
Hypothermia Shifts Neurodegeneration Phenotype in Neonatal Human Hypoxic–Ischemic Encephalopathy but Not in Related Piglet Models: Possible Relationship to Toxic Conformer and Intrinsically Disordered Prion-like Protein Accumulation

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Human Autopsy Brain Samples
2.2. Neonatal Piglet Brain Injury and Survival Models
2.3. Piglet Brain Harvesting
2.4. Human iPS Cell- and Embryonic Stem Cell-Derived Neural Cell Models of QA Excitotoxicity and TCP Proteinopathy
2.5. Western Blotting
2.6. IHC and Immunofluorescence
2.7. Quantification of H&E Neuropathology
2.8. Quantification of Proteinopathy in IHC Sections
2.9. Statistical Analysis
3. Results
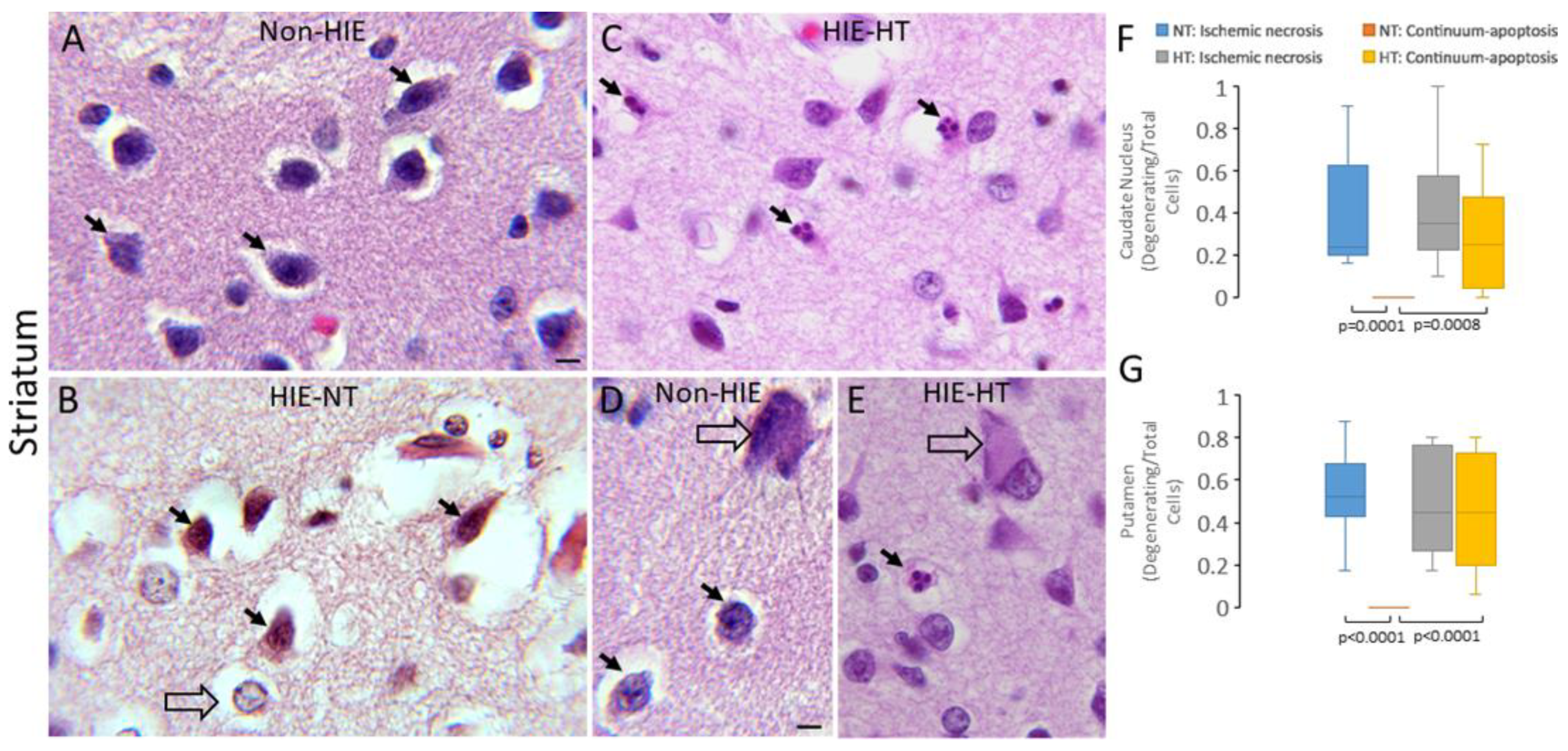
3.1. HT Shifts Neuronal Cell Death Pattern in Human HIE
3.2. HT Robustly Protects Cortical and Subcortical Neurons in Neonatal Piglet HI
3.3. Synucleinopathy Occurs Rapidly in Excitotoxically Injured Human Oligodendrocytes and Neurons in Cell Culture
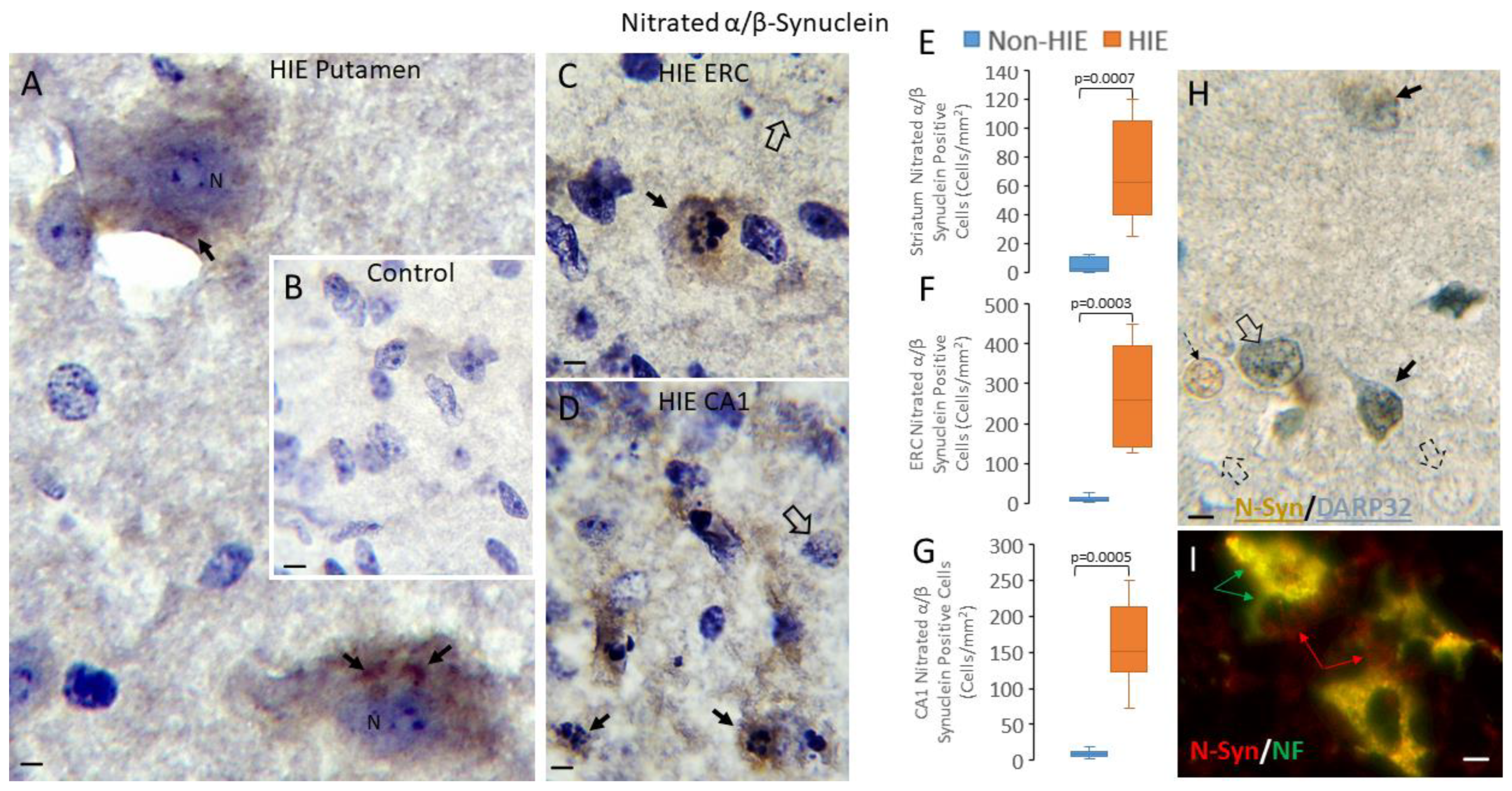
3.4. Synucleinopathy Occurs in Human Neonatal HIE Brain
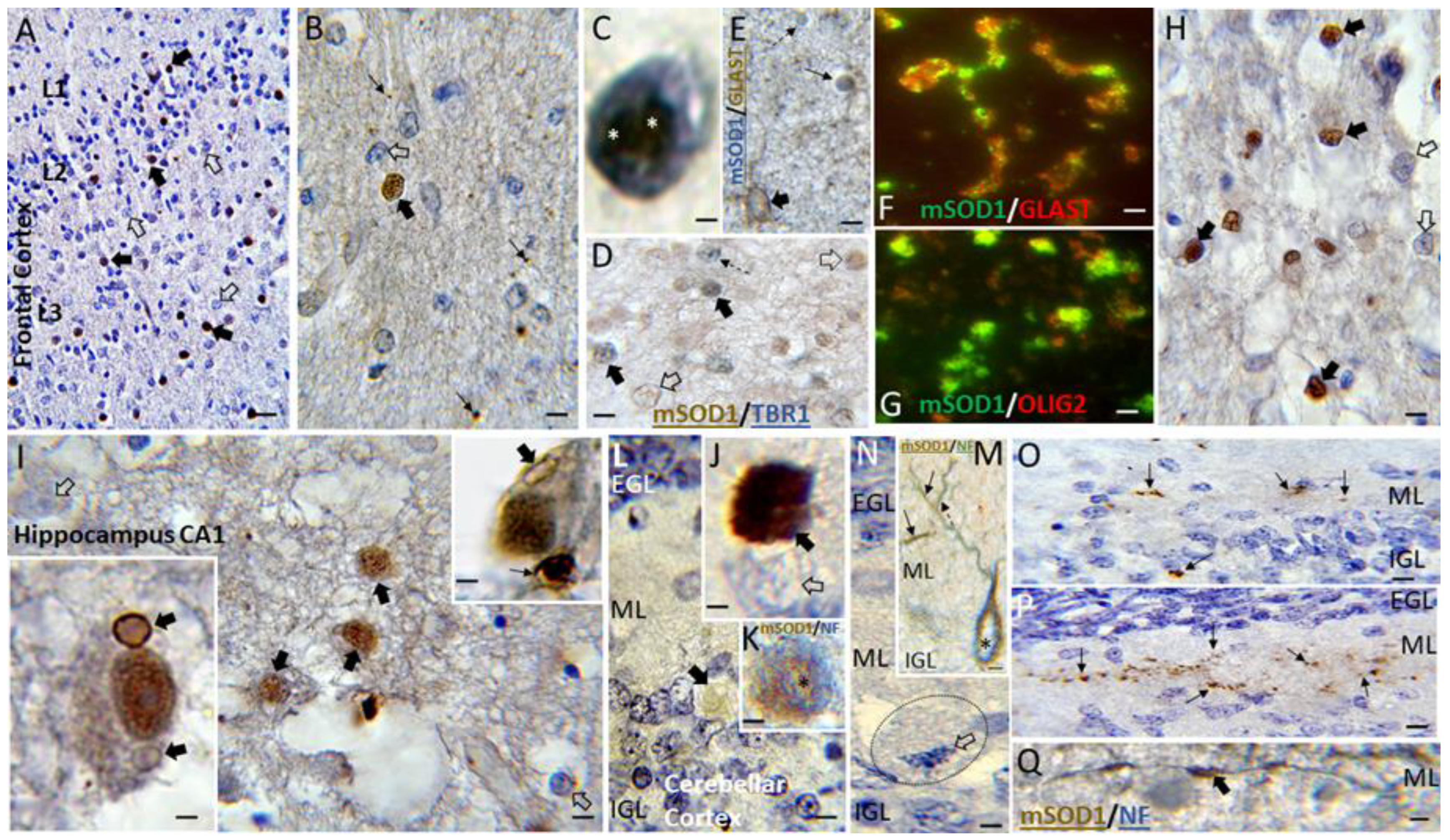
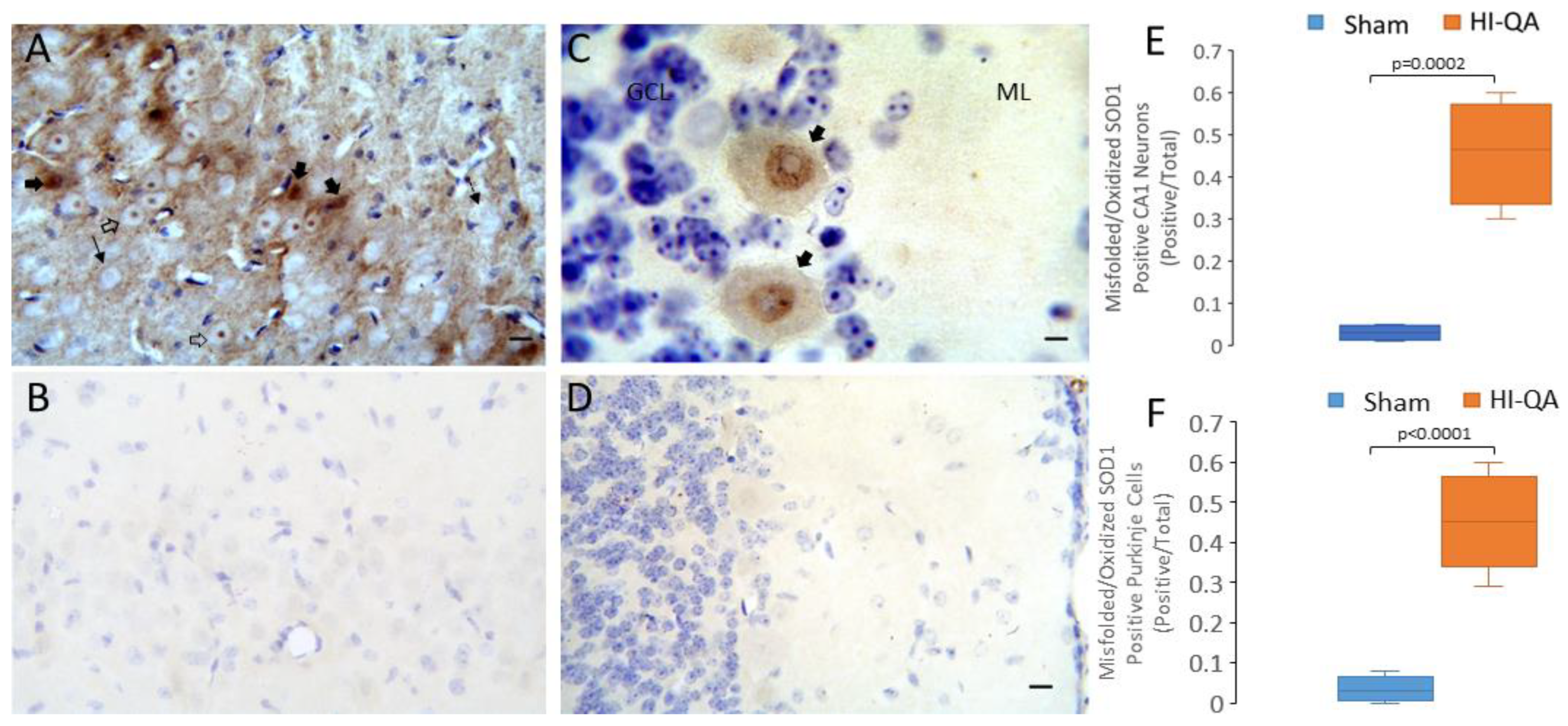
3.5. Putative Toxic Forms of SOD1 Accumulate in Human Neonatal HIE Brain
3.6. Synucleinopathy in Brain Occurs Acutely in Piglet HIE and Is Presynaptically Localized
3.7. Putative Toxic Forms of SOD1 Accumulate in Piglet HIE Brain
3.8. PrPopathy, Including Accumulation Proteinase K-Resistant PrP Immunoreactivity, Occurs in Neonatal Piglet Encephalopathies
4. Discussion
4.1. Experimental Design Considerations
4.2. HT Shifts Neurodegeneration Type in Human HIE
4.3. HT Does Not Shift Neurodegeneration Type in Piglet HI Models
4.4. Aberrant, Putatively Toxic, Proteins Can Accumulate Rapidly in Neonatal HIE and Piglet HI
4.5. Pathology in PrP, the Iconic TCP That Can Mediate Trans-Synaptic Spreading of Disease, Is Seen in Neonatal Brain HI
4.6. Proteinopathy as a Possible Mediator of Neurodegeneration and Delayed Trans-Synaptic Network Selective Vulnerability in Neonatal HIE
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oza, S.; Lawn, J.E.; Hogan, D.R.; Mathers, C.; Cousens, S.N. Neonatal cause-of-death estimates for the early and late neonatal periods for 194 countries: 2000–2013. Bull. World Health Organ. 2015, 93, 19–28. [Google Scholar] [CrossRef]
- Barkovich, A.J. MR and CT evaluation of profound neonatal and infantile asphyxia. Am. J. Neuroradiol. 1992, 13, 959–972. [Google Scholar]
- Miller, S.P.; Ramaswamy, V.; Michelson, D.; Barkovich, A.J.; Holshouser, B.; Wycliffe, N.; Glidden, D.V.; Deming, D.; Partridge, J.C.; Wu, Y.W.; et al. Patterns of brain injury in term neonatal encephalopathy. J. Pediatr. 2005, 146, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Roland, E.H.; Poskitt, K.; Rodriguez, E.; Lupton, B.A.; Hill, A. Perinatal hypoxic-ischemic thalamic injury: Clinical features and neuroimaging. Ann. Neurol. 1998, 44, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, M.A. The asphyxiated term infant. In MRI of the Neonatal Brain; WB Saunders: London, UK, 2002; pp. 99–128. [Google Scholar]
- Weeke, L.C.; Groenendaal, F.; Mudigonda, K.; Blennow, M.; Lequin, M.H.; Meiners, L.C.; van Haastert, I.C.; Benders, M.J.; Hallberg, B.; de Vries, L.S. A novel magnetic resonance imaging score predicts neurodevelopmental outcome after perinatal asphyxia and therapeutic hypothermia. J. Pediatr. 2018, 192, 33–40.e2. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Brambrink, A.; Koehler, R.C.; Traystman, R.J. Primary sensory and forebrain motor systems in the newborn brain are preferentially damaged by hypoxia-ischemia. J. Comp. Neurol. 1997, 377, 262–285. [Google Scholar] [CrossRef]
- Jacobs, S.E.; Berg, M.; Hunt, R.; Tarnow-Mordi, W.O.; Inder, T.E.; Davis, P.G. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst. Rev. 2013, 2013, CD003311. [Google Scholar] [CrossRef]
- Shankaran, S.; Laptook, A.R.; Ehrenkranz, R.A.; Tyson, J.E.; McDonald, S.A.; Donovan, E.F.; Fanaroff, A.A.; Poole, W.K.; Wright, L.L.; Higgins, R.D.; et al. Whole-body hypothermia for neonates with hypoxic–ischemic encephalopathy. N. Engl. J. Med. 2005, 353, 1574–1584. [Google Scholar] [CrossRef]
- Natarajan, G.; Pappas, A.; Shankaran, S. Outcomes in childhood following therapeutic hypothermia for neonatal hypoxic-ischemic encephalopathy (HIE). Semin. Perinatol. 2016, 40, 549–555. [Google Scholar] [CrossRef]
- Edwards, A.D.; Brocklehurst, P.; Gunn, A.J.; Halliday, H.; Juszczak, E.; Levene, M.; Strohm, B.; Thoresen, M.; Whitelaw, A. Neurological outcomes at 18 months of age after moderate hypothermia for perinatal hypoxic ischaemic encephalopathy: Synthesis and meta-analysis of trial data. BMJ 2010, 340, c363. [Google Scholar] [CrossRef]
- Higgins, R.D.; Raju, T.; Edwards, A.D.; Azzopardi, D.V.; Bose, C.L.; Clark, R.H.; Ferriero, D.M.; Guillet, R.; Gunn, A.J.; Hagberg, H.; et al. Hypothermia and other treatment options for neonatal encephalopathy: An executive summary of the Eunice Kennedy Shriver NICHD workshop. J. Pediatr. 2011, 159, 851–858.e1. [Google Scholar] [CrossRef] [PubMed]
- Laptook, A.R.; Corbett, R.J.; Sterett, R.; Burns, D.K.; Tollefsbol, G.; Garcia, D. Modest hypothermia provides partial neuroprotection for ischemic neonatal brain. Pediatr. Res. 1994, 35 Pt 1, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Towfighi, J.; Housman, C.; Heitjan, D.F.; Vannucci, R.C.; Yager, J.Y. The effect of focal cerebral cooling on perinatal hypoxic-ischemic brain damage. Acta Neuropathol. 1994, 87, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Haaland, K.; Løberg, E.M.; Steen, P.A.; Thoresen, M. Posthypoxic hypothermia in newborn piglets. Pediatr. Res. 1997, 41 Pt 1, 505–512. [Google Scholar] [CrossRef]
