
A Novel Model for Simultaneous Evaluation of Hyperoxia-Mediated Brain and Lung Injury in Neonatal Rats
 , , , , , ,
, , , , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Procedure
2.2. Lung Histomorphometric Analysis
2.3. Brain and Lung Immunohistochemistry
2.4. Confocal Microscopy
2.5. Immunoblotting
2.6. Real-Time PCR
2.7. Statistical Analysis
3. Results
3.1. Seven Days of Hyperoxia Induce Hypomyelination in the Immature Brain
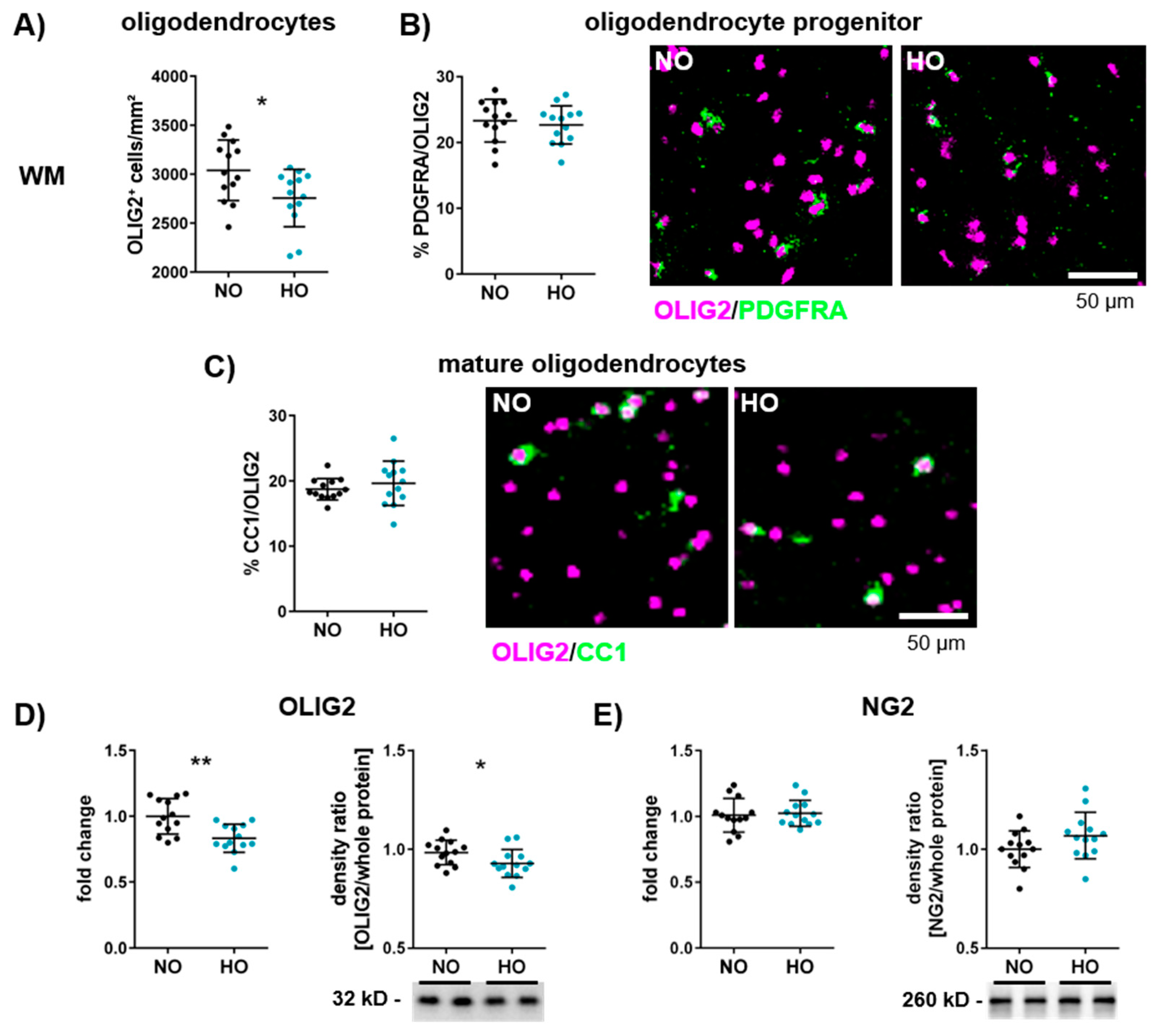
3.2. Hyperoxia Reduces the Number of Oligodendrocytes in the Developing Brain
3.3. Altered Microglia Activation After One Week of Hyperoxia in the White Matter
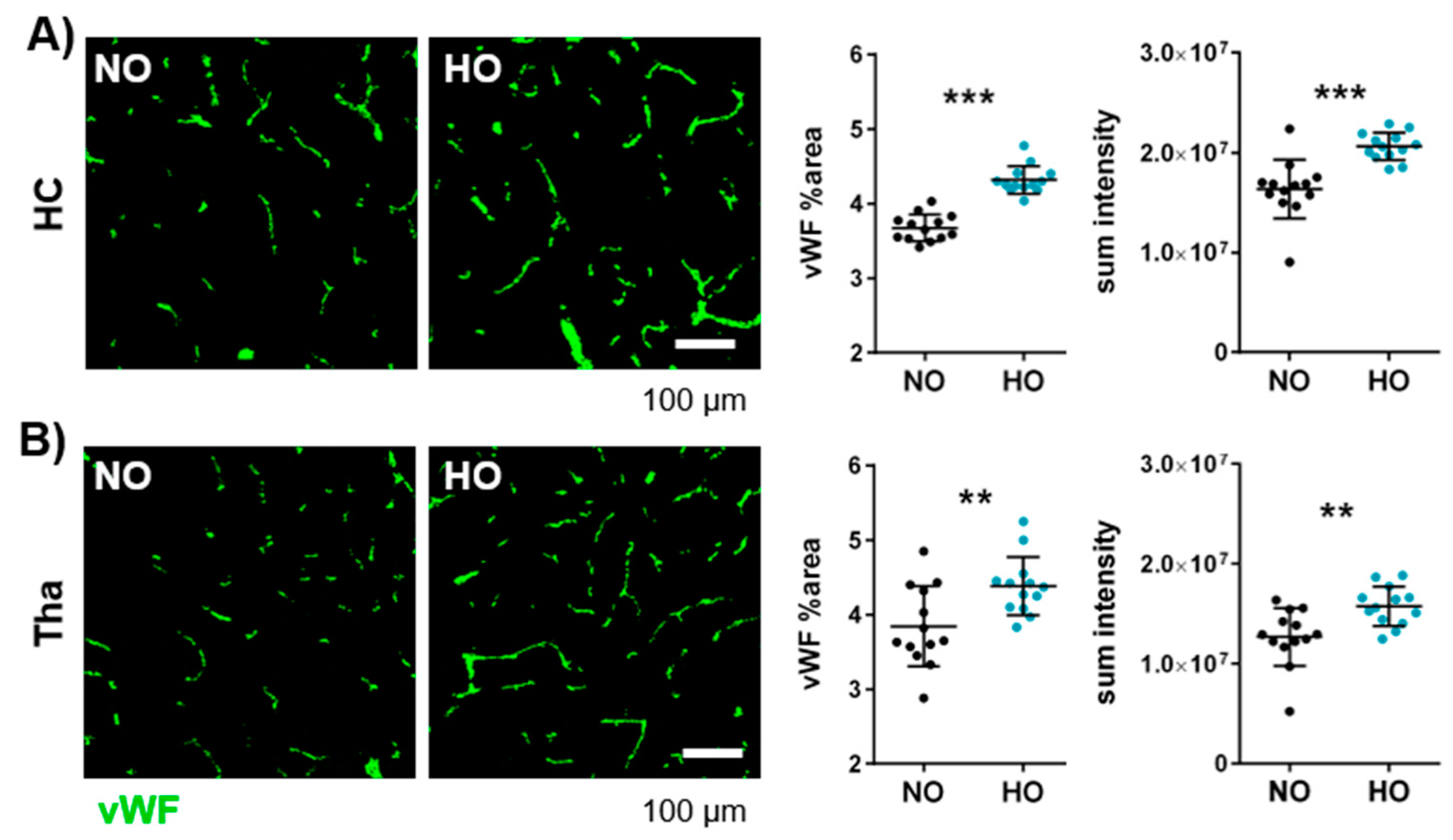
3.4. Postnatal Hyperoxia Leads to Hypervascularisation in the Immature Brain
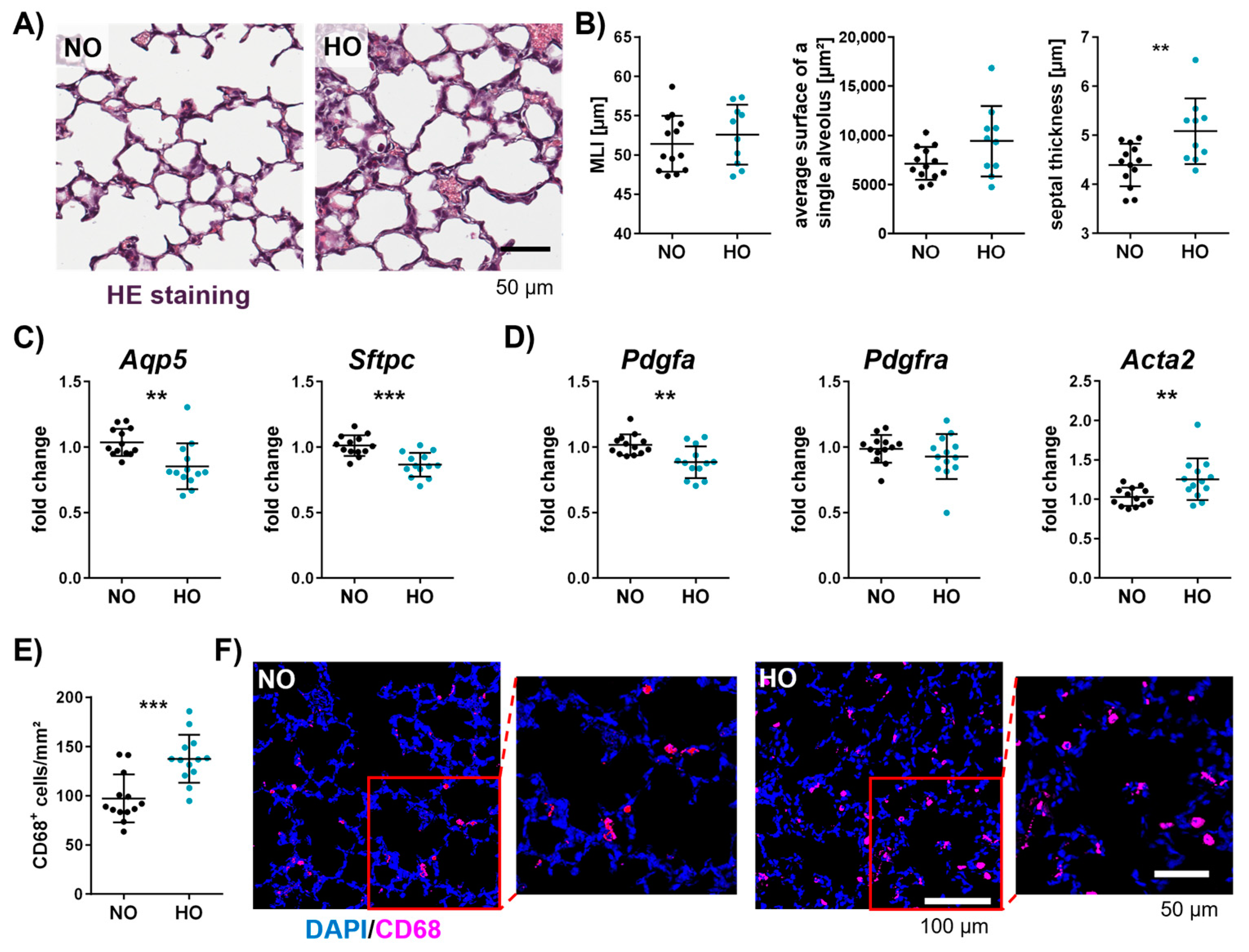
3.5. Neonatal Hyperoxia Causes an Arrest of Alveolarisation and Increases the Number of Macrophages in the Neonatal Lung
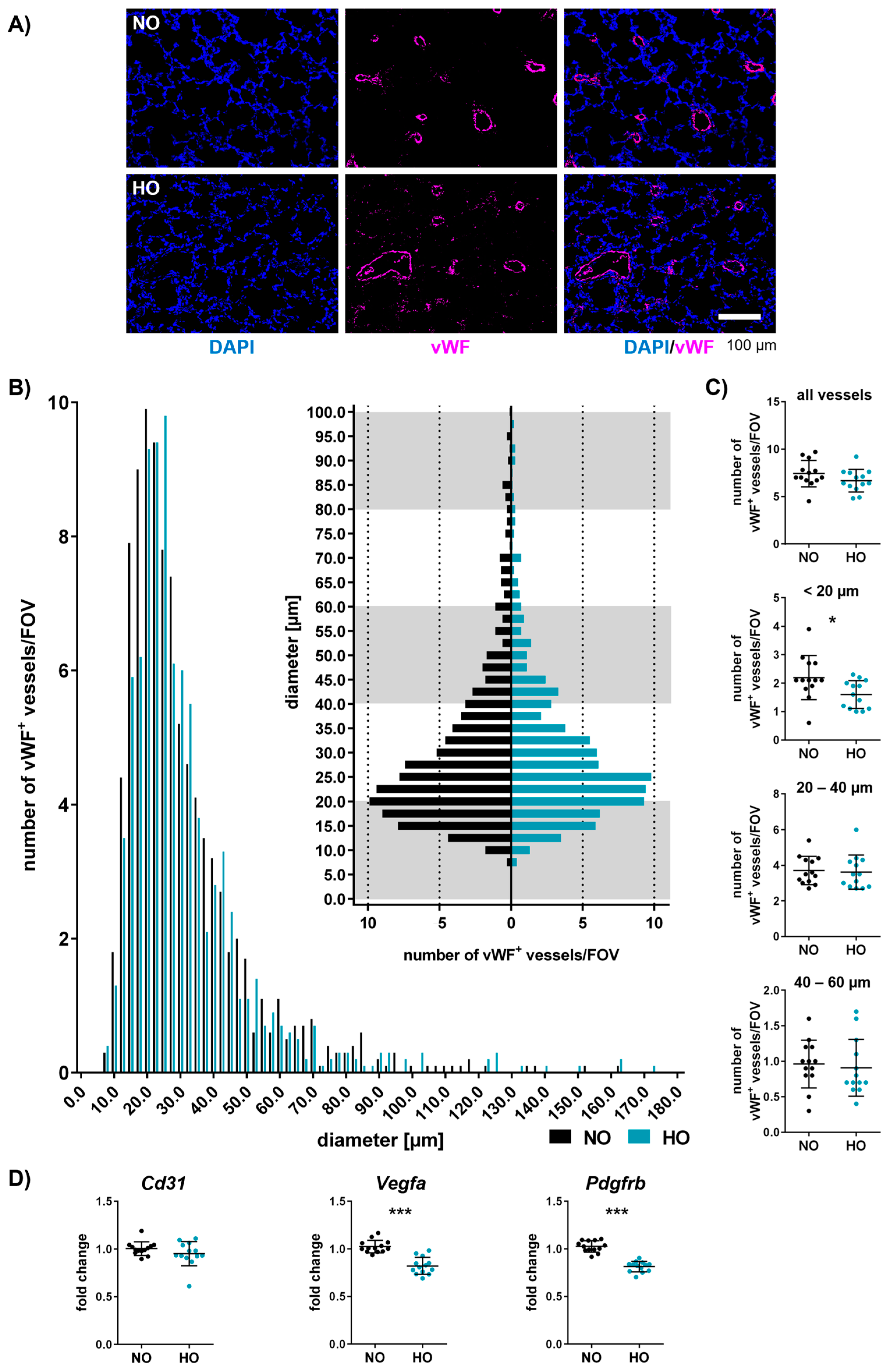
3.6. Neonatal Hyperoxia Induces Vascular Remodelling of Small Pulmonary Vessels
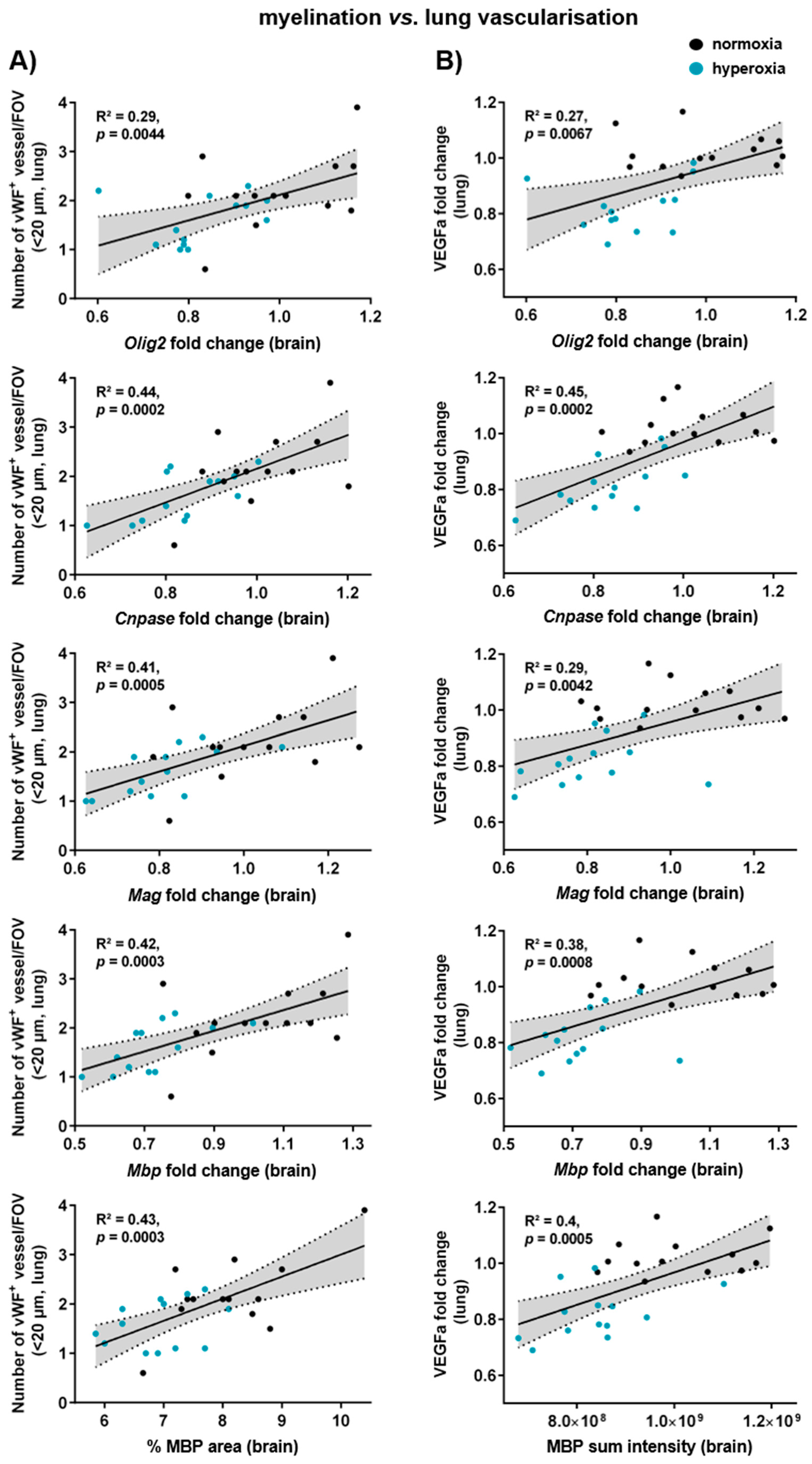
3.7. Myelination, Microglia Activation, and Vascularisation in the Brain Correlate with Lung Vascularisation and Gene Expression of Alveolar Markers
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACTA2 | Actin alpha 2 |
| AQP5 | Aquaporin 5 |
| BSA | Bovine serum albumin |
| CA | Cornu ammonis |
| CC1 | Adenomatous polyposis coli clone CC1 |
| CD206 | Cluster of differentiation 206 |
| CD31 | Cluster of differentiation 31 or platelet endothelial cell adhesion molecule 1 |
| CD68 | Cluster of differentiation 68 or macrosialin |
| CD86 | Cluster of differentiation 86 |
| Cing | Cingulum |
| CNPase | 2′,3′-cyclic-nucleotide 3′-phosphodiesterase |
| CTX | Cortex |
| Dapi | 4′,6-diamidino-2-phenylindol |
| DC | Deep cortical |
| DG | Dentate gyrus |
| EC | External capsule |
| FOV | Field of view |
| HC | Hippocampus |
| HE | Haematoxylin–eosin |
| HO | Hyperoxia |
| Iba1 | Ionised calcium-binding adapter molecule 1 |
| IL12 | Interleukin 12 |
| IntC | Internal capsule |
| MAG | Myelin-associated glycoprotein |
| MBP | Myelin basic protein |
| MLI | Mean linear intercept |
| MP | Non-fat milk powder |
| NG2 | Neural/glial antigen 2 |
| NO | Normoxia |
| Olig2 | Oligodendrocyte transcription factor 2 |
| P | Postnatal day |
| PBS | Phosphate-buffered saline |
| PDGFRA | Platelet-derived growth factor receptor alpha |
| PDGFRB | Platelet-derived growth factor receptor beta |
| PFA | Paraformaldehyde |
| PGDFa | Platelet-derived growth factor A |
| SFTPC | Pro-surfactant protein C |
| TBS | Tris-buffered saline |
| TBS-T | 0.1% Tween-20 in TBS |
| Tha | Thalamus |
| VEGFa | Vascular endothelial growth factor A |
| vWF | von Willebrand factor |
| WM | White matter |
References
- Ohuma, E.O.; Moller, A.B.; Bradley, E.; Chakwera, S.; Hussain-Alkhateeb, L.; Lewin, A.; Okwaraji, Y.B.; Mahanani, W.R.; Johansson, E.W.; Lavin, T.; et al. National, regional, and global estimates of preterm birth in 2020, with trends from 2010: A systematic analysis. Lancet 2023, 402, 1261–1271. [Google Scholar] [CrossRef] [PubMed]
