
Colorectal Organoids: Models, Imaging, Omics, Therapy, Immunology, and Ethics

 , , , ,
, , , ,  ,
, 
Abstract
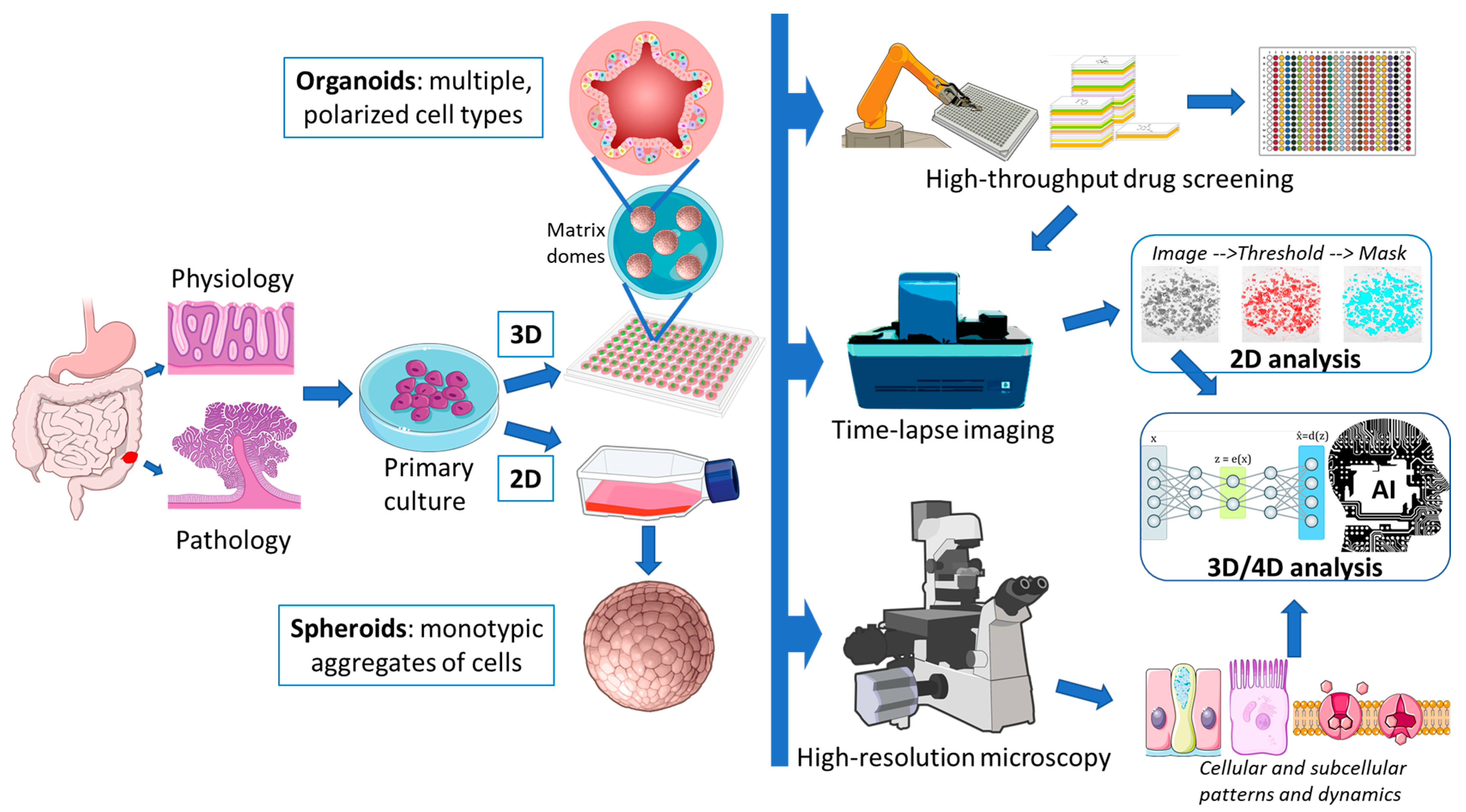
1. Organoids as Models
1.1. Sources and Propagation of Colorectal Tissue
1.2. Matrigel vs. Hydrogel
1.3. Matrigel vs. Collagen-I
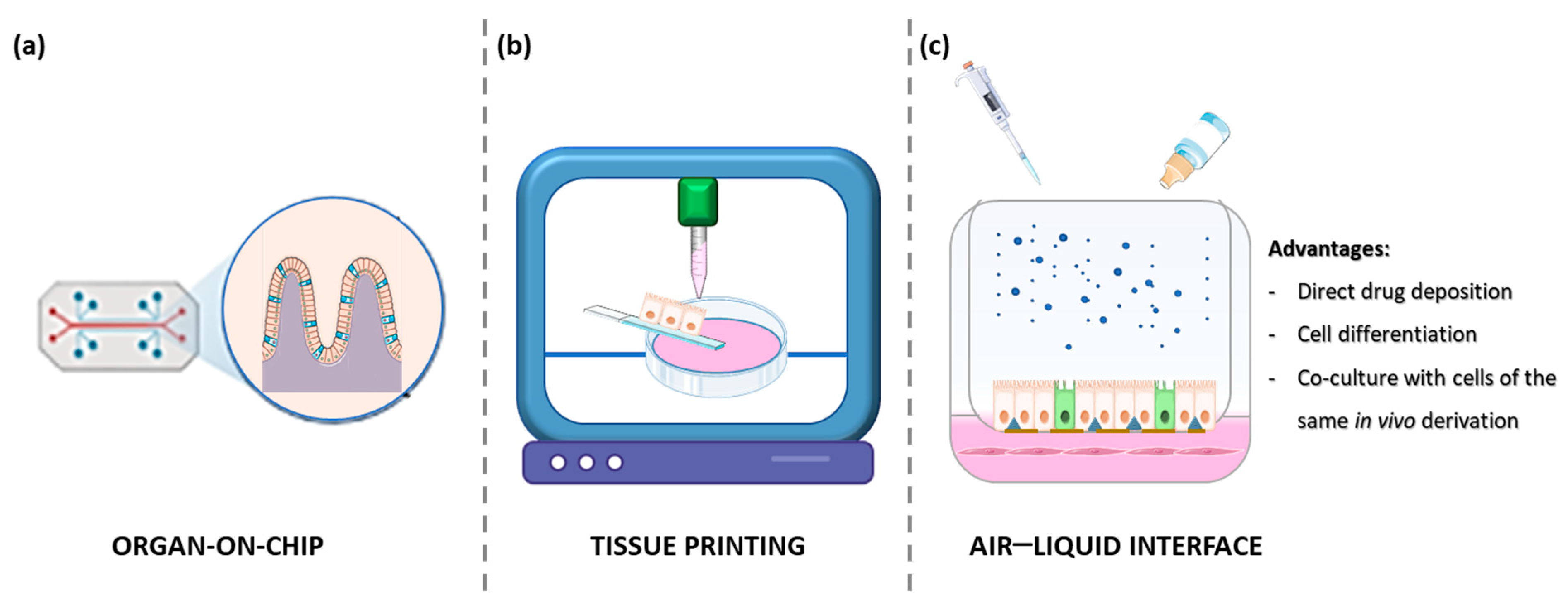
1.4. Organ on a Chip
1.5. Air–Liquid Interface (ALI) Cultures
1.6. Tissue Printing
1.7. Multicellular Assembloids
1.8. Multi-Organism Systems
2. Organoids Imaging and Image Analysis
3. Omics of Colorectal Organoids
3.1. Genomics
3.2. Epigenomics
3.3. Transcriptomics
3.4. Proteomics
3.5. Metabolomics
4. Organoids as Models for Therapy
Organoids for Drug Screening and Personalized Medicine
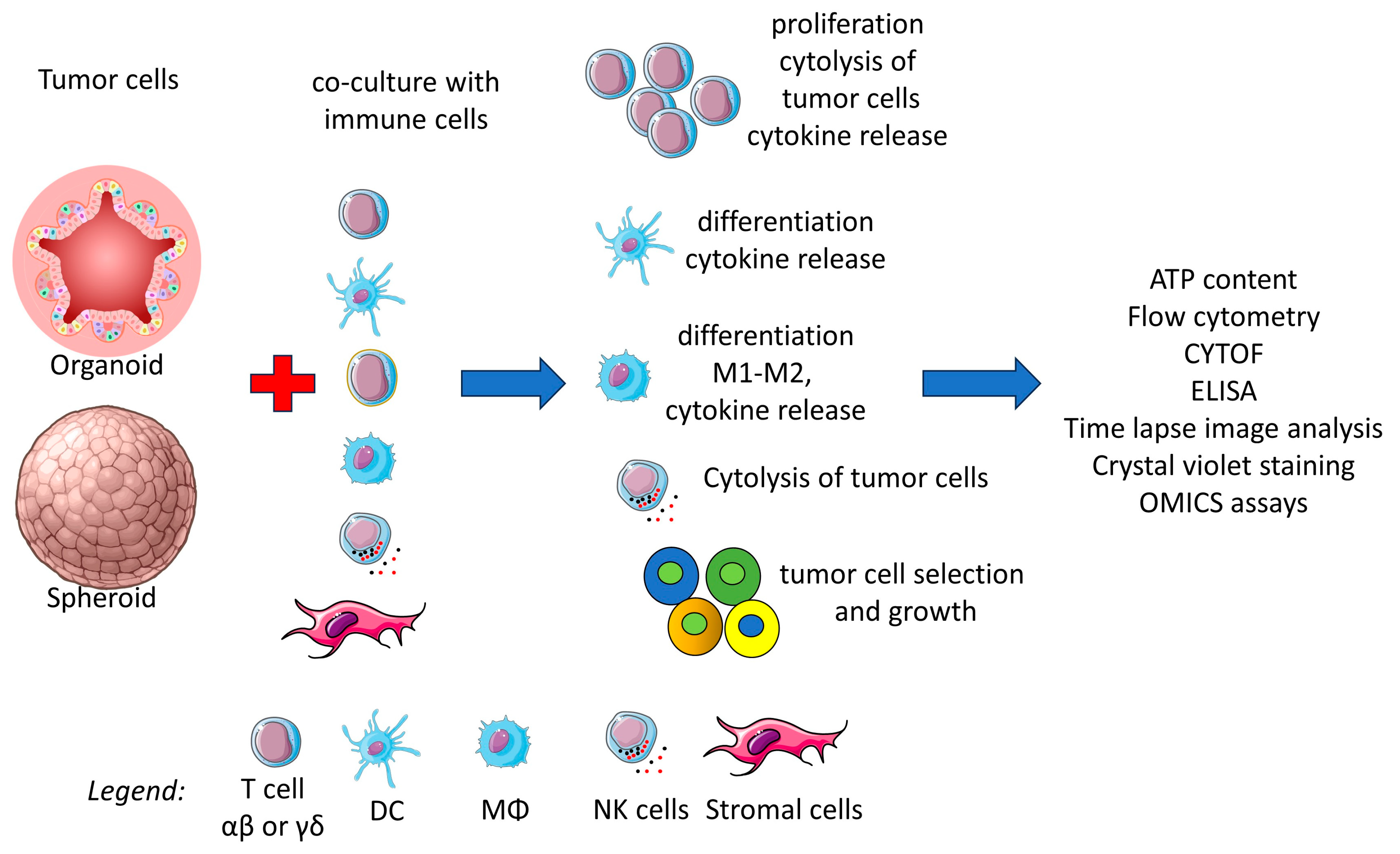
5. Organoids as Immunotherapy Models
5.1. Three-Dimensional Co-Cultures of T Lymphocytes and CRC
5.2. Interactions Between Antigen-Presenting Cells and CRC Organoids
5.3. Innate Cytolytic Cells Can Affect the Viability and Growth of CRC Organoids
6. Biobanks in the Era of Translational and Personalized Medicine
6.1. Living Biobanks for Organoids: Challenges for Standardization
6.2. Living Biobanks for Organoids: Ethical, Legal, and Social Issues (ELSIs)
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, L.; Yan, Q.; Chen, Z.; Wei, X.; Li, L.; Tang, D.; Tan, J.; Xu, C.; Yu, C.; Lai, Y.; et al. Overview of Research Progress and Application of Experimental Models of Colorectal Cancer. Front. Pharmacol. 2023, 14, 1193213. [Google Scholar] [CrossRef]
- Zhang, C.; Sui, Y.; Liu, S.; Yang, M. In Vitro and In Vivo Experimental Models for Cancer Immunotherapy Study. Curr. Res. Biotechnol. 2024, 7, 100210. [Google Scholar] [CrossRef]
- Jubelin, C.; Muñoz-Garcia, J.; Griscom, L.; Cochonneau, D.; Ollivier, E.; Heymann, M.-F.; Vallette, F.M.; Oliver, L.; Heymann, D. Three-Dimensional In Vitro Culture Models in Oncology Research. Cell Biosci. 2022, 12, 155. [Google Scholar] [CrossRef]
- Zhang, C.-Y.; Liu, S.; Yang, M. Regulatory T Cells and Their Associated Factors in Hepatocellular Carcinoma Development and Therapy. World J. Gastroenterol. 2022, 28, 3346–3358. [Google Scholar] [CrossRef]
- Centeno, E.G.Z.; Cimarosti, H.; Bithell, A. 2D versus 3D Human Induced Pluripotent Stem Cell-Derived Cultures for Neurodegenerative Disease Modelling. Mol. Neurodegener. 2018, 13, 27. [Google Scholar] [CrossRef]
- Fujii, M.; Sato, T. Somatic Cell-Derived Organoids as Prototypes of Human Epithelial Tissues and Diseases. Nat. Mater. 2021, 20, 156–169. [Google Scholar] [CrossRef]
- Rheinwald, J.G.; Green, H. Serial Cultivation of Strains of Human Epidermal Keratinocytes: The Formation of Keratinizing Colonies from Single Cells. Cell 1975, 6, 331–343. [Google Scholar]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.J.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-Term Expansion of Epithelial Organoids from Human Colon, Adenoma, Adenocarcinoma, and Barrett’s Epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef]
- Zhao, Z.; Chen, X.; Dowbaj, A.M.; Sljukic, A.; Bratlie, K.; Lin, L.; Fong, E.L.S.; Balachander, G.M.; Chen, Z.; Soragni, A.; et al. Organoids. Nat. Rev. Methods Primers 2022, 2, 94. [Google Scholar] [CrossRef]
- Liu, T.; Li, X.; Li, H.; Qin, J.; Xu, H.; Wen, J.; He, Y.; Zhang, C. Intestinal Organoid Modeling: Bridging the Gap from Experimental Model to Clinical Translation. Front. Oncol. 2024, 14, 1334631. [Google Scholar] [CrossRef]
- Dotti, I.; Mora-Buch, R.; Ferrer-Picón, E.; Planell, N.; Jung, P.; Masamunt, M.C.; Leal, R.F.; de Carpi, J.M.; Llach, J.; Ordás, I.; et al. Alterations in the Epithelial Stem Cell Compartment Could Contribute to Permanent Changes in the Mucosa of Patients with Ulcerative Colitis. Gut 2017, 66, 2069–2079. [Google Scholar] [CrossRef] [PubMed]
- Neyazi, M.; Bharadwaj, S.S.; Bullers, S.; Varenyiova, Z.; Oxford IBD Cohort Study Investigators; Travis, S.; Arancibia-Cárcamo, C.V.; Powrie, F.; Geremia, A. Overexpression of Cancer-Associated Stem Cell Gene OLFM4 in the Colonic Epithelium of Patients With Primary Sclerosing Cholangitis. Inflamm. Bowel Dis. 2021, 27, 1316–1327. [Google Scholar] [CrossRef] [PubMed]
- Howell, K.J.; Kraiczy, J.; Nayak, K.M.; Gasparetto, M.; Ross, A.; Lee, C.; Mak, T.N.; Koo, B.-K.; Kumar, N.; Lawley, T.; et al. DNA Methylation and Transcription Patterns in Intestinal Epithelial Cells From Pediatric Patients with Inflammatory Bowel Diseases Differentiate Disease Subtypes and Associate with Outcome. Gastroenterology 2018, 154, 585–598. [Google Scholar] [CrossRef]
- Freire, R.; Ingano, L.; Serena, G.; Cetinbas, M.; Anselmo, A.; Sapone, A.; Sadreyev, R.I.; Fasano, A.; Senger, S. Human Gut Derived-Organoids Provide Model to Study Gluten Response and Effects of Microbiota-Derived Molecules in Celiac Disease. Sci. Rep. 2019, 9, 7029. [Google Scholar] [CrossRef]
- Bierlaagh, M.C.; van Mourik, P.; Vonk, A.M.; Pott, J.; Muilwijk, D.; Berkers, G.; Aalbers, B.L.; Vleggaar, F.P.; Michel, S.; Boj, S.F.; et al. Centralized Intestinal Organoid Generation Is a Feasible and Safe Approach for Personalized Medicine as Demonstrated in the HIT-CF Europe Organoid Study. J. Cyst. Fibros. 2024, 23, 703–706. [Google Scholar] [CrossRef]
- Fujii, M.; Shimokawa, M.; Date, S.; Takano, A.; Matano, M.; Nanki, K.; Ohta, Y.; Toshimitsu, K.; Nakazato, Y.; Kawasaki, K.; et al. A Colorectal Tumor Organoid Library Demonstrates Progressive Loss of Niche Factor Requirements during Tumorigenesis. Cell Stem Cell 2016, 18, 827–838. [Google Scholar] [CrossRef]