- Gunn, A.J.; Gunn, T.R.; de Haan, H.H.; Williams, C.E.; Gluckman, P.D. Dramatic neuronal rescue with prolonged selective head cooling after ischemia in fetal lambs. J. Clin. Investig. 1997, 99, 248–256. [Google Scholar] [CrossRef]
- Agnew, D.M.; Koehler, R.C.; Guerguerian, A.M.; Shaffner, D.H.; Traystman, R.J.; Martin, L.J.; Ichord, R.N. Hypothermia for 24 hours after asphyxic cardiac arrest in piglets provides striatal neuroprotection that is sustained 10 days after rewarming. Pediatr. Res. 2003, 54, 253–262. [Google Scholar] [CrossRef]
- Primiani, C.T.; Lee, J.K.; O’Brien, C.E.; Chen, M.W.; Perin, J.; Kulikowicz, E.; Santos, P.; Adams, S.; Lester, B.; Rivera-Diaz, N.; et al. Hypothermic protection in neocortex is topographic and laminar, seizure unmitigating, and partially rescues neurons depleted of RNA splicing protein Rbfox3/NeuN in neonatal hypoxic-ischemic male piglets. Cells 2023, 12, 2454. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Chang, Q. DNA damage response and repair, DNA methylation, and cell death in human neurons and experimental animal neurons are different. J. Neuropathol. Exp. Neurol. 2018, 77, 636–655. [Google Scholar] [CrossRef]
- Masoli, S.; Sanchez-Ponce, D.; Vrieler, N.; Abu-Haya, K.; Lerner, V.; Shahar, T.; Nedelescu, H.; Rizza, M.F.; Benavides-Piccione, R.; DeFelipe, J.; et al. Human Purkinje cells outperform mouse Purkinje cells in dendritic complexity and computational capacity. Commun. Biol. 2024, 7, 5. [Google Scholar] [CrossRef]
- Turner, M.J.; Dietz, R.M. Potential adjuncts to therapeutic hypothermia to mitigate multiorgan injury in perinatal hypoxia-ischemia. Neoreviews 2023, 24, e771–e782. [Google Scholar] [CrossRef]
- Zhang, K.; Sejnowski, T.J. A universal scaling law between gray matter and white matter of cerebral cortex. Proc. Natl. Acad. Sci. USA 2000, 97, 5621–5626. [Google Scholar] [CrossRef] [PubMed]
- Massaro, A.N.; Evangelou, I.; Fatemi, A.; Vezina, G.; Mccarter, R.; Glass, P.; Limperopoulos, C. White matter tract integrity and developmental outcome in newborn infants with hypoxic-ischemic encephalopathy treated with hypothermia. Dev. Med. Child Neurol. 2015, 57, 441–448. [Google Scholar] [CrossRef]
- O’Brien, C.E.; Santos, P.T.; Kulikowicz, E.; Reyes, M.; Koehler, R.C.; Martin, L.J.; Lee, J.K. Hypoxia-ischemia and hypothermia independently and interactively affect neuronal pathology in neonatal piglets with short-term recovery. Dev. Neurosci. 2019, 41, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Arnautovic, T.; Sinha, S.; Laptook, A.R. Neonatal hypoxic-ischemic encephalopathy and hypothermia treatment. Obstet. Gynecol. 2024, 143, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Ferriero, D.M. Neonatal brain injury. N. Engl. J. Med. 2004, 351, 1985–1995. [Google Scholar] [CrossRef]
- Martin, L.J. Neuronal cell death in nervous system development, disease, and injury. Int. J. Mol. Med. 2001, 7, 455–478. [Google Scholar] [CrossRef]
- Raffray, M.; Cohen, G.M. Apoptosis and necrosis in toxicology: A continuum or distinct modes of cell death? Pharmacol. Ther. 1997, 75, 153–177. [Google Scholar] [CrossRef]
- Martin, L.J.; Brambrink, A.M.; Price, A.C.; Kaiser, A.; Agnew, D.M.; Ichord, R.N.; Traystman, R.J. Neuronal death in newborn striatum after hypoxia-ischemia is necrosis and evolves with oxidative stress. Neurobiol. Dis. 2000, 7, 169–191. [Google Scholar] [CrossRef]
- Northington, F.J.; Zelaya, M.E.; O’Riordan, D.P.; Blomgren, K.; Flock, D.L.; Hagberg, H.; Ferriero, D.M.; Martin, L.J. Failure to complete apoptosis following neonatal hypoxia-ischemia manifests as “continuum” phenotype of cell death and occurs with multiple manifestations of mitochondrial dysfunction in rodent forebrain. Neuroscience 2007, 149, 822–833. [Google Scholar] [CrossRef]
- Martin, L.J. Mitochondrial and cell death mechanisms in neurodegenerative diseases. Pharmaceuticals 2010, 3, 839–915. [Google Scholar] [CrossRef]
- El Demerdash, N.; Chen, M.W.; O’Brien, C.E.; Adams, S.; Kulikowicz, E.; Martin, L.J.; Lee, J.K. Oleuropein activates neonatal neocortical proteasomes, but proteasome gene targeting by AAV9 is variable in a clinically relevant piglet model of brain hypoxia-ischemia and hypothermia. Cells 2021, 10, 2120. [Google Scholar] [CrossRef] [PubMed]
- Brotherton, T.E.; Li, Y.; Cooper, D.; Gearing, M.; Julien, J.P.; Rothstein, J.D.; Boylan, K.; Glass, J.D. Localization of a toxic form of superoxide dismutase 1 protein to pathologically affected tissues in familial ALS. Proc. Natl. Acad. Sci. USA 2012, 109, 5505–5510. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Qiang, M.; Wei, Y.; He, R. A novel molecular mechanism for nitrated {alpha}-synuclein-induced cell death. J. Mol. Cell Biol. 2011, 3, 239–249. [Google Scholar] [CrossRef]
- Khalilov, R.A.; Dzhafarova, A.M.; Khizrieva, S.I.; Abdullaev, V.R. Thermostability of lactate dehydrogenase in rat brain under conditions of short-term moderate hypothermia. Bull. Exp. Biol. Med. 2020, 168, 326–329. [Google Scholar] [CrossRef]
- Ou, J.; Ball, J.M.; Luan, Y.; Zhao, T.; Miyagishima, K.J.; Xu, Y.; Zhou, H.; Chen, J.; Merriman, D.K.; Xie, Z.; et al. iPSCs from a hibernator provide a platform for studying cold adaptation and its potential medical applications. Cell 2018, 173, 851–863.e16. [Google Scholar] [CrossRef] [PubMed]
- Roilo, M.; Kullmann, M.K.; Hengst, L. Cold-inducible RNA-binding protein (CIRP) induces translation of the cell-cycle inhibitor p27Kip1. Nucleic Acids Res. 2018, 46, 3198–3210. [Google Scholar] [CrossRef]
- Roobol, A.; Carden, M.J.; Newsam, R.J.; Smales, C.M. Biochemical insights into the mechanisms central to the response of mammalian cells to cold stress and subsequent rewarming. FEBS J. 2009, 276, 286–302. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Braak, H.; Brettschneider, J.; Ludolph, A.C.; Lee, V.M.; Trojanowski, J.Q.; Del Tredici, K. Amyotrophic lateral sclerosis—A model of corticofugal axonal spread. Nat. Rev. Neurol. 2013, 9, 708–714. [Google Scholar] [CrossRef]