- Kusuda, S.; Fujimura, M.; Uchiyama, A.; Totsu, S.; Matsunami, K.; Neonatal Research Network, J. Trends in morbidity and mortality among very-low-birth-weight infants from 2003 to 2008 in Japan. Pediatr. Res. 2012, 72, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Sefidkar, R.; Zayeri, F.; Kazemi, E.; Salehi, M.; Dehnad, A.; Hafizi, M. A Trend Study of Preterm Infant Mortality Rate in Developed and Developing Countries over 1990 to 2017. Iran. J. Public Health 2021, 50, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Liu, J.; Liu, M. Global, Regional, and National Incidence and Mortality of Neonatal Preterm Birth, 1990–2019. JAMA Pediatr. 2022, 176, 787–796. [Google Scholar] [CrossRef]
- Stoll, B.J.; Hansen, N.I.; Bell, E.F.; Walsh, M.C.; Carlo, W.A.; Shankaran, S.; Laptook, A.R.; Sanchez, P.J.; Van Meurs, K.P.; Wyckoff, M.; et al. Trends in Care Practices, Morbidity, and Mortality of Extremely Preterm Neonates, 1993–2012. JAMA 2015, 314, 1039–1051. [Google Scholar] [CrossRef]
- Nakanishi, H.; Suenaga, H.; Uchiyama, A.; Kono, Y.; Kusuda, S.; Neonatal Research Network, Japan. Trends in the neurodevelopmental outcomes among preterm infants from 2003–2012: A retrospective cohort study in Japan. J. Perinatol. 2018, 38, 917–928. [Google Scholar] [CrossRef]
- Horbar, J.D.; Greenberg, L.T.; Buzas, J.S.; Ehret, D.E.Y.; Soll, R.F.; Edwards, E.M. Trends in Mortality and Morbidities for Infants Born 24 to 28 Weeks in the US: 1997-2021. Pediatrics 2024, 153, e2023064153. [Google Scholar] [CrossRef]
- Cheong, J.L.Y.; Olsen, J.E.; Lee, K.J.; Spittle, A.J.; Opie, G.F.; Clark, M.; Boland, R.A.; Roberts, G.; Josev, E.K.; Davis, N.; et al. Temporal Trends in Neurodevelopmental Outcomes to 2 Years After Extremely Preterm Birth. JAMA Pediatr. 2021, 175, 1035–1042. [Google Scholar] [CrossRef]
- Reich, B.; Hoeber, D.; Bendix, I.; Felderhoff-Mueser, U. Hyperoxia and the Immature Brain. Dev. Neurosci. 2016, 38, 311–330. [Google Scholar] [CrossRef]
- Day, C.L.; Ryan, R.M. Bronchopulmonary dysplasia: New becomes old again! Pediatr. Res. 2017, 81, 210–213. [Google Scholar] [CrossRef]
- Mathias, M.; Chang, J.; Perez, M.; Saugstad, O. Supplemental Oxygen in the Newborn: Historical Perspective and Current Trends. Antioxidants 2021, 10, 1879. [Google Scholar] [CrossRef] [PubMed]
- Panfoli, I.; Candiano, G.; Malova, M.; De Angelis, L.; Cardiello, V.; Buonocore, G.; Ramenghi, L.A. Oxidative Stress as a Primary Risk Factor for Brain Damage in Preterm Newborns. Front. Pediatr. 2018, 6, 369. [Google Scholar] [CrossRef] [PubMed]
- Perrone, S.; Tataranno, M.L.; Buonocore, G. Oxidative stress and bronchopulmonary dysplasia. J. Clin. Neonatol. 2012, 1, 109–114. [Google Scholar] [CrossRef]
- Coalson, J.J. Pathology of bronchopulmonary dysplasia. Semin. Perinatol. 2006, 30, 179–184. [Google Scholar] [CrossRef]
- Jobe, A.H.; Kallapur, S.G. Long term consequences of oxygen therapy in the neonatal period. Semin. Fetal Neonatal Med. 2010, 15, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.L.; Chen, H.L. New developments in neonatal respiratory management. Pediatr. Neonatol. 2022, 63, 341–347. [Google Scholar] [CrossRef]