- van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective Derivation of a Living Organoid Biobank of Colorectal Cancer Patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Lee, I.-T.; Takahashi, Y.; Sasaki, T.; Yamauchi, Y.; Sato, R. Human Colon Organoid Differentiation from Induced Pluripotent Stem Cells Using an Improved Method. FEBS Lett. 2024. [Google Scholar] [CrossRef]
- Yoshida, S.; Miwa, H.; Kawachi, T.; Kume, S.; Takahashi, K. Generation of Intestinal Organoids Derived from Human Pluripotent Stem Cells for Drug Testing. Sci. Rep. 2020, 10, 5989. [Google Scholar] [CrossRef]
- Daoud, A.; Múnera, J.O. Generation of Human Colonic Organoids from Human Pluripotent Stem Cells. Methods Cell Biol. 2020, 159, 201–227. [Google Scholar] [CrossRef] [PubMed]
- Brassard, J.A.; Lutolf, M.P. Engineering Stem Cell Self-Organization to Build Better Organoids. Cell Stem Cell 2019, 24, 860–876. [Google Scholar] [CrossRef] [PubMed]
- McCracken, K.W.; Aihara, E.; Martin, B.; Crawford, C.M.; Broda, T.; Treguier, J.; Zhang, X.; Shannon, J.M.; Montrose, M.H.; Wells, J.M. Wnt/β-Catenin Promotes Gastric Fundus Specification in Mice and Humans. Nature 2017, 541, 182–187. [Google Scholar] [CrossRef]
- Wang, E.; Xiang, K.; Zhang, Y.; Wang, X.-F. Patient-Derived Organoids (PDOs) and PDO-Derived Xenografts (PDOXs): New Opportunities in Establishing Faithful Pre-Clinical Cancer Models. J. Natl. Cancer Cent. 2022, 2, 263–276. [Google Scholar] [CrossRef]
- Janitri, V.; ArulJothi, K.N.; Ravi Mythili, V.M.; Singh, S.K.; Prasher, P.; Gupta, G.; Dua, K.; Hanumanthappa, R.; Karthikeyan, K.; Anand, K. The Roles of Patient-Derived Xenograft Models and Artificial Intelligence toward Precision Medicine. MedComm 2024, 5, e745. [Google Scholar] [CrossRef]
- Tosca, E.M.; Ronchi, D.; Rocchetti, M.; Magni, P. Predicting Tumor Volume Doubling Time and Progression-Free Survival in Untreated Patients from Patient-Derived-Xenograft (PDX) Models: A Translational Model-Based Approach. AAPS J. 2024, 26, 92. [Google Scholar] [CrossRef]
- Aisenbrey, E.A.; Murphy, W.L. Synthetic Alternatives to Matrigel. Nat. Rev. Mater. 2020, 5, 539–551. [Google Scholar] [CrossRef]
- Scholz, J.A.C.; Garg, R.; Compton, S.R.; Allore, H.G.; Zeiss, C.J.; Uchio, E.M. Poliomyelitis in MuLV-Infected ICR-SCID Mice after Injection of Basement Membrane Matrix Contaminated with Lactate Dehydrogenase-Elevating Virus. Comp. Med. 2011, 61, 404–411. [Google Scholar]
- Hirokawa, Y.; Clarke, J.; Palmieri, M.; Tan, T.; Mouradov, D.; Li, S.; Lin, C.; Li, F.; Luo, H.; Wu, K.; et al. Low-Viscosity Matrix Suspension Culture Enables Scalable Analysis of Patient-Derived Organoids and Tumoroids from the Large Intestine. Commun. Biol. 2021, 4, 1067. [Google Scholar] [CrossRef]
- Kim, S.; Min, S.; Choi, Y.S.; Jo, S.-H.; Jung, J.H.; Han, K.; Kim, J.; An, S.; Ji, Y.W.; Kim, Y.-G.; et al. Tissue Extracellular Matrix Hydrogels as Alternatives to Matrigel for Culturing Gastrointestinal Organoids. Nat. Commun. 2022, 13, 1692. [Google Scholar] [CrossRef]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; van de Haar, J.; Fanchi, L.F.; Slagter, M.; van der Velden, D.L.; Kaing, S.; Kelderman, S.; et al. Generation of Tumor-Reactive T Cells by Co-Culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018, 174, 1586–1598.e12. [Google Scholar] [CrossRef] [PubMed]
- Gjorevski, N.; Lutolf, M.P. Synthesis and Characterization of Well-Defined Hydrogel Matrices and Their Application to Intestinal Stem Cell and Organoid Culture. Nat. Protoc. 2017, 12, 2263–2274. [Google Scholar] [CrossRef] [PubMed]
- Hirota, A.; AlMusawi, S.; Nateri, A.S.; Ordóñez-Morán, P.; Imajo, M. Biomaterials for Intestinal Organoid Technology and Personalized Disease Modeling. Acta Biomater. 2021, 132, 272–287. [Google Scholar] [CrossRef]
- Kozlowski, M.T.; Crook, C.J.; Ku, H.T. Towards Organoid Culture without Matrigel. Commun. Biol. 2021, 4, 1387. [Google Scholar] [CrossRef]
- Hernandez-Gordillo, V.; Kassis, T.; Lampejo, A.; Choi, G.; Gamboa, M.E.; Gnecco, J.S.; Brown, A.; Breault, D.T.; Carrier, R.; Griffith, L.G. Fully Synthetic Matrices for in Vitro Culture of Primary Human Intestinal Enteroids and Endometrial Organoids. Biomaterials 2020, 254, 120125. [Google Scholar] [CrossRef]
- Bergenheim, F.; Fregni, G.; Buchanan, C.F.; Riis, L.B.; Heulot, M.; Touati, J.; Seidelin, J.B.; Rizzi, S.C.; Nielsen, O.H. A Fully Defined 3D Matrix for Ex Vivo Expansion of Human Colonic Organoids from Biopsy Tissue. Biomaterials 2020, 262, 120248. [Google Scholar] [CrossRef]
- Jabaji, Z.; Brinkley, G.J.; Khalil, H.A.; Sears, C.M.; Lei, N.Y.; Lewis, M.; Stelzner, M.; Martín, M.G.; Dunn, J.C.Y. Type I Collagen as an Extracellular Matrix for the In Vitro Growth of Human Small Intestinal Epithelium. PLoS ONE 2014, 9, e107814. [Google Scholar] [CrossRef]
- Jee, J.H.; Lee, D.H.; Ko, J.; Hahn, S.; Jeong, S.Y.; Kim, H.K.; Park, E.; Choi, S.Y.; Jeong, S.; Lee, J.W.; et al. Development of Collagen-Based 3D Matrix for Gastrointestinal Tract-Derived Organoid Culture. Stem Cells Int. 2019, 2019, 8472712. [Google Scholar] [CrossRef]
- Sachs, N.; Tsukamoto, Y.; Kujala, P.; Peters, P.J.; Clevers, H. Intestinal Epithelial Organoids Fuse to Form Self-Organizing Tubes in Floating Collagen Gels. Development 2017, 144, 1107–1112. [Google Scholar] [CrossRef]