- Woerman, A.L.; Watts, J.C.; Aoyagi, A.; Giles, K.; Middleton, L.T.; Prusiner, S.B. α-Synuclein: Multiple system atrophy prions. Cold Spring Harb. Perspect. Med. 2018, 8, a024588. [Google Scholar] [CrossRef]
- Abounit, S.; Wu, J.W.; Duff, K.; Victoria, G.S.; Zurzolo, C. Tunneling nanotubes: A possible highway in the spreading of tau and other prion-like proteins in neurodegenerative diseases. Prion 2016, 10, 344–351. [Google Scholar] [CrossRef]
- Ayers, J.I.; Paras, N.A.; Prusiner, S.B. Expanding spectrum of prion diseases. Emerg. Top. Life Sci. 2020, 4, 155–167. [Google Scholar] [CrossRef]
- Furukawa, Y.; Tokuda, E. Does wild-type Cu/Zn-superoxide dismutase have pathogenic roles in amyotrophic lateral sclerosis? Transl. Neurodegener. 2020, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- Kovač, V.; Čurin Šerbec, V. Prion protein: The molecule of many forms and faces. Int. J. Mol. Sci. 2022, 23, 1232. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: Introducing the D2 concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef]
- Jones, C.L.; Tepe, J.J. Proteasome activation to combat proteotoxicity. Molecules 2019, 24, 2841. [Google Scholar] [CrossRef]
- Lambert-Smith, I.A.; Saunders, D.N.; Yerbury, J.J. Proteostasis impairment and ALS. Prog. Biophys. Mol. Biol. 2022, 174, 3–27. [Google Scholar] [CrossRef]
- Riemenschneider, H.; Guo, Q.; Bader, J.; Frottin, F.; Farny, D.; Kleinberger, G.; Haass, C.; Mann, M.; Hartl, F.U.; Baumeister, W.; et al. Gel-like inclusions of C-terminal fragments of TDP-43 sequester stalled proteasomes in neurons. EMBO Rep. 2022, 23, e53890. [Google Scholar] [CrossRef]
- Portera-Cailliau, C.; Price, D.L.; Martin, L.J. Excitotoxic neuronal death in the immature brain is an apoptosis-necrosis morphological continuum. J. Comp. Neurol. 1997, 378, 70–87. [Google Scholar] [CrossRef]
- Portera-Cailliau, C.; Price, D.L.; Martin, L.J. Non-NMDA and NMDA receptor-mediated excitotoxic neuronal deaths in adult brain are morphologically distinct: Further evidence for an apoptosis-necrosis continuum. J. Comp. Neurol. 1997, 378, 88–104. [Google Scholar] [CrossRef]
- Santos, P.T.; O’Brien, C.E.; Chen, M.W.; Hopkins, C.D.; Adams, S.; Kulikowicz, E.; Singh, R.; Koehler, R.C.; Martin, L.J.; Lee, J.K. Proteasome biology is compromised in white matter after asphyxic cardiac arrest in neonatal piglets. J. Am. Heart Assoc. 2018, 7, e009415. [Google Scholar] [CrossRef] [PubMed]
- Brás, I.C.; Xylaki, M.; Outeiro, T.F. Mechanisms of alpha-synuclein toxicity: An update and outlook. Prog. Brain Res. 2019, 252, 91–129. [Google Scholar] [CrossRef]
- Duda, J.E.; Giasson, B.I.; Chen, Q.; Gur, T.L.; Hurtig, H.I.; Stern, M.B.; Gollomp, S.M.; Ischiropoulos, H.; Lee, V.M.; Trojanowski, J.Q. Widespread nitration of pathological inclusions in neurodegenerative synucleinopathies. Am. J. Pathol. 2000, 157, 1439–1445. [Google Scholar] [CrossRef]
- Giasson, B.I.; Jakes, R.; Goedert, M.; Duda, J.E.; Leight, S.; Trojanowski, J.Q.; Lee, V.M. A panel of epitope-specific antibodies detects protein domains distributed throughout human alpha-synuclein in Lewy bodies of Parkinson’s disease. J. Neurosci. Res. 2000, 59, 528–533. [Google Scholar] [CrossRef]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 2000, 290, 985–989. [Google Scholar] [CrossRef]
- Martin, L.J. Neuronal death in amyotrophic lateral sclerosis is apoptosis: Possible contribution of a programmed cell death mechanism. J. Neuropathol. Exp. Neurol. 1999, 58, 459–471. [Google Scholar] [CrossRef]
- Martin, L.J.; Gertz, B.; Pan, Y.; Price, A.C.; Molkentin, J.D.; Chang, Q. The mitochondrial permeability transition pore in motor neurons: Involvement in the pathobiology of ALS mice. Exp. Neurol. 2009, 218, 333–346. [Google Scholar] [CrossRef]
- Martin, L.J.; Koh, S.J.; Price, A.; Park, D.; Kim, B.W. Nuclear localization of human SOD1 in motor neurons in mouse model and patient amyotrophic lateral sclerosis: Possible links to cholinergic phenotype, NADPH oxidase, oxidative stress, and DNA damage. Int. J. Mol. Sci. 2024, 25, 9106. [Google Scholar] [CrossRef]
- Chen, M.W.; Santos, P.; Kulikowicz, E.; Koehler, R.C.; Lee, J.K.; Martin, L.J. Targeting the mitochondrial permeability transition pore for neuroprotection in a piglet model of neonatal hypoxic-ischemic encephalopathy. J. Neurosci. Res. 2021, 99, 1550–1564. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Wang, B.; Reyes, M.; Armstrong, J.S.; Kulikowicz, E.; Santos, P.T.; Lee, J.H.; Koehler, R.C.; Martin, L.J. Hypothermia and rewarming activate a macroglial unfolded protein response independent of hypoxic-ischemic brain injury in neonatal piglets. Dev. Neurosci. 2016, 38, 277–294. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Santos, P.T.; Chen, M.W.; O’Brien, C.E.; Kulikowicz, E.; Adams, S.; Hardart, H.; Koehler, R.C.; Martin, L.J. Combining hypothermia and oleuropein subacutely protects subcortical white matter in a swine model of neonatal hypoxic-ischemic encephalopathy. J. Neuropathol. Exp. Neurol. 2021, 80, 182–198. [Google Scholar] [CrossRef]
- Salinas-Zeballos, M.E.; Zeballos, G.A.; Gootman, P.M. A stereotaxic atlas of the developing swine (Sus scrofa) forebrain. In Swine in Biomedical Research; Tumbleson, M.E., Ed.; Plenum Press: New York, NY, USA, 1986; Volume 2, pp. 887–906. [Google Scholar]
- Lee, J.K.; Liu, D.; Raven, E.P.; Jiang, D.; Liu, P.; Qin, Q.; Kulikowicz, E.; Santos, P.T.; Adams, S.; Zhang, J.; et al. Mean diffusivity in striatum correlates with acute neuronal death but not lesser neuronal injury in a pilot study of neonatal piglets with encephalopathy. J. Magn. Reson. Imaging 2020, 52, 1216–1226. [Google Scholar] [CrossRef]
- Amrein Almira, A.; Chen, M.W.; El Demerdash, N.; Javdan, C.; Park, D.; Lee, J.K.; Martin, L.J. Proteasome localization and activity in pig brain and in vivo small molecule screening for activators. Front. Cell. Neurosci. 2024, 18, 1353542. [Google Scholar] [CrossRef]
