/cells14010011