- Ramadan, R.; Wouters, V.M.; van Neerven, S.M.; de Groot, N.E.; Garcia, T.M.; Muncan, V.; Franklin, O.D.; Battle, M.; Carlson, K.S.; Leach, J.; et al. The Extracellular Matrix Controls Stem Cell Specification and Crypt Morphology in the Developing and Adult Mouse Gut. Biol. Open 2022, 11, bio059544. [Google Scholar] [CrossRef]
- Leung, C.M.; de Haan, P.; Ronaldson-Bouchard, K.; Kim, G.-A.; Ko, J.; Rho, H.S.; Chen, Z.; Habibovic, P.; Jeon, N.L.; Takayama, S.; et al. A Guide to the Organ-on-a-Chip. Nat. Rev. Methods Primers 2022, 2, 33. [Google Scholar] [CrossRef]
- Xiang, T.; Wang, J.; Li, H. Current Applications of Intestinal Organoids: A Review. Stem Cell Res. Ther. 2024, 15, 155. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Huang, Y.-J.; Trapecar, M.; Wright, C.; Schneider, K.; Kemmitt, J.; Hernandez-Gordillo, V.; Yoon, J.Y.; Poyet, M.; Alm, E.J.; et al. An Immune-Competent Human Gut Microphysiological System Enables Inflammation-Modulation by Faecalibacterium prausnitzii. NPJ Biofilms Microbiomes 2024, 10, 31. [Google Scholar] [CrossRef]
- Apostolou, A.; Panchakshari, R.A.; Banerjee, A.; Manatakis, D.V.; Paraskevopoulou, M.D.; Luc, R.; Abu-Ali, G.; Dimitriou, A.; Lucchesi, C.; Kulkarni, G.; et al. A Novel Microphysiological Colon Platform to Decipher Mechanisms Driving Human Intestinal Permeability. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 1719–1741. [Google Scholar] [CrossRef]
- Palikuqi, B.; Nguyen, D.-H.T.; Li, G.; Schreiner, R.; Pellegata, A.F.; Liu, Y.; Redmond, D.; Geng, F.; Lin, Y.; Gómez-Salinero, J.M.; et al. Adaptable Haemodynamic Endothelial Cells for Organogenesis and Tumorigenesis. Nature 2020, 585, 426–432. [Google Scholar] [CrossRef]
- Rajasekar, S.; Lin, D.S.Y.; Abdul, L.; Liu, A.; Sotra, A.; Zhang, F.; Zhang, B. IFlowPlate—A Customized 384-Well Plate for the Culture of Perfusable Vascularized Colon Organoids. Adv. Mater. 2020, 32, e2002974. [Google Scholar] [CrossRef]
- Mitrofanova, O.; Nikolaev, M.; Xu, Q.; Broguiere, N.; Cubela, I.; Camp, J.G.; Bscheider, M.; Lutolf, M.P. Bioengineered Human Colon Organoids with in Vivo-like Cellular Complexity and Function. Cell Stem Cell 2024, 31, 1175–1186.e7. [Google Scholar] [CrossRef]
- Ootani, A.; Li, X.; Sangiorgi, E.; Ho, Q.T.; Ueno, H.; Toda, S.; Sugihara, H.; Fujimoto, K.; Weissman, I.L.; Capecchi, M.R.; et al. Sustained in Vitro Intestinal Epithelial Culture within a Wnt-Dependent Stem Cell Niche. Nat. Med. 2009, 15, 701–706. [Google Scholar] [CrossRef]
- Dotti, I.; Álvarez, I.; Esteller, M.; Ricart, E.; Salas, A. P043 ALI Culture Derived from Human Organoids Recapitulates Cell Composition during Intestinal Crypt Formation. J. Crohn’s Colitis 2023, 17, i209–i210. [Google Scholar] [CrossRef]
- Sabapaty, A.; Lin, P.-Y.; Dunn, J.C.Y. Effect of Air-Liquid Interface on Cultured Human Intestinal Epithelial Cells. FASEB BioAdv. 2024, 6, 41–52. [Google Scholar] [CrossRef]
- Wang, Y.; Chiang, I.-L.; Ohara, T.E.; Fujii, S.; Cheng, J.; Muegge, B.D.; Heul, A.V.; Han, N.D.; Lu, Q.; Xiong, S.; et al. Long-Term Culture Captures Injury-Repair Cycles of Colonic Stem Cells. Cell 2019, 179, 1144–1159.e15. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Nadauld, L.; Ootani, A.; Corney, D.C.; Pai, R.K.; Gevaert, O.; Cantrell, M.A.; Rack, P.G.; Neal, J.T.; Chan, C.W.-M.; et al. Oncogenic Transformation of Diverse Gastrointestinal Tissues in Primary Organoid Culture. Nat. Med. 2014, 20, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Drost, J.; van Jaarsveld, R.H.; Ponsioen, B.; Zimberlin, C.; van Boxtel, R.; Buijs, A.; Sachs, N.; Overmeer, R.M.; Offerhaus, G.J.; Begthel, H.; et al. Sequential Cancer Mutations in Cultured Human Intestinal Stem Cells. Nature 2015, 521, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wen, H.; Li, F.; Xue, X.; Sun, X.; Li, F.; Hu, R.; Xi, H.; Boccellato, F.; Meyer, T.F.; et al. Revealing the Pathogenesis of Gastric Intestinal Metaplasia Based on the Mucosoid Air-Liquid Interface. J. Transl. Med. 2024, 22, 468. [Google Scholar] [CrossRef]
- Kim, R.; Allbritton, N.L. A Microphysiological System with an Anaerobic Air-Liquid Interface and Functional Mucus Layer for Coculture of Intestinal Bacteria and Primary Human Colonic Epithelium. Adv. Mater. Interfaces 2024, 11, 2400093. [Google Scholar] [CrossRef]
- Ren, Y.; Yang, X.; Ma, Z.; Sun, X.; Zhang, Y.; Li, W.; Yang, H.; Qiang, L.; Yang, Z.; Liu, Y.; et al. Developments and Opportunities for 3D Bioprinted Organoids. Int. J. Bioprinting 2021, 7, 364. [Google Scholar] [CrossRef]
- Brassard, J.A.; Nikolaev, M.; Hübscher, T.; Hofer, M.; Lutolf, M.P. Recapitulating Macro-Scale Tissue Self-Organization through Organoid Bioprinting. Nat. Mater. 2021, 20, 22–29. [Google Scholar] [CrossRef]
- Han, H.; Park, Y.; Choi, Y.-M.; Yong, U.; Kang, B.; Shin, W.; Min, S.; Kim, H.J.; Jang, J. A Bioprinted Tubular Intestine Model Using a Colon-Specific Extracellular Matrix Bioink. Adv. Healthc. Mater. 2022, 11, e2101768. [Google Scholar] [CrossRef]
- Cadamuro, F.; Marongiu, L.; Marino, M.; Tamini, N.; Nespoli, L.; Zucchini, N.; Terzi, A.; Altamura, D.; Gao, Z.; Giannini, C.; et al. 3D Bioprinted Colorectal Cancer Models Based on Hyaluronic Acid and Signalling Glycans. Carbohydr. Polym. 2023, 302, 120395. [Google Scholar] [CrossRef]
- Sun, H.; Sun, L.; Ke, X.; Liu, L.; Li, C.; Jin, B.; Wang, P.; Jiang, Z.; Zhao, H.; Yang, Z.; et al. Prediction of Clinical Precision Chemotherapy by Patient-Derived 3D Bioprinting Models of Colorectal Cancer and Its Liver Metastases. Adv. Sci. 2024, 11, e2304460. [Google Scholar] [CrossRef]