- Lee, J.K.; Liu, D.; Jiang, D.; Kulikowicz, E.; Tekes, A.; Liu, P.; Qin, Q.; Koehler, R.C.; Aggarwal, M.; Zhang, J.; et al. Fractional anisotropy from diffusion tensor imaging correlates with acute astrocyte and myelin swelling in neonatal swine models of excitotoxic and hypoxic-ischemic brain injury. J. Comp. Neurol. 2021, 529, 2750–2770. [Google Scholar] [CrossRef] [PubMed]
- Baxter, P.S.; Márkus, N.M.; Dando, O.; He, X.; Al-Mubarak, B.R.; Qiu, J.; Hardingham, G.E. Targeted de-repression of neuronal Nrf2 inhibits α-synuclein accumulation. Cell Death Dis. 2021, 12, 218. [Google Scholar] [CrossRef]
- Ruesink, H.; Reimer, L.; Gregersen, E.; Moellerm, A.; Betzer, C.; Jensen, P.H. Stabilization of α-synuclein oligomers using formaldehyde. PLoS ONE 2019, 14, e0216764. [Google Scholar] [CrossRef]
- Kovacs, G.G.; Wagner, U.; Dumont, B.; Pikkarainen, M.; Osman, A.A.; Streichenberger, N.; Leisser, I.; Verchère, J.; Baron, T.; Alafuzoff, I.; et al. An antibody with high reactivity for disease-associated α-synuclein reveals extensive brain pathology. Acta Neuropathol. 2012, 124, 37–50. [Google Scholar] [CrossRef]
- Kumar, S.T.; Jagannath, S.; Francois, C.; Vanderstichele, H.; Stoops, E.; Lashuel, H.A. How specific are the conformation-specific α-synuclein antibodies? Characterization and validation of 16 α-synuclein conformation-specific antibodies using well-characterized preparations of α-synuclein monomers, fibrils and oligomers with distinct structures and morphology. Neurobiol. Dis. 2020, 146, 105086. [Google Scholar] [CrossRef]
- Sengupta, U.; Guerrero-Muñoz, M.J.; Castillo-Carranza, D.L.; Lasagna-Reeves, C.A.; Gerson, J.E.; Paulucci-Holthauzen, A.A.; Krishnamurthy, S.; Farhed, M.; Jackson, G.R.; Kayed, R. Pathological interface between oligomeric alpha-synuclein and tau in synucleinopathies. Biol. Psychiatry 2015, 78, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Urushitani, M.; Ezzi, S.A.; Julien, J.P. Therapeutic effects of immunization with mutant superoxide dismutase in mice models of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2007, 104, 2495–2500. [Google Scholar] [CrossRef] [PubMed]
- Pickles, S.; Semmler, S.; Broom, H.R.; Destroismaisons, L.; Legroux, L.; Arbour, N.; Meiering, E.; Cashman, N.R.; Vande Velde, C. ALS-linked misfolded SOD1 species have divergent impacts on mitochondria. Acta Neuropathol. Commun. 2016, 4, 43. [Google Scholar] [CrossRef]
- O’Rourke, K.I.; Baszler, T.V.; Miller, J.M.; Spraker, T.R.; Sadler-Riggleman, I.; Knowles, D.P. Monoclonal antibody F89/160.1.5 defines a conserved epitope on the ruminant prion protein. J. Clin. Microbiol. 1998, 36, 1750–1755. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.R.; Chaiwun, B.; Young, L.; Cote, R.J.; Taylor, C.R. Antigen retrieval technique utilizing citrate buffer or urea solution for immunohistochemical demonstration of androgen receptor in formalin-fixed paraffin sections. J. Histochem. Cytochem. 1993, 41, 1599–1604. [Google Scholar] [CrossRef]
- Kitamoto, T.; Ogomori, K.; Tateishi, J.; Prusiner, S.B. Formic acid pretreatment enhances immunostaining of cerebral and systemic amyloids. Lab. Investig. 1987, 57, 230–236. [Google Scholar]
- Imberdis, T.; Ayrolles-Torro, A.; Duarte Rodrigues, A.; Torrent, J.; Alvarez-Martinez, M.T.; Kovacs, G.G.; Verdier, J.M.; Robitzer, M.; Perrier, V.A. Fluorescent Oligothiophene-Bis-Triazine ligand interacts with PrP fibrils and detects SDS-resistant oligomers in human prion diseases. Mol. Neurodegener. 2016, 11, 11. [Google Scholar] [CrossRef]
- Russelakis-Carneiro, M.; Saborio, G.P.; Anderes, L.; Soto, C. Changes in the glycosylation pattern of prion protein in murine scrapie. Implications for the mechanism of neurodegeneration in prion diseases. J. Biol. Chem. 2002, 277, 36872–36877. [Google Scholar] [CrossRef]
- Martin, L.J.; Adams, D.A.; Niedzwiecki, M.V.; Wong, M. Aberrant DNA and RNA methylation occur in spinal cord and skeletal muscle of human SOD1 mouse models of ALS and in human ALS: Targeting DNA methylation is therapeutic. Cells 2022, 11, 3448. [Google Scholar] [CrossRef]
- Martin, L.J.; Brambrink, A.M.; Lehmann, C.; Portera-Cailliau, C.; Koehler, R.; Rothstein, J.; Traystman, R.J. Hypoxia-ischemia causes abnormalities in glutamate transporters and death of astroglia and neurons in newborn striatum. Ann. Neurol. 1997, 42, 335–348. [Google Scholar] [CrossRef]
- Martin, L.J.; Al-Abdulla, N.A.; Brambrink, A.M.; Kirsch, J.R.; Sieber, F.E.; Portera-Cailliau, C. Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: A perspective on the contributions of apoptosis and necrosis. Brain Res. Bull. 1998, 46, 281–309. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuronal types in the striatum of man. Cell Tissue Res. 1982, 227, 319–342. [Google Scholar] [CrossRef]
- Lieberman, A.R. The axon reaction: A review of the principal features of perikaryal responses to axon injury. Int. Rev. Neurobiol. 1971, 14, 49–124. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Wang, H.W.; Fu, X.H.; Zhang, W.Q.; Wu, X.Y.; Guo, Q.Y.; Wang, X.M. Expression of N-methyl-d-aspartate receptor 1 and its phosphorylated state in basal ganglia of a neonatal piglet hypoxic-ischemic brain injury model: A controlled study of (1)H MRS. Eur. J. Paediatr. Neurol. 2012, 16, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Wong, M.; Hanaford, A. Neonatal brain injury and genetic causes of adult-onset neurodegenerative disease in mice interact with effects on acute and late outcomes. Front. Neurol. 2019, 10, 635. [Google Scholar] [CrossRef]
- Souza, J.M.; Giasson, B.I.; Chen, Q.; Lee, V.M.; Ischiropoulos, H. Dityrosine cross-linking promotes formation of stable alpha-synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J. Biol. Chem. 2000, 275, 18344–18349. [Google Scholar] [CrossRef]