- Kiger, J. Neonatal ventilation. Semin. Pediatr. Surg. 2022, 31, 151199. [Google Scholar] [CrossRef]
- Ozer, E.A. Lung-protective Ventilation in Neonatal Intensive Care Unit. J. Clin. Neonatol. 2020, 9, 1–7. [Google Scholar] [CrossRef]
- van Kaam, A.H.; De Luca, D.; Hentschel, R.; Hutten, J.; Sindelar, R.; Thome, U.; Zimmermann, L.J.I. Modes and strategies for providing conventional mechanical ventilation in neonates. Pediatr. Res. 2021, 90, 957–962. [Google Scholar] [CrossRef]
- Vaucher, Y.E.; Peralta-Carcelen, M.; Finer, N.N.; Carlo, W.A.; Gantz, M.G.; Walsh, M.C.; Laptook, A.R.; Yoder, B.A.; Faix, R.G.; Das, A.; et al. Neurodevelopmental outcomes in the early CPAP and pulse oximetry trial. N. Engl. J. Med. 2012, 367, 2495–2504. [Google Scholar] [CrossRef]
- Donlon, J.; Bhat, V.; Hunter, K.; Kushnir, A.; Bhandari, V. Impact of severity and age with variable definitions of bronchopulmonary dysplasia on neurodevelopmental outcomes. Pediatr. Res. 2024, 96, 1243–1250. [Google Scholar] [CrossRef]
- Katz, T.A.; van Kaam, A.H.; Mugie, S.M.; Aarnoudse-Moens, C.S.H.; de Groof, F.; van Kempen, A.; van den Heuvel, M.E.N.; Vogelzang, J.; Rijpert, M.; Schiering, I.A.; et al. Risk Factors for Neurodevelopmental Impairment at 2- and 5-Years Corrected Age in Preterm Infants with Established Bronchopulmonary Dysplasia. Neonatology 2024, 121, 125–132. [Google Scholar] [CrossRef]
- Obst, S.; Herz, J.; Alejandre Alcazar, M.A.; Endesfelder, S.; Mobius, M.A.; Rudiger, M.; Felderhoff-Muser, U.; Bendix, I. Perinatal Hyperoxia and Developmental Consequences on the Lung-Brain Axis. Oxid. Med. Cell. Longev. 2022, 2022, 5784146. [Google Scholar] [CrossRef]
- Chang, Y.S.; Choi, S.J.; Sung, D.K.; Kim, S.Y.; Oh, W.; Yang, Y.S.; Park, W.S. Intratracheal transplantation of human umbilical cord blood-derived mesenchymal stem cells dose-dependently attenuates hyperoxia-induced lung injury in neonatal rats. Cell Transplant. 2011, 20, 1843–1854. [Google Scholar] [CrossRef] [PubMed]
- Alejandre-Alcazar, M.A.; Kwapiszewska, G.; Reiss, I.; Amarie, O.V.; Marsh, L.M.; Sevilla-Perez, J.; Wygrecka, M.; Eul, B.; Kobrich, S.; Hesse, M.; et al. Hyperoxia modulates TGF-beta/BMP signaling in a mouse model of bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L537–L549. [Google Scholar] [CrossRef] [PubMed]
- Vadivel, A.; Alphonse, R.S.; Etches, N.; van Haaften, T.; Collins, J.J.; O’Reilly, M.; Eaton, F.; Thebaud, B. Hypoxia-inducible factors promote alveolar development and regeneration. Am. J. Respir. Cell Mol. Biol. 2014, 50, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Felderhoff-Mueser, U.; Bittigau, P.; Sifringer, M.; Jarosz, B.; Korobowicz, E.; Mahler, L.; Piening, T.; Moysich, A.; Grune, T.; Thor, F.; et al. Oxygen causes cell death in the developing brain. Neurobiol. Dis. 2004, 17, 273–282. [Google Scholar] [CrossRef]
- Brehmer, F.; Bendix, I.; Prager, S.; van de Looij, Y.; Reinboth, B.S.; Zimmermanns, J.; Schlager, G.W.; Brait, D.; Sifringer, M.; Endesfelder, S.; et al. Interaction of inflammation and hyperoxia in a rat model of neonatal white matter damage. PLoS ONE 2012, 7, e49023. [Google Scholar] [CrossRef]