- Harter, M.F.; Recaldin, T.; Gerard, R.; Avignon, B.; Bollen, Y.; Esposito, C.; Guja-Jarosz, K.; Kromer, K.; Filip, A.; Aubert, J.; et al. Analysis of Off-Tumour Toxicities of T-Cell-Engaging Bispecific Antibodies via Donor-Matched Intestinal Organoids and Tumouroids. Nat. Biomed. Eng. 2024, 8, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Workman, M.J.; Mahe, M.M.; Trisno, S.; Poling, H.M.; Watson, C.L.; Sundaram, N.; Chang, C.-F.; Schiesser, J.; Aubert, P.; Stanley, E.G.; et al. Engineered Human Pluripotent-Stem-Cell-Derived Intestinal Tissues with a Functional Enteric Nervous System. Nat. Med. 2017, 23, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Ihara, S.; Hirata, Y.; Hikiba, Y.; Yamashita, A.; Tsuboi, M.; Hata, M.; Konishi, M.; Suzuki, N.; Sakitani, K.; Kinoshita, H.; et al. Adhesive Interactions between Mononuclear Phagocytes and Intestinal Epithelium Perturb Normal Epithelial Differentiation and Serve as a Therapeutic Target in Inflammatory Bowel Disease. J. Crohn’s Colitis 2018, 12, 1219–1231. [Google Scholar] [CrossRef]
- Lin, M.; Hartl, K.; Heuberger, J.; Beccaceci, G.; Berger, H.; Li, H.; Liu, L.; Müllerke, S.; Conrad, T.; Heymann, F.; et al. Establishment of Gastrointestinal Assembloids to Study the Interplay between Epithelial Crypts and Their Mesenchymal Niche. Nat. Commun. 2023, 14, 3025. [Google Scholar] [CrossRef]
- Eicher, A.K.; Kechele, D.O.; Sundaram, N.; Berns, H.M.; Poling, H.M.; Haines, L.E.; Sanchez, J.G.; Kishimoto, K.; Krishnamurthy, M.; Han, L.; et al. Functional Human Gastrointestinal Organoids Can Be Engineered from Three Primary Germ Layers Derived Separately from Pluripotent Stem Cells. Cell Stem Cell 2022, 29, 36–51.e6. [Google Scholar] [CrossRef]
- Teijeira, A.; Migueliz, I.; Garasa, S.; Karanikas, V.; Luri, C.; Cirella, A.; Olivera, I.; Cañamero, M.; Alvarez, M.; Ochoa, M.C.; et al. Three-Dimensional Colon Cancer Organoids Model the Response to CEA-CD3 T-Cell Engagers. Theranostics 2022, 12, 1373–1387. [Google Scholar] [CrossRef]
- Sasaki, N.; Miyamoto, K.; Maslowski, K.M.; Ohno, H.; Kanai, T.; Sato, T. Development of a Scalable Coculture System for Gut Anaerobes and Human Colon Epithelium. Gastroenterology 2020, 159, 388–390.e5. [Google Scholar] [CrossRef]
- Thomas, A.M.; Manghi, P.; Asnicar, F.; Pasolli, E.; Armanini, F.; Zolfo, M.; Beghini, F.; Manara, S.; Karcher, N.; Pozzi, C.; et al. Metagenomic Analysis of Colorectal Cancer Datasets Identifies Cross-Cohort Microbial Diagnostic Signatures and a Link with Choline Degradation. Nat. Med. 2019, 25, 667–678. [Google Scholar] [CrossRef]
- Wirbel, J.; Pyl, P.T.; Kartal, E.; Zych, K.; Kashani, A.; Milanese, A.; Fleck, J.S.; Voigt, A.Y.; Palleja, A.; Ponnudurai, R.; et al. Meta-Analysis of Fecal Metagenomes Reveals Global Microbial Signatures That Are Specific for Colorectal Cancer. Nat. Med. 2019, 25, 679–689. [Google Scholar] [CrossRef]
- Cuevas-Ramos, G.; Petit, C.R.; Marcq, I.; Boury, M.; Oswald, E.; Nougayrède, J.-P. Escherichia Coli Induces DNA Damage in Vivo and Triggers Genomic Instability in Mammalian Cells. Proc. Natl. Acad. Sci. USA 2010, 107, 11537–11542. [Google Scholar] [CrossRef]
- Wilson, M.R.; Jiang, Y.; Villalta, P.W.; Stornetta, A.; Boudreau, P.D.; Carrá, A.; Brennan, C.A.; Chun, E.; Ngo, L.; Samson, L.D.; et al. The Human Gut Bacterial Genotoxin Colibactin Alkylates DNA. Science 2019, 363, eaar7785. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Kim, C.S.; Healy, A.R.; Wernke, K.M.; Wang, Z.; Frischling, M.C.; Shine, E.E.; Wang, W.; Herzon, S.B.; Crawford, J.M. Structure Elucidation of Colibactin and Its DNA Cross-Links. Science 2019, 365, eaax2685. [Google Scholar] [CrossRef] [PubMed]
- Iftekhar, A.; Berger, H.; Bouznad, N.; Heuberger, J.; Boccellato, F.; Dobrindt, U.; Hermeking, H.; Sigal, M.; Meyer, T.F. Genomic Aberrations after Short-Term Exposure to Colibactin-Producing E. coli Transform Primary Colon Epithelial Cells. Nat. Commun. 2021, 12, 1003. [Google Scholar] [CrossRef] [PubMed]
- Pleguezuelos-Manzano, C.; Puschhof, J.; Rosendahl Huber, A.; van Hoeck, A.; Wood, H.M.; Nomburg, J.; Gurjao, C.; Manders, F.; Dalmasso, G.; Stege, P.B.; et al. Mutational Signature in Colorectal Cancer Caused by Genotoxic Pks+ E. coli. Nature 2020, 580, 269–273. [Google Scholar] [CrossRef]
- Rodvold, J.J.; Grimmer, M.; Ruiz, K.; Marsters, S.A.; Oikonomidi, I.; Tan-Aristy, E.; Pham, V.C.; Sarkar, T.; Harnoss, J.M.; Shatz-Binder, W.; et al. ATF6 Promotes Colorectal Cancer Growth and Stemness by Regulating the Wnt Pathway. Cancer Res. Commun. 2024, 4, 2734–2755. [Google Scholar] [CrossRef]
- Xiao, D.; Deng, Q.; Guo, Y.; Huang, X.; Zou, M.; Zhong, J.; Rao, P.; Xu, Z.; Liu, Y.; Hu, Y.; et al. Generation of Self-Organized Sensory Ganglion Organoids and Retinal Ganglion Cells from Fibroblasts. Sci. Adv. 2020, 6, eaaz5858. [Google Scholar] [CrossRef]
- Bian, X.; Li, G.; Wang, C.; Liu, W.; Lin, X.; Chen, Z.; Cheung, M.; Luo, X. A Deep Learning Model for Detection and Tracking in High-Throughput Images of Organoid. Comput. Biol. Med. 2021, 134, 104490. [Google Scholar] [CrossRef]
- Gritti, N.; Lim, J.L.; Anlaş, K.; Pandya, M.; Aalderink, G.; Martínez-Ara, G.; Trivedi, V. MOrgAna: Accessible Quantitative Analysis of Organoids with Machine Learning. Development 2021, 148, dev199611. [Google Scholar] [CrossRef]