- Cheung, H.H.; Teves, L.; Wallace, M.C.; Gurd, J.W. Increased phosphorylation of the NR1 subunit of the NMDA receptor following cerebral ischemia. J. Neurochem. 2001, 78, 1179–1182. [Google Scholar] [CrossRef]
- Liu, H.N.; Tjostheim, S.; Dasilva, K.; Taylor, D.; Zhao, B.; Rakhit, R.; Brown, M.; Chakrabartty, A.; McLaurin, J.; Robertson, J. Targeting of monomer/misfolded SOD1 as a therapeutic strategy for amyotrophic lateral sclerosis. J. Neurosci. 2012, 32, 8791–8799. [Google Scholar] [CrossRef]
- Beck, E.; Daniel, P.M.; Parry, H.B. Degeneration of the cerebellar and hypothalamoneurohypophysial systems in sheep with scrapie; and its relationship to human systems degenerations. Brain 1964, 87, 153–176. [Google Scholar] [CrossRef]
- Borchelt, D.R.; Koliatsos, V.E.; Guarnieri, M.; Pardo, C.A.; Sisodia, S.S.; Price, D.L. Rapid anterograde axonal transport of the cellular prion glycoprotein in the peripheral and central nervous systems. J. Biol. Chem. 1994, 269, 14711–14714. [Google Scholar] [CrossRef]
- Fraser, H. Neuronal spread of scrapie agent and targeting of lesions within the retino-tectal pathway. Nature 1982, 295, 149–150. [Google Scholar] [CrossRef]
- Ito, M. The neuronal mechanism of the cerebellar efferent system. Proc. Aust. Assoc. Neurol. 1968, 5, 13–18. [Google Scholar] [PubMed]
- Jansen, J.; Brodal, A. Experimental studies on the intrinsic fibers of the cerebellum II. The cortico-nuclear projection. Cerebellum 1940, 10, 126–180. [Google Scholar] [CrossRef]
- Johnston, M.V. Hypoxic and ischemic disorders of infants and children. Lecture for 38th Meeting of Japanese Society of Child Neurology, Tokyo, Japan, July 1996. Brain Dev. 1997, 19, 235–239. [Google Scholar] [CrossRef]
- Mueller-Burke, D.; Koehler, R.C.; Martin, L.J. Rapid NMDA receptor phosphorylation and oxidative stress precede striatal neurodegeneration after hypoxic ischemia in newborn piglets and are attenuated with hypothermia. Int. J. Dev. Neurosci. 2008, 26, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Tavassoly, O.; Sade, D.; Bera, S.; Shaham-Niv, S.; Vocadlo, D.J.; Gazit, E. Quinolinic acid amyloid-like fibrillar assemblies seed α-synuclein aggregation. J. Mol. Biol. 2018, 430, 3847–3862. [Google Scholar] [CrossRef] [PubMed]
- Friede, R.L. Ponto-subicular lesions in perinatal anoxia. Arch. Pathol. 1972, 94, 343–354. [Google Scholar]
- Janzer, R.C.; Friede, R.L. Hypotensive brain stem necrosis or cardiac arrest encephalopathy? Acta Neuropathol. 1980, 50, 53–56. [Google Scholar] [CrossRef]
- Nakamura, Y.; Nakashima, T.; Fukuda, S.; Nakashima, H.; Hashimoto, T. Hypoxic-ischemic brain lesions found in asphyxiating neonates. Acta Pathol. Jpn. 1986, 36, 551–563. [Google Scholar] [CrossRef]
- Schiering, I.A.; de Haan, T.R.; Niermeijer, J.M.; Koelman, J.H.; Majoie, C.B.; Reneman, L.; Aronica, E. Correlation between clinical and histologic findings in the human neonatal hippocampus after perinatal asphyxia. J. Neuropathol. Exp. Neurol. 2014, 73, 324–334. [Google Scholar] [CrossRef]
- Vlasyuk, V.V. Hypoxic-ischemic injuries of the brain. In Birth Trauma and Perinatal Brain Damage; Springer International Publishing: Berlin/Heidelberg, Germany, 2019; pp. 211–269. [Google Scholar] [CrossRef]
- Gorman, A.M. Neuronal cell death in neurodegenerative diseases: Recurring themes around protein handling. J. Cell. Mol. Med. 2008, 12, 2263–2280. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Green, D.R. Necroptosis. N. Engl. J. Med. 2014, 370, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.L.; Siesjö, B.K.; Hu, B.R. Pathogenesis of hippocampal neuronal death after hypoxia-ischemia changes during brain development. Neuroscience 2004, 127, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Lemyre, B.; Chau, V. Hypothermia for newborns with hypoxic-ischemic encephalopathy. Paediatr. Child Health 2018, 23, 285–291. [Google Scholar] [CrossRef]
- Griffiths, A.D.; Laurence, K.M. The effect of hypoxia and hypoglycaemia on the brain of the newborn human infant. Dev. Med. Child Neurol. 1974, 16, 308–319. [Google Scholar] [CrossRef]
- Hamburger, V.; Levi-Montalcini, R. Proliferation, differentiation and degeneration in the spinal ganglia of the chick embryo under normal and experimental conditions. J. Exp. Zool. 1949, 111, 457–501. [Google Scholar] [CrossRef]
- Pittman, R.N.; Wang, S.; DiBenedetto, A.J.; Mills, J.C. A system for characterizing cellular and molecular events in programmed neuronal cell death. J. Neurosci. 1993, 13, 3669–3680. [Google Scholar] [CrossRef]
- Sanders, E.J.; Wride, M.A. Programmed cell death in development. Int. Rev. Cytol. 1995, 163, 105–173. [Google Scholar] [CrossRef]
- Saunders, J.W., Jr. Death in embryonic systems. Science 1966, 154, 604–612. [Google Scholar] [CrossRef]
- Demarest, T.G.; Schuh, R.A.; Waddell, J.; McKenna, M.C.; Fiskum, G. Sex-dependent mitochondrial respiratory impairment and oxidative stress in a rat model of neonatal hypoxic-ischemic encephalopathy. J. Neurochem. 2016, 137, 714–729. [Google Scholar] [CrossRef]
- Bosco, D.A.; Morfini, G.; Karabacak, N.M.; Song, Y.; Gros-Louis, F.; Pasinelli, P.; Goolsby, H.; Fontaine, B.A.; Lemay, N.; McKenna-Yasek, D.; et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 2010, 13, 1396–1403. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.; Sengupta, U.; Puangmalai, N.; Bhatt, N.; Kayed, R. Polymorphic alpha-synuclein oligomers: Characterization and differential detection with novel corresponding antibodies. Mol. Neurobiol. 2023, 60, 2691–2705. [Google Scholar] [CrossRef] [PubMed]
- Emin, D.; Zhang, Y.P.; Lobanova, E.; Miller, A.; Li, X.; Xia, Z.; Dakin, H.; Sideris, D.I.; Lam, J.Y.L.; Ranasinghe, R.T.; et al. Small soluble α-synuclein aggregates are the toxic species in Parkinson’s disease. Nat. Commun. 2022, 13, 5512. [Google Scholar] [CrossRef]