- Sifringer, M.; Bendix, I.; Borner, C.; Endesfelder, S.; von Haefen, C.; Kalb, A.; Holifanjaniaina, S.; Prager, S.; Schlager, G.W.; Keller, M.; et al. Prevention of neonatal oxygen-induced brain damage by reduction of intrinsic apoptosis. Cell Death Dis. 2012, 3, e250. [Google Scholar] [CrossRef]
- Endesfelder, S.; Weichelt, U.; Strauss, E.; Schlor, A.; Sifringer, M.; Scheuer, T.; Buhrer, C.; Schmitz, T. Neuroprotection by Caffeine in Hyperoxia-Induced Neonatal Brain Injury. Int. J. Mol. Sci. 2017, 18, 187. [Google Scholar] [CrossRef]
- Serdar, M.; Herz, J.; Kempe, K.; Lumpe, K.; Reinboth, B.S.; Sizonenko, S.V.; Hou, X.; Herrmann, R.; Hadamitzky, M.; Heumann, R.; et al. Fingolimod protects against neonatal white matter damage and long-term cognitive deficits caused by hyperoxia. Brain Behav. Immun. 2016, 52, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Serdar, M.; Herz, J.; Kempe, K.; Winterhager, E.; Jastrow, H.; Heumann, R.; Felderhoff-Muser, U.; Bendix, I. Protection of Oligodendrocytes Through Neuronal Overexpression of the Small GTPase Ras in Hyperoxia-Induced Neonatal Brain Injury. Front. Neurol. 2018, 9, 175. [Google Scholar] [CrossRef] [PubMed]
- Gerstner, B.; Buhrer, C.; Rheinlander, C.; Polley, O.; Schuller, A.; Berns, M.; Obladen, M.; Felderhoff-Mueser, U. Maturation-dependent oligodendrocyte apoptosis caused by hyperoxia. J. Neurosci. Res. 2006, 84, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Crump, C.; Sundquist, J.; Sundquist, K. Preterm or early term birth and risk of attention-deficit/hyperactivity disorder: A national cohort and co-sibling study. Ann. Epidemiol. 2023, 86, 119–125.e4. [Google Scholar] [CrossRef]
- Montagna, A.; Karolis, V.; Batalle, D.; Counsell, S.; Rutherford, M.; Arulkumaran, S.; Happe, F.; Edwards, D.; Nosarti, C. ADHD symptoms and their neurodevelopmental correlates in children born very preterm. PLoS ONE 2020, 15, e0224343. [Google Scholar] [CrossRef]
- Doyle, L.W.; Anderson, P.J. Long-term outcomes of bronchopulmonary dysplasia. Semin. Fetal Neonatal Med. 2009, 14, 391–395. [Google Scholar] [CrossRef]
- Landry, J.S.; Chan, T.; Lands, L.; Menzies, D. Long-term impact of bronchopulmonary dysplasia on pulmonary function. Can. Respir. J. 2011, 18, 265–270. [Google Scholar] [CrossRef]
- Alvira, C.M. Aberrant Pulmonary Vascular Growth and Remodeling in Bronchopulmonary Dysplasia. Front. Med. 2016, 3, 21. [Google Scholar] [CrossRef]
- Ali, A.; Zambrano, R.; Duncan, M.R.; Chen, S.; Luo, S.; Yuan, H.; Chen, P.; Benny, M.; Schmidt, A.; Young, K.; et al. Hyperoxia-activated circulating extracellular vesicles induce lung and brain injury in neonatal rats. Sci. Rep. 2021, 11, 8791. [Google Scholar] [CrossRef]
- Dapaah-Siakwan, F.; Zambrano, R.; Luo, S.; Duncan, M.R.; Kerr, N.; Donda, K.; de Rivero Vaccari, J.P.; Keane, R.W.; Dietrich, W.D.; Benny, M.; et al. Caspase-1 Inhibition Attenuates Hyperoxia-induced Lung and Brain Injury in Neonatal Mice. Am. J. Respir. Cell Mol. Biol. 2019, 61, 341–354. [Google Scholar] [CrossRef]