- Powell, R.T.; Moussalli, M.J.; Guo, L.; Bae, G.; Singh, P.; Stephan, C.; Shureiqi, I.; Davies, P.J. DeepOrganoid: A Brightfield Cell Viability Model for Screening Matrix-Embedded Organoids. SLAS Discov. 2022, 27, 175–184. [Google Scholar] [CrossRef]
- Park, T.; Kim, T.K.; Han, Y.D.; Kim, K.-A.; Kim, H.; Kim, H.S. Development of a Deep Learning Based Image Processing Tool for Enhanced Organoid Analysis. Sci. Rep. 2023, 13, 19841. [Google Scholar] [CrossRef]
- Matthews, J.M.; Schuster, B.; Kashaf, S.S.; Liu, P.; Ben-Yishay, R.; Ishay-Ronen, D.; Izumchenko, E.; Shen, L.; Weber, C.R.; Bielski, M.; et al. OrganoID: A Versatile Deep Learning Platform for Tracking and Analysis of Single-Organoid Dynamics. PLoS Comput. Biol. 2022, 18, e1010584. [Google Scholar] [CrossRef]
- Abdul, L.; Xu, J.; Sotra, A.; Chaudary, A.; Gao, J.; Rajasekar, S.; Anvari, N.; Mahyar, H.; Zhang, B. D-CryptO: Deep Learning-Based Analysis of Colon Organoid Morphology from Brightfield Images. Lab. Chip 2022, 22, 4118–4128. [Google Scholar] [CrossRef] [PubMed]
- Lefferts, J.W.; Kroes, S.; Smith, M.B.; Niemöller, P.J.; Nieuwenhuijze, N.D.A.; van Kooten, H.N.S.; van der Ent, C.K.; Beekman, J.M.; van Beuningen, S.F.B. OrgaSegment: Deep-Learning Based Organoid Segmentation to Quantify CFTR Dependent Fluid Secretion. Commun. Biol. 2024, 7, 319. [Google Scholar] [CrossRef]
- Dekkers, J.F.; Wiegerinck, C.L.; de Jonge, H.R.; Bronsveld, I.; Janssens, H.M.; de Winter-de Groot, K.M.; Brandsma, A.M.; de Jong, N.W.M.; Bijvelds, M.J.C.; Scholte, B.J.; et al. A Functional CFTR Assay Using Primary Cystic Fibrosis Intestinal Organoids. Nat. Med. 2013, 19, 939–945. [Google Scholar] [CrossRef]
- Tomer, R.; Ye, L.; Hsueh, B.; Deisseroth, K. Advanced CLARITY for Rapid and High-Resolution Imaging of Intact Tissues. Nat. Protoc. 2014, 9, 1682–1697. [Google Scholar] [CrossRef]
- Ke, M.-T.; Fujimoto, S.; Imai, T. SeeDB: A Simple and Morphology-Preserving Optical Clearing Agent for Neuronal Circuit Reconstruction. Nat. Neurosci. 2013, 16, 1154–1161. [Google Scholar] [CrossRef]
- Unnersjö-Jess, D.; Scott, L.; Blom, H.; Brismar, H. Super-Resolution Stimulated Emission Depletion Imaging of Slit Diaphragm Proteins in Optically Cleared Kidney Tissue. Kidney Int. 2016, 89, 243–247. [Google Scholar] [CrossRef]
- Edwards, S.J.; Carannante, V.; Kuhnigk, K.; Ring, H.; Tararuk, T.; Hallböök, F.; Blom, H.; Önfelt, B.; Brismar, H. High-Resolution Imaging of Tumor Spheroids and Organoids Enabled by Expansion Microscopy. Front. Mol. Biosci. 2020, 7, 208. [Google Scholar] [CrossRef]
- Dekkers, J.F.; Alieva, M.; Wellens, L.M.; Ariese, H.C.R.; Jamieson, P.R.; Vonk, A.M.; Amatngalim, G.D.; Hu, H.; Oost, K.C.; Snippert, H.J.G.; et al. High-Resolution 3D Imaging of Fixed and Cleared Organoids. Nat. Protoc. 2019, 14, 1756–1771. [Google Scholar] [CrossRef]
- Krzic, U.; Gunther, S.; Saunders, T.E.; Streichan, S.J.; Hufnagel, L. Multiview Light-Sheet Microscope for Rapid in Toto Imaging. Nat. Methods 2012, 9, 730–733. [Google Scholar] [CrossRef]
- Eismann, B.; Krieger, T.G.; Beneke, J.; Bulkescher, R.; Adam, L.; Erfle, H.; Herrmann, C.; Eils, R.; Conrad, C. Automated 3D Light-Sheet Screening with High Spatiotemporal Resolution Reveals Mitotic Phenotypes. J. Cell Sci. 2020, 133, jcs245043. [Google Scholar] [CrossRef] [PubMed]
- Hof, L.; Moreth, T.; Koch, M.; Liebisch, T.; Kurtz, M.; Tarnick, J.; Lissek, S.M.; Verstegen, M.M.A.; van der Laan, L.J.W.; Huch, M.; et al. Long-Term Live Imaging and Multiscale Analysis Identify Heterogeneity and Core Principles of Epithelial Organoid Morphogenesis. BMC Biol. 2021, 19, 37. [Google Scholar] [CrossRef] [PubMed]
- de Medeiros, G.; Ortiz, R.; Strnad, P.; Boni, A.; Moos, F.; Repina, N.; Challet Meylan, L.; Maurer, F.; Liberali, P. Multiscale Light-Sheet Organoid Imaging Framework. Nat. Commun. 2022, 13, 4864. [Google Scholar] [CrossRef]
- Schöneberg, J.; Dambournet, D.; Liu, T.-L.; Forster, R.; Hockemeyer, D.; Betzig, E.; Drubin, D.G. 4D Cell Biology: Big Data Image Analytics and Lattice Light-Sheet Imaging Reveal Dynamics of Clathrin-Mediated Endocytosis in Stem Cell–Derived Intestinal Organoids. Mol. Biol. Cell 2018, 29, 2959–2968. [Google Scholar] [CrossRef]
- Boehnke, K.; Iversen, P.W.; Schumacher, D.; Lallena, M.J.; Haro, R.; Amat, J.; Haybaeck, J.; Liebs, S.; Lange, M.; Schäfer, R.; et al. Assay Establishment and Validation of a High-Throughput Screening Platform for Three-Dimensional Patient-Derived Colon Cancer Organoid Cultures. J. Biomol. Screen. 2016, 21, 931–941. [Google Scholar] [CrossRef]
- Du, Y.; Li, X.; Niu, Q.; Mo, X.; Qui, M.; Ma, T.; Kuo, C.J.; Fu, H. Development of a Miniaturized 3D Organoid Culture Platform for Ultra-High-Throughput Screening. J. Mol. Cell Biol. 2020, 12, 630–643. [Google Scholar] [CrossRef]
- Luo, Z.; Wang, B.; Luo, F.; Guo, Y.; Jiang, N.; Wei, J.; Wang, X.; Tseng, Y.; Chen, J.; Zhao, B.; et al. Establishment of a Large-Scale Patient-Derived High-Risk Colorectal Adenoma Organoid Biobank for High-Throughput and High-Content Drug Screening. BMC Med. 2023, 21, 336. [Google Scholar] [CrossRef]
- Bozal, S.B.; Sjogren, G.; Costa, A.P.; Brown, J.S.; Roberts, S.; Baker, D.; Gabriel, P.; Ristau, B.T.; Samuels, M.; Flynn, W.F.; et al. Development of an Automated 3D High Content Cell Screening Platform for Organoid Phenotyping. SLAS Discov. 2024, 29, 100182. [Google Scholar] [CrossRef]