- Zunke, F.; Moise, A.C.; Belur, N.R.; Gelyana, E.; Stojkovska, I.; Dzaferbegovic, H.; Toker, N.J.; Jeon, S.; Fredriksen, K.; Mazzulli, J.R. Reversible conformational conversion of α-synuclein into toxic assemblies by glucosylceramide. Neuron 2018, 97, 92–107.e10. [Google Scholar] [CrossRef] [PubMed]
- Eiserich, J.P.; Estévez, A.G.; Bamberg, T.V.; Ye, Y.Z.; Chumley, P.H.; Beckman, J.S.; Freeman, B.A. Microtubule dysfunction by posttranslational nitrotyrosination of alpha-tubulin: A nitric oxide-dependent mechanism of cellular injury. Proc. Natl. Acad. Sci. USA 1999, 96, 6365–6370. [Google Scholar] [CrossRef]
- Paré, B.; Lehmann, M.; Beaudin, M.; Nordström, U.; Saikali, S.; Julien, J.P.; Gilthorpe, J.D.; Marklund, S.L.; Cashman, N.R.; Andersen, P.M.; et al. Misfolded SOD1 pathology in sporadic amyotrophic lateral sclerosis. Sci. Rep. 2018, 8, 14223. [Google Scholar] [CrossRef]
- Kim, B.W.; Ryu, J.; Jeong, Y.E.; Kim, J.; Martin, L.J. Human motor neurons with SOD1-G93A mutation generated from CRISPR/Cas9 gene-edited iPSCs develop pathological features of amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2020, 14, 604171. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R.; Spillantini, M.G. The synucleinopathies: Twenty years on. J. Park. Dis. 2017, 7 (Suppl. S1), S51–S69. [Google Scholar] [CrossRef]
- Murphy, D.D.; Rueter, S.M.; Trojanowski, J.Q.; Lee, V.M. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 2000, 20, 3214–3220. [Google Scholar] [CrossRef]
- Norris, E.H.; Giasson, B.I.; Lee, V.M. Alpha-synuclein: Normal function and role in neurodegenerative diseases. Curr. Top. Dev. Biol. 2004, 60, 17–54. [Google Scholar] [CrossRef]
- Sang, J.C.; Lee, J.E.; Dear, A.J.; De, S.; Meisl, G.; Thackray, A.M.; Bujdoso, R.; Knowles, T.P.J.; Klenerman, D. Direct observation of prion protein oligomer formation reveals an aggregation mechanism with multiple conformationally distinct species. Chem. Sci. 2019, 10, 4588–4597. [Google Scholar] [CrossRef] [PubMed]
- Simoneau, S.; Rezaei, H.; Salès, N.; Kaiser-Schulz, G.; Lefebvre-Roque, M.; Vidal, C.; Fournier, J.G.; Comte, J.; Wopfner, F.; Grosclaude, J.; et al. In vitro and in vivo neurotoxicity of prion protein oligomers. PLoS Pathog. 2007, 3, e125. [Google Scholar] [CrossRef] [PubMed]
- Salès, N.; Rodolfo, K.; Hässig, R.; Faucheux, B.; Di Giamberardino, L.; Moya, K.L. Cellular prion protein localization in rodent and primate brain. Eur. J. Neurosci. 1998, 10, 2464–2471. [Google Scholar] [CrossRef] [PubMed]
- Fanardzhyan, V.V.; Oganesyan, E.A.; Melik-Mus’yan, A.B. Morphological and functional organization of cortico-nuclear cerebellar projections. Neurosci. Behav. Physiol. 1973, 6, 206–217. [Google Scholar] [CrossRef]
- Glatzel, M.; Stoeck, K.; Seeger, H.; Lührs, T.; Aguzzi, A. Human prion diseases: Molecular and clinical aspects. Arch. Neurol. 2005, 62, 545–552. [Google Scholar] [CrossRef]
- Dammermann, A.; Merdes, A. Assembly of centrosomal proteins and microtubule organization depends on PCM-1. J. Cell. Biol. 2002, 159, 255–266. [Google Scholar] [CrossRef]
- Yu, W.; Centonze, V.E.; Ahmad, F.J.; Baas, P.W. Microtubule nucleation and release from the neuronal centrosome. J. Cell Biol. 1993, 122, 349–359. [Google Scholar] [CrossRef]
- Tajes, M.; Guivernau, B.; Ramos-Fernández, E.; Bosch-Morató, M.; Palomer, E.; Guix, F.X.; Muñoz, F.J. The pathophysiology of triose phosphate isomerase dysfunction in Alzheimer’s disease. Histol. Histopathol. 2013, 28, 43–51. [Google Scholar] [CrossRef]
- Dunker, A.K.; Babu, M.M.; Barbar, E.; Blackledge, M.; Bondos, S.E.; Dosztányi, Z.; Dyson, H.J.; Forman-Kay, J.; Fuxreiter, M.; Gsponer, J.; et al. What’s in a name? Why these proteins are intrinsically disordered: Why these proteins are intrinsically disordered. Intrinsically Disord. Proteins 2013, 1, e24157. [Google Scholar] [CrossRef]
- Masuda, Y.; Uemura, S.; Ohashi, R.; Nakanishi, A.; Takegoshi, K.; Shimizu, T.; Shirasawa, T.; Irie, K. Identification of physiological and toxic conformations in Abeta42 aggregates. Chembiochem 2009, 10, 287–295. [Google Scholar] [CrossRef]
- Cushman, M.; Johnson, B.S.; King, O.D.; Gitler, A.D.; Shorter, J. Prion-like disorders: Blurring the divide between transmissibility and infectivity. J. Cell. Sci. 2010, 123, 1191–1201. [Google Scholar] [CrossRef]
- Wong, Y.C.; Krainc, D. α-Synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Yu, Z.; Chen, S. Alpha-synuclein nitration and its implications in Parkinson’s disease. ACS Chem. Neurosci. 2019, 10, 777–782. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wang, F.; Yung, K.K.L.; Zhang, S.; Qu, S. Effects of α-synuclein-associated post-translational modifications in Parkinson’s disease. ACS Chem. Neurosci. 2021, 12, 1061–1071. [Google Scholar] [CrossRef]
- Miller, S.P.; Ferriero, D.M. From selective vulnerability to connectivity: Insights from newborn brain imaging. Trends Neurosci. 2009, 32, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; El-Metwally, D.; Sours Rhodes, C.; Zhuo, J.; Almardawi, R.; Medina, A.E.; Wang, L.; Gullapalli, R.P.; Raghavan, P. Alterations in motor functional connectivity in neonatal hypoxic ischemic encephalopathy. Brain Inj. 2022, 36, 287–294. [Google Scholar] [CrossRef]
- Hosoya, Y.; Ohkanda, J. Intrinsically disordered proteins as regulators of transient biological processes and as untapped drug targets. Molecules 2021, 26, 2118. [Google Scholar] [CrossRef]
- Singh, R.; Kulikowicz, E.; Santos, P.T.; Koehler, R.C.; Martin, L.J.; Lee, J. Spatial T-maze identifies cognitive deficits in piglets 1 month after hypoxia-ischemia in a model of hippocampal pyramidal neuron loss and interneuron attrition. Behav. Brain Res. 2019, 369, 111921. [Google Scholar] [CrossRef]