- Kim, Y.E.; Park, W.S.; Sung, D.K.; Ahn, S.Y.; Sung, S.I.; Yoo, H.S.; Chang, Y.S. Intratracheal transplantation of mesenchymal stem cells simultaneously attenuates both lung and brain injuries in hyperoxic newborn rats. Pediatr. Res. 2016, 80, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Lithopoulos, M.A.; Strueby, L.; O’Reilly, M.; Zhong, S.; Mobius, M.A.; Eaton, F.; Fung, M.; Hurskainen, M.; Cyr-Depauw, C.; Suen, C.; et al. Pulmonary and Neurologic Effects of Mesenchymal Stromal Cell Extracellular Vesicles in a Multifactorial Lung Injury Model. Am. J. Respir. Crit. Care Med. 2022, 205, 1186–1201. [Google Scholar] [CrossRef] [PubMed]
- Lithopoulos, M.A.; Toussay, X.; Zhong, S.; Xu, L.; Mustafa, S.B.; Ouellette, J.; Freitas-Andrade, M.; Comin, C.H.; Bassam, H.A.; Baker, A.N.; et al. Neonatal hyperoxia in mice triggers long-term cognitive deficits via impairments in cerebrovascular function and neurogenesis. J. Clin. Investig. 2022, 132, e146095. [Google Scholar] [CrossRef]
- Pham, H.; Vottier, G.; Pansiot, J.; Duong-Quy, S.; Bollen, B.; Dalous, J.; Gallego, J.; Mercier, J.C.; Dinh-Xuan, A.T.; Bonnin, P.; et al. Inhaled NO prevents hyperoxia-induced white matter damage in neonatal rats. Exp. Neurol. 2014, 252, 114–123. [Google Scholar] [CrossRef]
- Delage, C.; Taib, T.; Mamma, C.; Lerouet, D.; Besson, V.C. Traumatic Brain Injury: An Age-Dependent View of Post-Traumatic Neuroinflammation and Its Treatment. Pharmaceutics 2021, 13, 1624. [Google Scholar] [CrossRef] [PubMed]
- Hirani, D.; Alvira, C.M.; Danopoulos, S.; Milla, C.; Donato, M.; Tian, L.; Mohr, J.; Dinger, K.; Vohlen, C.; Selle, J.; et al. Macrophage-derived IL-6 trans-signalling as a novel target in the pathogenesis of bronchopulmonary dysplasia. Eur. Respir. J. 2022, 59, 2002248. [Google Scholar] [CrossRef] [PubMed]
- Ladner-Keay, C.L.; Turner, R.J.; Edwards, R.A. Fluorescent Protein Visualization Immediately After Gel Electrophoresis Using an In-Gel Trichloroethanol Photoreaction with Tryptophan. Methods Mol. Biol. 2018, 1853, 179–190. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Semple, B.D.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 2013, 106–107, 1–16. [Google Scholar] [CrossRef]
- Kuhn, S.; Gritti, L.; Crooks, D.; Dombrowski, Y. Oligodendrocytes in Development, Myelin Generation and Beyond. Cells 2019, 8, 1424. [Google Scholar] [CrossRef]
- Kent, S.A.; Miron, V.E. Microglia regulation of central nervous system myelin health and regeneration. Nat. Rev. Immunol. 2024, 24, 49–63. [Google Scholar] [CrossRef]
- Thebaud, B.; Abman, S.H. Bronchopulmonary dysplasia: Where have all the vessels gone? Roles of angiogenic growth factors in chronic lung disease. Am. J. Respir. Crit. Care Med. 2007, 175, 978–985. [Google Scholar] [CrossRef]
- Hsia, C.C.; Hyde, D.M.; Ochs, M.; Weibel, E.R. An official research policy statement of the American Thoracic Society/European Respiratory Society: Standards for quantitative assessment of lung structure. Am. J. Respir. Crit. Care Med. 2010, 181, 394–418. [Google Scholar] [CrossRef] [PubMed]
- Solinc, J.; Ribot, J.; Soubrier, F.; Pavoine, C.; Dierick, F.; Nadaud, S. The Platelet-Derived Growth Factor Pathway in Pulmonary Arterial Hypertension: Still an Interesting Target? Life 2022, 12, 658. [Google Scholar] [CrossRef]