- Sakshaug, B.C.; Folkesson, E.; Haukaas, T.H.; Visnes, T.; Flobak, Å. Systematic Review: Predictive Value of Organoids in Colorectal Cancer. Sci. Rep. 2023, 13, 18124. [Google Scholar] [CrossRef]
- Qu, S.; Xu, R.; Yi, G.; Li, Z.; Zhang, H.; Qi, S.; Huang, G. Patient-Derived Organoids in Human Cancer: A Platform for Fundamental Research and Precision Medicine. Mol. Biomed. 2024, 5, 6. [Google Scholar] [CrossRef]
- Mao, Y.; Wang, W.; Yang, J.; Zhou, X.; Lu, Y.; Gao, J.; Wang, X.; Wen, L.; Fu, W.; Tang, F. Drug Repurposing Screening and Mechanism Analysis Based on Human Colorectal Cancer Organoids. Protein Cell 2024, 15, 285–304. [Google Scholar] [CrossRef] [PubMed]
- Obreque, J.; Vergara-Gómez, L.; Venegas, N.; Weber, H.; Owen, G.I.; Pérez-Moreno, P.; Leal, P.; Roa, J.C.; Bizama, C. Advances towards the Use of Gastrointestinal Tumor Patient-Derived Organoids as a Therapeutic Decision-Making Tool. Biol. Res. 2023, 56, 63. [Google Scholar] [CrossRef]
- Jose, A.; Kulkarni, P.; Thilakan, J.; Munisamy, M.; Malhotra, A.G.; Singh, J.; Kumar, A.; Rangnekar, V.M.; Arya, N.; Rao, M. Integration of Pan-Omics Technologies and Three-Dimensional In Vitro Tumor Models: An Approach toward Drug Discovery and Precision Medicine. Mol. Cancer 2024, 23, 50. [Google Scholar] [CrossRef]
- Kretzschmar, K. Cancer Research Using Organoid Technology. J. Mol. Med. 2021, 99, 501–515. [Google Scholar] [CrossRef]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-Derived Organoids Model Treatment Response of Metastatic Gastrointestinal Cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Cho, E.J.; Kim, M.; Jo, D.; Kim, J.; Oh, J.-H.; Chung, H.C.; Lee, S.-H.; Kim, D.; Chun, S.-M.; Kim, J.; et al. Immuno-Genomic Classification of Colorectal Cancer Organoids Reveals Cancer Cells with Intrinsic Immunogenic Properties Associated with Patient Survival. J. Exp. Clin. Cancer Res. 2021, 40, 230. [Google Scholar] [CrossRef]
- Drost, J.; van Boxtel, R.; Blokzijl, F.; Mizutani, T.; Sasaki, N.; Sasselli, V.; de Ligt, J.; Behjati, S.; Grolleman, J.E.; van Wezel, T.; et al. Use of CRISPR-Modified Human Stem Cell Organoids to Study the Origin of Mutational Signatures in Cancer. Science 2017, 358, 234–238. [Google Scholar] [CrossRef]
- Christensen, S.; Van der Roest, B.; Besselink, N.; Janssen, R.; Boymans, S.; Martens, J.W.M.; Yaspo, M.-L.; Priestley, P.; Kuijk, E.; Cuppen, E.; et al. 5-Fluorouracil Treatment Induces Characteristic T>G Mutations in Human Cancer. Nat. Commun. 2019, 10, 4571. [Google Scholar] [CrossRef]
- Tindle, C.; Fonseca, A.G.; Taheri, S.; Katkar, G.D.; Lee, J.; Maity, P.; Sayed, I.M.; Ibeawuchi, S.-R.; Vidales, E.; Pranadinata, R.F.; et al. A Living Organoid Biobank of Patients with Crohn’s Disease Reveals Molecular Subtypes for Personalized Therapeutics. Cell Rep. Med. 2024, 5, 101748. [Google Scholar] [CrossRef]
- Geurts, M.H.; de Poel, E.; Amatngalim, G.D.; Oka, R.; Meijers, F.M.; Kruisselbrink, E.; van Mourik, P.; Berkers, G.; de Groot, K.M.W.; Michel, S.; et al. CRISPR-Based Adenine Editors Correct Nonsense Mutations in a Cystic Fibrosis Organoid Biobank. Cell Stem Cell 2020, 26, 503–510.e7. [Google Scholar] [CrossRef] [PubMed]
- Toshimitsu, K.; Takano, A.; Fujii, M.; Togasaki, K.; Matano, M.; Takahashi, S.; Kanai, T.; Sato, T. Organoid Screening Reveals Epigenetic Vulnerabilities in Human Colorectal Cancer. Nat. Chem. Biol. 2022, 18, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Devall, M.A.; Eaton, S.; Ali, M.W.; Dampier, C.H.; Weisenberger, D.; Powell, S.M.; Li, L.; Casey, G. DNA Methylation Analysis of Normal Colon Organoids from Familial Adenomatous Polyposis Patients Reveals Novel Insight into Colon Cancer Development. Clin. Epigenetics 2022, 14, 104. [Google Scholar] [CrossRef] [PubMed]
- Devall, M.; Ali, M.W.; Eaton, S.; Weisenberger, D.J.; Reilley, M.J.; Powell, S.M.; Li, L.; Casey, G. Multi-Omic Analysis in Normal Colon Organoids Highlights MSH4 as a Novel Marker of Defective Mismatch Repair in Lynch Syndrome and Microsatellite Instability. Cancer Med. 2023, 12, 13551–13572. [Google Scholar] [CrossRef]
- Della Chiara, G.; Gervasoni, F.; Fakiola, M.; Godano, C.; D’Oria, C.; Azzolin, L.; Bonnal, R.J.P.; Moreni, G.; Drufuca, L.; Rossetti, G.; et al. Epigenomic Landscape of Human Colorectal Cancer Unveils an Aberrant Core of Pan-Cancer Enhancers Orchestrated by YAP/TAZ. Nat. Commun. 2021, 12, 2340. [Google Scholar] [CrossRef]
- Dennison, T.W.; Edgar, R.D.; Payne, F.; Nayak, K.M.; Ross, A.D.B.; Cenier, A.; Glemas, C.; Giachero, F.; Foster, A.R.; Harris, R.; et al. Patient-Derived Organoid Biobank Identifies Epigenetic Dysregulation of Intestinal Epithelial MHC-I as a Novel Mechanism in Severe Crohn’s Disease. Gut 2024, 73, 1464–1477. [Google Scholar] [CrossRef]
- Kraiczy, J.; Nayak, K.M.; Howell, K.J.; Ross, A.; Forbester, J.; Salvestrini, C.; Mustata, R.; Perkins, S.; Andersson-Rolf, A.; Leenen, E.; et al. DNA Methylation Defines Regional Identity of Human Intestinal Epithelial Organoids and Undergoes Dynamic Changes during Development. Gut 2019, 68, 49–61. [Google Scholar] [CrossRef]
- Wang, R.; Mao, Y.; Wang, W.; Zhou, X.; Wang, W.; Gao, S.; Li, J.; Wen, L.; Fu, W.; Tang, F. Systematic Evaluation of Colorectal Cancer Organoid System by Single-Cell RNA-Seq Analysis. Genome Biol. 2022, 23, 106. [Google Scholar] [CrossRef]
- Tung, K.-L.; Chen, K.-Y.; Negrete, M.; Chen, T.; Safi, A.; Aljamal, A.A.; Song, L.; Crawford, G.E.; Ding, S.; Hsu, D.S.; et al. Integrated Chromatin and Transcriptomic Profiling of Patient-Derived Colon Cancer Organoids Identifies Personalized Drug Targets to Overcome Oxaliplatin Resistance. Genes Dis. 2021, 8, 203–214. [Google Scholar] [CrossRef]