- Fiolek, T.J.; Magyar, C.L.; Wall, T.J.; Davies, S.B.; Campbell, M.V.; Savich, C.J.; Tepe, J.J.; Mosey, R.A. Dihydroquinazolines enhance 20S proteasome activity and induce degradation of α-synuclein, an intrinsically disordered protein associated with neurodegeneration. Bioorg. Med. Chem. Lett. 2021, 36, 127821. [Google Scholar] [CrossRef]
- Malik, R.; Meng, H.; Wongkongkathep, P.; Corrales, C.I.; Sepanj, N.; Atlasi, R.S.; Klärner, F.G.; Schrader, T.; Spencer, M.J.; Loo, J.A.; et al. The molecular tweezer CLR01 inhibits aberrant superoxide dismutase 1 (SOD1) self-assembly in vitro and in the G93A-SOD1 mouse model of ALS. J. Biol. Chem. 2019, 294, 3501–3513. [Google Scholar] [CrossRef]
- Prabhudesai, S.; Sinha, S.; Attar, A.; Kotagiri, A.; Fitzmaurice, A.G.; Lakshmanan, R.; Ivanova, M.I.; Loo, J.A.; Klärner, F.G.; Schrader, T.; et al. A novel “molecular tweezer” inhibitor of α-synuclein neurotoxicity in vitro and in vivo. Neurotherapeutics 2012, 9, 464–476. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case Identifier | Age at Birth (Weeks) | Putative Clinical Insult | Apgar (When at Birth) | Therapeutic Hypothermia | Last Recorded Blood pH | Age (at Death) | Postmortem Delay (Hours) |
|---|---|---|---|---|---|---|---|
| A15-7 | 36 (37) | Fetal deceleration and emergency C-section. Resuscitation involving chest compressions, gas of 6.9, and clinical seizures | 1 (1 min) | Yes | 7.283 arterial; 7.251 capillary | 7 days | 24 |
| A16-13 | 41.6 (42.9) | Shoulder dystocia; prolonged ruptured membranes | 1, 1, 5 (1, 5 and 10 min) | Yes | 7.425 arterial; 7.259 capillary | 9 days | 12 |
| A16-30 | 34 (34.3) | Car collision, placental abruption, emergency C-section, DIC in newborn, neonatal respiratory failure | 1, 1 (1 and 5 min) | Yes | 7.017 capillary; 6.971 arterial | 2 days | 144 |
| A17-1 | 39.3 (39.9) | Rupture of membranes, C-section, required chest compressions, cord gas of 6.9 | 1, 3, 4 (1, 5, and 10 min) | Yes | 7.221 arterial | 4 days | 12 |
| A17-3 | 38 (38.4) | Uncontrolled insulin-dependent diabetes (mother), emergency C-section, perinatal asphyxia, chest compressions | 0, 1, 3 (1, 5, and 10 min) | Yes | 7.271 arterial; 7.279 capillary | 3 days | 24 |
| A17-14 | 24 (48.6) | Chronic lung disease with secondary pulmonary hypertension, hypoxemic respiratory failure, worsening hypotension | 8, 9 (1 and 5 min) | 0 (missed therapeutic time window) | 7.439 arterial; 7.317 capillary | 172 days | 25 |
| A18-2 | 35.3 (35.9) | Diamniotic dichorionic twins, premature rupture of membranes, emergency C-section, Traumatic delivery (cephalohematoma, focal subdural hemorrhage, subarachnoid hemorrhages), chest compressions | 1, 0, 0 (1, 5, and 10 min) | Yes | 7.459 arterial; 7.283 capillary | 4 days | 16 |
| A18-3 | 39.9 (40.4) | Secondary apnea, respiratory failure, multiorgan failure, possible septic shock | 5, 2 (1, and 5 min) | Yes | 7.312 arterial | 4 days | 24 |
| A18-17 | 34.3 (36.4) | HIV+ mother via emergency C section for fetal deceleration for systole, chest compressions | 0, 0, 1 (1, 5, and 10 min) | 0 (missed therapeutic time window) | 7.217 arterial; 7.391 capillary | 15 days | 48 |
| A18-28 | 40.3 (40.4) | Non-reassuring fetal heart rate, acute phlebitis of umbilical cord, acute chorioamnionitis of membranes, chorionic plate had acute subchorionitis, tight nuchal chord, placental SGA | 0, 0, 1 (1, 5, and 10 min) | Yes | 6.6; BD > 20 | 1 day | 408 |
| 4314 | 38 | Respiratory insufficiency, anoxic encephalopathy | NA | No | NA | 4 days | 21 |
| 667 | 38 | Acute cardiac arrhythmia-arrest (Non-HIE control) | No | No | NA | 353 days | 13 |
| 828 | 36 | Meconium aspiration, seizure disorder | 8, 9 | No | NA | 90 days | 10 |
| 731 | 36 | Severe birth anoxia, anoxic encephalopathy, seizure disorder | NA | No | NA | 360 days | 14 |
| A54802 | 39 | Rupture of membranes, cardiorespiratory arrest | 1, 0, 0, 2, 3 (1, 5, 10, 15, 20 min) | Yes | NA | 4 days | 24 |
| A56447 | 35 | Non-reassuring fetal statue, C-section, immediate apnea, respiratory arrest. Anoxic encephalopathy | 2, 3, 3 (1, 5, 10) | No (missed therapeutic time window) | NA | 5 days | 24 |
| A54854 | 34.5 | Maternal gestational diabetes, late decelerations during cerclage removal, emergency C-section | 1, 1, 1, 1, 2 (1, 5, 10, 15, 20) | Yes | NA | 1 day | 22 |
| A54550 | 36 | SMA (Non-HIE control) | NA | NA | NA | 14 days | 18 |
| 4358 | 39 | Non-HIE control, accidental death | 8, 9 | NA | NA | 9 days | 25 |
| 4360 | 40 | Non-HIE control, accidental death | 8, 9 | NA | NA | 202 days | 33 |
| 4361 | 40 | Non-HIE control, undetermined | 8, 9 | NA | NA | 236 days | 27 |
| 4388 | 40 | Non-HIE control, accidental death | 8, 9 | NA | NA | 147 days | 53 |
| 4389 | 34 | Non-HIE control, accidental death | 8, 9 | NA | NA | 79 days | 27 |
| 4415 | 40 | Non-HIE control, undetermined | 8, 9 | NA | NA | 146 days | 46 |
| 4418 | 39 | Non-HIE control, accidental death | 8, 9 | NA | NA | 76 days | 24 |
| 4421 | 39 | Non-HIE control, accidental death | 8, 9 | NA | NA | 84 days | 40 |
| 4460 | 39 | Non-HIE control, accidental death | 8, 9 | NA | NA | 21 days | 25 |
| 5947 | 40 | Non-HIE control, accidental death | 8, 9 | NA | NA | 179 days | 11 |
| Injury/Insult | Survival and Group Sizes | Experimental Use | Justification | |
|---|---|---|---|---|
| Piglet (2–3 days old male) | Global hypoxia-ischemia (HI) or sham with normothermic (NT) or hypothermic (HT) recovery | 2–7 days. Sham-NT (n = 6), Sham-HT (n = 10), HI-NT (n = 8), HI-HT (n = 10) | Histology: H&E staining for neuronal counting and cell death morphology | Clinically relevant with therapeutic; Survival can be limited by seizures. |
| Piglet (2–4 days old male) | Global hypoxia-ischemia (HI) with NT or HT recovery | 29 h. Sham-NT (n = 4), Sham-HT (n = 4), HI-NT (n = 4), HI-HT (n = 4), naïve (n = 4) | Western blotting for toxic conformer proteins | Clinically relevant with therapeutic; short survival to avoid seizures. |