- Humayun, J.; Lofqvist, C.; Ley, D.; Hellstrom, A.; Gyllensten, H. Systematic review of the healthcare cost of bronchopulmonary dysplasia. BMJ Open 2021, 11, e045729. [Google Scholar] [CrossRef]
- Agut, T.; Alarcon, A.; Cabañas, F.; Bartocci, M. Preterm white matter injury: Ultrasound diagnosis and classification. Pediatr. Res. 2020, 87 (Suppl. S1), 37–49. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, T.; Ritter, J.; Mueller, S.; Felderhoff-Mueser, U.; Chew, L.J.; Gallo, V. Cellular changes underlying hyperoxia-induced delay of white matter development. J. Neurosci. 2011, 31, 4327–4344. [Google Scholar] [CrossRef] [PubMed]
- Hoeber, D.; Sifringer, M.; van de Looij, Y.; Herz, J.; Sizonenko, S.V.; Kempe, K.; Serdar, M.; Palasz, J.; Hadamitzky, M.; Endesfelder, S.; et al. Erythropoietin Restores Long-Term Neurocognitive Function Involving Mechanisms of Neuronal Plasticity in a Model of Hyperoxia-Induced Preterm Brain Injury. Oxid. Med. Cell. Longev. 2016, 2016, 9247493. [Google Scholar] [CrossRef]
- Schmitz, T.; Krabbe, G.; Weikert, G.; Scheuer, T.; Matheus, F.; Wang, Y.; Mueller, S.; Kettenmann, H.; Matyash, V.; Buhrer, C.; et al. Minocycline protects the immature white matter against hyperoxia. Exp. Neurol. 2014, 254, 153–165. [Google Scholar] [CrossRef]
- Hagemeyer, N.; Hanft, K.M.; Akriditou, M.A.; Unger, N.; Park, E.S.; Stanley, E.R.; Staszewski, O.; Dimou, L.; Prinz, M. Microglia contribute to normal myelinogenesis and to oligodendrocyte progenitor maintenance during adulthood. Acta Neuropathol. 2017, 134, 441–458. [Google Scholar] [CrossRef]
- McNamara, N.B.; Munro, D.A.D.; Bestard-Cuche, N.; Uyeda, A.; Bogie, J.F.J.; Hoffmann, A.; Holloway, R.K.; Molina-Gonzalez, I.; Askew, K.E.; Mitchell, S.; et al. Microglia regulate central nervous system myelin growth and integrity. Nature 2023, 613, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Vottier, G.; Pham, H.; Pansiot, J.; Biran, V.; Gressens, P.; Charriaut-Marlangue, C.; Baud, O. Deleterious effect of hyperoxia at birth on white matter damage in the newborn rat. Dev. Neurosci. 2011, 33, 261–269. [Google Scholar] [CrossRef]
- Mehl, L.C.; Manjally, A.V.; Bouadi, O.; Gibson, E.M.; Tay, T.L. Microglia in brain development and regeneration. Development 2022, 149, dev200425. [Google Scholar] [CrossRef]
- Lier, J.; Streit, W.J.; Bechmann, I. Beyond Activation: Characterizing Microglial Functional Phenotypes. Cells 2021, 10, 2236. [Google Scholar] [CrossRef]
- Yeo, H.G.; Hong, J.J.; Lee, Y.; Yi, K.S.; Jeon, C.Y.; Park, J.; Won, J.; Seo, J.; Ahn, Y.J.; Kim, K.; et al. Increased CD68/TGFbeta Co-expressing Microglia/Macrophages after Transient Middle Cerebral Artery Occlusion in Rhesus Monkeys. Exp. Neurobiol. 2019, 28, 458–473. [Google Scholar] [CrossRef] [PubMed]
- Coelho-Santos, V.; Shih, A.Y. Postnatal development of cerebrovascular structure and the neurogliovascular unit. Wiley Interdiscip. Rev. Dev. Biol. 2020, 9, e363. [Google Scholar] [CrossRef]
- Sirinyan, M.; Sennlaub, F.; Dorfman, A.; Sapieha, P.; Gobeil, F., Jr.; Hardy, P.; Lachapelle, P.; Chemtob, S. Hyperoxic exposure leads to nitrative stress and ensuing microvascular degeneration and diminished brain mass and function in the immature subject. Stroke 2006, 37, 2807–2815. [Google Scholar] [CrossRef] [PubMed]