- Ryu, K.-B.; Seo, J.-A.; Lee, K.; Choi, J.; Yoo, G.; Ha, J.-H.; Ahn, M.R. Drug-Resistance Biomarkers in Patient-Derived Colorectal Cancer Organoid and Fibroblast Co-Culture System. Curr. Issues Mol. Biol. 2024, 46, 5794–5811. [Google Scholar] [CrossRef]
- Papaccio, F.; García-Mico, B.; Gimeno-Valiente, F.; Cabeza-Segura, M.; Gambardella, V.; Gutiérrez-Bravo, M.F.; Alfaro-Cervelló, C.; Martinez-Ciarpaglini, C.; Rentero-Garrido, P.; Zúñiga-Trejos, S.; et al. Proteotranscriptomic Analysis of Advanced Colorectal Cancer Patient Derived Organoids for Drug Sensitivity Prediction. J. Exp. Clin. Cancer Res. 2023, 42, 8. [Google Scholar] [CrossRef] [PubMed]
- Murthy, S.; Anbazhagan, M.; Maddipatla, S.C.; Kolachala, V.L.; Dodd, A.; Pelia, R.; Cutler, D.J.; Matthews, J.D.; Kugathasan, S. Single-Cell Transcriptomics of Rectal Organoids from Individuals with Perianal Fistulizing Crohn’s Disease Reveals Patient-Specific Signatures. Sci. Rep. 2024, 14, 26142. [Google Scholar] [CrossRef]
- Gonneaud, A.; Jones, C.; Turgeon, N.; Lévesque, D.; Asselin, C.; Boudreau, F.; Boisvert, F.-M. A SILAC-Based Method for Quantitative Proteomic Analysis of Intestinal Organoids. Sci. Rep. 2016, 6, 38195. [Google Scholar] [CrossRef]
- Cristobal, A.; van den Toorn, H.W.P.; van de Wetering, M.; Clevers, H.; Heck, A.J.R.; Mohammed, S. Personalized Proteome Profiles of Healthy and Tumor Human Colon Organoids Reveal Both Individual Diversity and Basic Features of Colorectal Cancer. Cell Rep. 2017, 18, 263–274. [Google Scholar] [CrossRef]
- Pratscher, B.; Kuropka, B.; Csukovich, G.; Doulidis, P.G.; Spirk, K.; Kramer, N.; Freund, P.; Rodríguez-Rojas, A.; Burgener, I.A. Traces of Canine Inflammatory Bowel Disease Reflected by Intestinal Organoids. Int. J. Mol. Sci. 2024, 25, 576. [Google Scholar] [CrossRef]
- van der Kemp, W.J.M.; Grinde, M.T.; Malvik, J.O.; van Laarhoven, H.W.M.; Prompers, J.J.; Klomp, D.W.J.; Burgering, B.; Bathen, T.F.; Moestue, S.A. Metabolic Profiling of Colorectal Cancer Organoids: A Comparison between High-Resolution Magic Angle Spinning Magnetic Resonance Spectroscopy and Solution Nuclear Magnetic Resonance Spectroscopy of Polar Extracts. NMR Biomed. 2023, 36, e4882. [Google Scholar] [CrossRef]
- Neef, S.K.; Janssen, N.; Winter, S.; Wallisch, S.K.; Hofmann, U.; Dahlke, M.H.; Schwab, M.; Mürdter, T.E.; Haag, M. Metabolic Drug Response Phenotyping in Colorectal Cancer Organoids by LC-QTOF-MS. Metabolites 2020, 10, 494. [Google Scholar] [CrossRef]
- Bozzi, F.; Mogavero, A.; Varinelli, L.; Belfiore, A.; Manenti, G.; Caccia, C.; Volpi, C.C.; Beznoussenko, G.V.; Milione, M.; Leoni, V.; et al. MIF/CD74 Axis Is a Target for Novel Therapies in Colon Carcinomatosis. J. Exp. Clin. Cancer Res. 2017, 36, 16. [Google Scholar] [CrossRef]
- Díaz-Gay, M.; Alexandrov, L.B. Chapter Ten—Unraveling the Genomic Landscape of Colorectal Cancer through Mutational Signatures. In Advances in Cancer Research; Berger, F.G., Boland, C.R., Eds.; Novel Approaches to Colorectal Cancer; Academic Press: Cambridge, MA, USA, 2021; Volume 151, pp. 385–424. [Google Scholar]
- Günther, C.; Winner, B.; Neurath, M.F.; Stappenbeck, T.S. Organoids in Gastrointestinal Diseases: From Experimental Models to Clinical Translation. Gut 2022, 71, 1892–1908. [Google Scholar] [CrossRef]
- Nakayama, M.; Wang, D.; Kok, S.Y.; Oshima, H.; Oshima, M. Genetic Alterations and Microenvironment That Drive Malignant Progression of Colorectal Cancer: Lessons from Mouse and Organoid Models. J. Cancer Prev. 2022, 27, 1–6. [Google Scholar] [CrossRef]
- Kuipers, E.J.; Grady, W.M.; Lieberman, D.; Seufferlein, T.; Sung, J.J.; Boelens, P.G.; van de Velde, C.J.H.; Watanabe, T. Colorectal Cancer. Nat. Rev. Dis. Primers 2015, 1, 15065. [Google Scholar] [CrossRef] [PubMed]
- Le Berre, C.; Honap, S.; Peyrin-Biroulet, L. Ulcerative Colitis. Lancet 2023, 402, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Dolinger, M.; Torres, J.; Vermeire, S. Crohn’s Disease. Lancet 2024, 403, 1177–1191. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic Plasticity and the Hallmarks of Cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef]
- Yang, X.; Zhong, J.; Zhang, Q.; Feng, L.; Zheng, Z.; Zhang, J.; Lu, S. Advances and Insights of APC-Asef Inhibitors for Metastatic Colorectal Cancer Therapy. Front. Mol. Biosci. 2021, 8, 662579. [Google Scholar] [CrossRef]
- Roudko, V.; Cimen Bozkus, C.; Greenbaum, B.; Lucas, A.; Samstein, R.; Bhardwaj, N. Lynch Syndrome and MSI-H Cancers: From Mechanisms to “Off-The-Shelf” Cancer Vaccines. Front. Immunol. 2021, 12, 757804. [Google Scholar] [CrossRef]
- Roe, J.-S.; Hwang, C.-I.; Somerville, T.D.D.; Milazzo, J.P.; Lee, E.J.; Silva, B.D.; Maiorino, L.; Tiriac, H.; Young, C.M.; Miyabayashi, K.; et al. Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell 2017, 170, 875–888.e20. [Google Scholar] [CrossRef]
- Kobayashi, K.S.; van den Elsen, P.J. NLRC5: A Key Regulator of MHC Class I-Dependent Immune Responses. Nat. Rev. Immunol. 2012, 12, 813–820. [Google Scholar] [CrossRef]
- Fabbri, L.; Chakraborty, A.; Robert, C.; Vagner, S. The Plasticity of MRNA Translation during Cancer Progression and Therapy Resistance. Nat. Rev. Cancer 2021, 21, 558–577. [Google Scholar] [CrossRef]