| Piglet (2–3 days old male) | Global HI (no temperature management) | 96 h (4 days). Sham (n = 6), HI (n = 6) | Histology: immunohistochemistry and immunofluorescence | Gold standard historical model. Much information on neuropathology without cooling and need to control for cooling effects on brain. Ideal balance of survival time and requirement for animal care. |
| Piglet (2–3 days old male) | Global HI (no temperature management) | 96 h (4 days). Sham (n = 4), HI (n = 4) | Western blotting for toxic conformer proteins | As above |
| Piglet (2–3 days old male) | Global HI plus quinolinic acid (QA) excitotoxic lesion (2-hit protocol, no temperature management) | 48 h. Sham (n = 4), HI-QA (n = 4), Vehicle (n = 4), naïve (n = 4) | Western blotting for toxic conformer proteins | Newest model that bridges in vivo and human cell culture QA experiments. Exquisite regionally specific white and gray matter molecular profiling attractive. |
| Piglet (2–3 days old male) | Global HI plus quinolinic acid (QA) excitotoxic lesion (2-hit protocol, no temperature management) | 15 days. Sham (n = 5), HI-QA (n = 6) | Histology: immunohistochemistry and immunofluorescence | Newest model that capitalizes on lesser asphyxic heart damage for longer survival without need to control for cooling. Short and long-term survival attractive. |
| Antibody | Characterization | Target/Antigen | IgG Type | Source |
|---|---|---|---|---|
| α-Synuclein (Syn) Aggregate, clone MJFR-14-6-4-2 | [69,70] | α-Syn conformation-specific/full-length α-Syn | Rabbit monoclonal | Abcam, Waltham, MA, USA |
| α-Syn Aggregate, clone 5G4 | [71,72] | Aggregated α-Syn/KLH-conjugated peptide corresponding to human aggregated α-Syn | Mouse monoclonal | Millipore-Sigma, St. Louis, MO, USA |
| α-Syn Oligomer (Syn33) | [73] | Wildtype full-length α-Syn oligomers | Rabbit polyclonal | Millipore-Sigma |
| Nitrated Syn, clone Syn12 | [57] | Nitrated Syn/α-Syn nitrated at Tyr125 and Tyr136 (β-Syn nitrated at Tyr130) | Mouse monoclonal | Santa Cruz Biotechnology, Dallas, TX, USA |
| Misfolded-Aggregated SOD1, clone C4F6 | [74] | SOD1/full-length SOD1 apoenzyme | Mouse monoclonal | Medimabs, Montreal, Quebec, Canada |
| Misfolded-Aggregated SOD1, clone B8H10 | [75] | SOD1/full-length SOD1 apoenzyme | Mouse monoclonal | Medimabs |
| PrP, clone F89/160.1.5 | [76] | PrP n-IHFG-n | Mouse monoclonal | Invitrogen-ThermoFisher Scientific, Waltham, MA, USA |
| Cell Type Target | Identification Purpose | IgG Type | Source |
|---|---|---|---|
| Neurofilament 68 (NF68) | Neuron cell body and axon | Rabbit polyclonal | Abcam Ab9035 |
| NeuN | Neuron cell body | Rabbit polyclonal | Millipore ABN78 |
| Ubiquitin | Inclusions | Mouse monoclonal | Abcam Ab7254 |
| CNPase | Oligodendrocytes | Mouse monoclonal, clone 11-5B | Millipore-Sigma |
| Olig2 | Oligodendrocytes | Rabbit monoclonal, EPR2673 | Abcam, Ab109186 |
| GLAST | Astrocytes | Rabbit polyclonal | Proteintech, 20785-1-AP |
| Tyrosine hydroxylase | Midbrain dopaminergic neurons | Rabbit polyclonal | Novus Biologicals, NB300-109 |
| DARP32 | Striatal neurons | Rabbit monoclonal, EP720Y | Abcam Ab40801 |
| TBR1 | Cortical neurons | Rabbit polyclonal | Abcam Ab31940 |
| Synaptophysin | Presynaptic terminals | Mouse monoclonal. 4E12C4 | Proteintech, 67864-1-Ig |
| SV2a | Presynaptic terminals | Rabbit polyclonal | Synaptic Systems, Gottingen, Germany, 119003 |
| Synapsin 1 and 2 | Presynaptic terminals | Rabbit polyclonal | Synaptic Systems, 106002 |
| Cysteine String Protein (CSP) | Presynaptic terminals | Rabbit polyclonal | Stressgen Bioreagents, Victoria, British Columbia, Canada, VAP-SV003 |
| Munc18 | Presynaptic terminals | Mouse monoclonal, 31/munc-18 | BD Transduction Laboratories, San Diego, CA, USA, 610336 |
| β-synuclein | Neurons and presynaptic terminals | Rabbit monoclonal, EP1537Y | Epitomics, Burlingame, CA, USA, 1977-1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, L.J.; Lee, J.K.; Niedzwiecki, M.V.; Amrein Almira, A.; Javdan, C.; Chen, M.W.; Olberding, V.; Brown, S.M.; Park, D.; Yohannan, S.; et al. Hypothermia Shifts Neurodegeneration Phenotype in Neonatal Human Hypoxic–Ischemic Encephalopathy but Not in Related Piglet Models: Possible Relationship to Toxic Conformer and Intrinsically Disordered Prion-like Protein Accumulation. Cells 2025, 14, 586. https://doi.org/10.3390/cells14080586
Martin LJ, Lee JK, Niedzwiecki MV, Amrein Almira A, Javdan C, Chen MW, Olberding V, Brown SM, Park D, Yohannan S, et al. Hypothermia Shifts Neurodegeneration Phenotype in Neonatal Human Hypoxic–Ischemic Encephalopathy but Not in Related Piglet Models: Possible Relationship to Toxic Conformer and Intrinsically Disordered Prion-like Protein Accumulation. Cells. 2025; 14(8):586. https://doi.org/10.3390/cells14080586
Chicago/Turabian StyleMartin, Lee J., Jennifer K. Lee, Mark V. Niedzwiecki, Adriana Amrein Almira, Cameron Javdan, May W. Chen, Valerie Olberding, Stephen M. Brown, Dongseok Park, Sophie Yohannan, and et al. 2025. "Hypothermia Shifts Neurodegeneration Phenotype in Neonatal Human Hypoxic–Ischemic Encephalopathy but Not in Related Piglet Models: Possible Relationship to Toxic Conformer and Intrinsically Disordered Prion-like Protein Accumulation" Cells 14, no. 8: 586. https://doi.org/10.3390/cells14080586
APA StyleMartin, L. J., Lee, J. K., Niedzwiecki, M. V., Amrein Almira, A., Javdan, C., Chen, M. W., Olberding, V., Brown, S. M., Park, D., Yohannan, S., Putcha, H., Zheng, B., Garrido, A., Benderoth, J., Kisner, C., Ghaemmaghami, J., Northington, F. J., & Kratimenos, P. (2025). Hypothermia Shifts Neurodegeneration Phenotype in Neonatal Human Hypoxic–Ischemic Encephalopathy but Not in Related Piglet Models: Possible Relationship to Toxic Conformer and Intrinsically Disordered Prion-like Protein Accumulation. Cells, 14(8), 586. https://doi.org/10.3390/cells14080586