- Morken, T.S.; Nyman, A.K.; Sandvig, I.; Torp, S.H.; Skranes, J.; Goa, P.E.; Brubakk, A.M.; Wideroe, M. Brain development after neonatal intermittent hyperoxia-hypoxia in the rat studied by longitudinal MRI and immunohistochemistry. PLoS ONE 2013, 8, e84109. [Google Scholar] [CrossRef]
- Hellstrom, A.; Smith, L.E.; Dammann, O. Retinopathy of prematurity. Lancet 2013, 382, 1445–1457. [Google Scholar] [CrossRef]
- Nakano, A.; Kondo, R.; Kaneko, Y.; Arima, S.; Asano, D.; Morita, A.; Sakamoto, K.; Nagamitsu, T.; Nakahara, T. Changes in components of the neurovascular unit in the retina in a rat model of retinopathy of prematurity. Cell Tissue Res. 2020, 379, 473–486. [Google Scholar] [CrossRef]
- Shaffer, S.G.; O’Neill, D.; Bradt, S.K.; Thibeault, D.W. Chronic vascular pulmonary dysplasia associated with neonatal hyperoxia exposure in the rat. Pediatr. Res. 1987, 21, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, H.; Sugiura, T.; Streisand, J.B.; Lonning, S.M.; Roberts, J.D., Jr. TGF-beta-neutralizing antibodies improve pulmonary alveologenesis and vasculogenesis in the injured newborn lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L151–L161. [Google Scholar] [CrossRef] [PubMed]
- Thebaud, B.; Ladha, F.; Michelakis, E.D.; Sawicka, M.; Thurston, G.; Eaton, F.; Hashimoto, K.; Harry, G.; Haromy, A.; Korbutt, G.; et al. Vascular endothelial growth factor gene therapy increases survival, promotes lung angiogenesis, and prevents alveolar damage in hyperoxia-induced lung injury: Evidence that angiogenesis participates in alveolarization. Circulation 2005, 112, 2477–2486. [Google Scholar] [CrossRef] [PubMed]
- Alphonse, R.S.; Vadivel, A.; Fung, M.; Shelley, W.C.; Critser, P.J.; Ionescu, L.; O’Reilly, M.; Ohls, R.K.; McConaghy, S.; Eaton, F.; et al. Existence, functional impairment, and lung repair potential of endothelial colony-forming cells in oxygen-induced arrested alveolar growth. Circulation 2014, 129, 2144–2157. [Google Scholar] [CrossRef]
- Wilson, W.L.; Mullen, M.; Olley, P.M.; Rabinovitch, M. Hyperoxia-induced pulmonary vascular and lung abnormalities in young rats and potential for recovery. Pediatr. Res. 1985, 19, 1059–1067. [Google Scholar] [CrossRef]
- Jakkula, M.; Le Cras, T.D.; Gebb, S.; Hirth, K.P.; Tuder, R.M.; Voelkel, N.F.; Abman, S.H. Inhibition of angiogenesis decreases alveolarization in the developing rat lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L600–L607. [Google Scholar] [CrossRef]
- Kato, K.; Dieguez-Hurtado, R.; Park, D.Y.; Hong, S.P.; Kato-Azuma, S.; Adams, S.; Stehling, M.; Trappmann, B.; Wrana, J.L.; Koh, G.Y.; et al. Pulmonary pericytes regulate lung morphogenesis. Nat. Commun. 2018, 9, 2448. [Google Scholar] [CrossRef]
- Popova, A.P.; Bentley, J.K.; Cui, T.X.; Richardson, M.N.; Linn, M.J.; Lei, J.; Chen, Q.; Goldsmith, A.M.; Pryhuber, G.S.; Hershenson, M.B. Reduced platelet-derived growth factor receptor expression is a primary feature of human bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L231–L239. [Google Scholar] [CrossRef]
- Oak, P.; Pritzke, T.; Thiel, I.; Koschlig, M.; Mous, D.S.; Windhorst, A.; Jain, N.; Eickelberg, O.; Foerster, K.; Schulze, A.; et al. Attenuated PDGF signaling drives alveolar and microvascular defects in neonatal chronic lung disease. EMBO Mol. Med. 2017, 9, 1504–1520. [Google Scholar] [CrossRef]
- Wagenaar, G.T.M.; Laghmani, E.H.; de Visser, Y.P.; Sengers, R.M.A.; Steendijk, P.; Baelde, H.J.; Walther, F.J.; Silva, D.M.G.; Nardiello, C.; Pozarska, A.; et al. Ambrisentan reduces pulmonary arterial hypertension but does not stimulate alveolar and vascular development in neonatal rats with hyperoxic lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L264–L275. [Google Scholar] [CrossRef]
- Hirani, D.V.; Thielen, F.; Mansouri, S.; Danopoulos, S.; Vohlen, C.; Haznedar-Karakaya, P.; Mohr, J.; Wilke, R.; Selle, J.; Grosch, T.; et al. CXCL10 deficiency limits macrophage infiltration, preserves lung matrix, and enables lung growth in bronchopulmonary dysplasia. Inflamm. Regen. 2023, 43, 52. [Google Scholar] [CrossRef] [PubMed]
- Heydarian, M.; Schulz, C.; Stoeger, T.; Hilgendorff, A. Association of immune cell recruitment and BPD development. Mol. Cell. Pediatr. 2022, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Michael, Z.; Spyropoulos, F.; Ghanta, S.; Christou, H. Bronchopulmonary Dysplasia: An Update of Current Pharmacologic Therapies and New Approaches. Clin. Med. Insights Pediatr. 2018, 12, 1179556518817322. [Google Scholar] [CrossRef] [PubMed]
- Ophelders, D.; Gussenhoven, R.; Klein, L.; Jellema, R.K.; Westerlaken, R.J.J.; Hutten, M.C.; Vermeulen, J.; Wassink, G.; Gunn, A.J.; Wolfs, T. Preterm Brain Injury, Antenatal Triggers, and Therapeutics: Timing Is Key. Cells 2020, 9, 1871. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Obst, S.; Serdar, M.; Herz, J.; Kempe, K.; Assili, M.; Rizazad, M.; Hirani, D.; Alejandre Alcazar, M.A.; Endesfelder, S.; Möbius, M.A.; et al. A Novel Model for Simultaneous Evaluation of Hyperoxia-Mediated Brain and Lung Injury in Neonatal Rats. Cells 2025, 14, 443. https://doi.org/10.3390/cells14060443
Obst S, Serdar M, Herz J, Kempe K, Assili M, Rizazad M, Hirani D, Alejandre Alcazar MA, Endesfelder S, Möbius MA, et al. A Novel Model for Simultaneous Evaluation of Hyperoxia-Mediated Brain and Lung Injury in Neonatal Rats. Cells. 2025; 14(6):443. https://doi.org/10.3390/cells14060443
Chicago/Turabian StyleObst, Stefanie, Meray Serdar, Josephine Herz, Karina Kempe, Meriem Assili, Mandana Rizazad, Dharmesh Hirani, Miguel A. Alejandre Alcazar, Stefanie Endesfelder, Marius A. Möbius, and et al. 2025. "A Novel Model for Simultaneous Evaluation of Hyperoxia-Mediated Brain and Lung Injury in Neonatal Rats" Cells 14, no. 6: 443. https://doi.org/10.3390/cells14060443
APA StyleObst, S., Serdar, M., Herz, J., Kempe, K., Assili, M., Rizazad, M., Hirani, D., Alejandre Alcazar, M. A., Endesfelder, S., Möbius, M. A., Rüdiger, M., Felderhoff-Müser, U., & Bendix, I. (2025). A Novel Model for Simultaneous Evaluation of Hyperoxia-Mediated Brain and Lung Injury in Neonatal Rats. Cells, 14(6), 443. https://doi.org/10.3390/cells14060443