- Gonneaud, A.; Asselin, C.; Boudreau, F.; Boisvert, F.-M. Phenotypic Analysis of Organoids by Proteomics. Proteomics 2017, 17, 1700023. [Google Scholar] [CrossRef]
- Lindhorst, P.H.; Hummon, A.B. Proteomics of Colorectal Cancer: Tumors, Organoids, and Cell Cultures—A Minireview. Front. Mol. Biosci. 2020, 7, 604492. [Google Scholar] [CrossRef]
- Hedberg Oldfors, C.; Dios, D.G.; Linder, A.; Visuttijai, K.; Samuelson, E.; Karlsson, S.; Nilsson, S.; Behboudi, A. Analysis of an Independent Tumor Suppressor Locus Telomeric to Tp53 Suggested Inpp5k and Myo1c as Novel Tumor Suppressor Gene Candidates in This Region. BMC Genet. 2015, 16, 80. [Google Scholar] [CrossRef]
- Funakoshi, S.; Ezaki, T.; Kong, J.; Guo, R.J.; Lynch, J.P. Repression of the Desmocollin 2 Gene Expression in Human Colon Cancer Cells Is Relieved by the Homeodomain Transcription Factors Cdx1 and Cdx2. Mol. Cancer Res. 2008, 6, 1478–1490. [Google Scholar] [CrossRef]
- Xie, C.; Powell, C.; Yao, M.; Wu, J.; Dong, Q. Ubiquitin-Conjugating Enzyme E2C: A Potential Cancer Biomarker. Int. J. Biochem. Cell Biol. 2014, 47, 113–117. [Google Scholar] [CrossRef]
- Raynes, D.A.; Graner, M.W.; Bagatell, R.; McLellan, C.; Guerriero, V. Increased Expression of the Hsp70 Cochaperone HspBP1 in Tumors. Tumour Biol. 2003, 24, 281–285. [Google Scholar] [CrossRef]
- Yao, Y.; Xu, X.; Yang, L.; Zhu, J.; Wan, J.; Shen, L.; Xia, F.; Fu, G.; Deng, Y.; Pan, M.; et al. Patient-Derived Organoids Predict Chemoradiation Responses of Locally Advanced Rectal Cancer. Cell Stem Cell 2020, 26, 17–26.e6. [Google Scholar] [CrossRef]
- Weeber, F.; van de Wetering, M.; Hoogstraat, M.; Dijkstra, K.K.; Krijgsman, O.; Kuilman, T.; Gadellaa-van Hooijdonk, C.G.M.; van der Velden, D.L.; Peeper, D.S.; Cuppen, E.P.J.G.; et al. Preserved Genetic Diversity in Organoids Cultured from Biopsies of Human Colorectal Cancer Metastases. Proc. Natl. Acad. Sci. USA 2015, 112, 13308–13311. [Google Scholar] [CrossRef]
- Porpora, M.; Conte, M.; Lania, G.; Bellomo, C.; Rapacciuolo, L.; Chirdo, F.G.; Auricchio, R.; Troncone, R.; Auricchio, S.; Barone, M.V.; et al. Inflammation Is Present, Persistent and More Sensitive to Proinflammatory Triggers in Celiac Disease Enterocytes. Int. J. Mol. Sci. 2022, 23, 1973. [Google Scholar] [CrossRef]
- He, G.-W.; Lin, L.; DeMartino, J.; Zheng, X.; Staliarova, N.; Dayton, T.; Begthel, H.; van de Wetering, W.J.; Bodewes, E.; van Zon, J.; et al. Optimized Human Intestinal Organoid Model Reveals Interleukin-22-Dependency of Paneth Cell Formation. Cell Stem Cell 2022, 29, 1333–1345.e6. [Google Scholar] [CrossRef]
- Martini, G.; Belli, V.; Napolitano, S.; Ciaramella, V.; Ciardiello, D.; Belli, A.; Izzo, F.; Avallone, A.; Selvaggi, F.; Menegon Tasselli, F.; et al. Establishment of Patient-Derived Tumor Organoids to Functionally Inform Treatment Decisions in Metastatic Colorectal Cancer. ESMO Open 2023, 8, 101198. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, J.; Lin, D.; Kang, H.; Luo, Y.; Wang, X.; Wang, L.; Liu, D. Forming Single-Cell-Derived Colon Cancer Organoid Arrays on a Microfluidic Chip for High Throughput Tumor Heterogeneity Analysis. ACS Biomater. Sci. Eng. 2024, 10, 5265–5273. [Google Scholar] [CrossRef] [PubMed]
- Farin, H.F.; Mosa, M.H.; Ndreshkjana, B.; Grebbin, B.M.; Ritter, B.; Menche, C.; Kennel, K.B.; Ziegler, P.K.; Szabó, L.; Bollrath, J.; et al. Colorectal Cancer Organoid-Stroma Biobank Allows Subtype-Specific Assessment of Individualized Therapy Responses. Cancer Discov. 2023, 13, 2192–2211. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Kobayashi, S.; Ogasawara, N.; Okamoto, R.; Nakamura, T.; Watanabe, M.; Jensen, K.B.; Yui, S. Transplantation of Intestinal Organoids into a Mouse Model of Colitis. Nat. Protoc. 2022, 17, 649–671. [Google Scholar] [CrossRef]
- Hou, Q.; Huang, J.; Ayansola, H.; Masatoshi, H.; Zhang, B. Intestinal Stem Cells and Immune Cell Relationships: Potential Therapeutic Targets for Inflammatory Bowel Diseases. Front. Immunol. 2020, 11, 623691. [Google Scholar] [CrossRef]
- Qu, M.; Xiong, L.; Lyu, Y.; Zhang, X.; Shen, J.; Guan, J.; Chai, P.; Lin, Z.; Nie, B.; Li, C.; et al. Establishment of Intestinal Organoid Cultures Modeling Injury-Associated Epithelial Regeneration. Cell Res. 2021, 31, 259–271. [Google Scholar] [CrossRef]
- Wilks, M. Bacteria and Early Human Development. Early Hum. Dev. 2007, 83, 165–170. [Google Scholar] [CrossRef]
- Wu, H.; Xie, S.; Miao, J.; Li, Y.; Wang, Z.; Wang, M.; Yu, Q. Lactobacillus Reuteri Maintains Intestinal Epithelial Regeneration and Repairs Damaged Intestinal Mucosa. Gut Microbes 2020, 11, 997–1014. [Google Scholar] [CrossRef]
- Spelier, S.; de Poel, E.; Ithakisiou, G.N.; Suen, S.W.F.; Hagemeijer, M.C.; Muilwijk, D.; Vonk, A.M.; Brunsveld, J.E.; Kruisselbrink, E.; van der Ent, C.K.; et al. High-Throughput Functional Assay in Cystic Fibrosis Patient-Derived Organoids Allows Drug Repurposing. ERJ Open Res. 2023, 9, 00495–02022. [Google Scholar] [CrossRef]
- Bulcaen, M.; Kortleven, P.; Liu, R.B.; Maule, G.; Dreano, E.; Kelly, M.; Ensinck, M.M.; Thierie, S.; Smits, M.; Ciciani, M.; et al. Prime Editing Functionally Corrects Cystic Fibrosis-Causing CFTR Mutations in Human Organoids and Airway Epithelial Cells. Cell Rep. Med. 2024, 5, 101544. [Google Scholar] [CrossRef]
- Shen, X.; Zhang, Y.; Xu, Z.; Gao, H.; Feng, W.; Li, W.; Miao, Y.; Xu, Z.; Zong, Y.; Zhao, J.; et al. KLF5 Inhibition Overcomes Oxaliplatin Resistance in Patient-Derived Colorectal Cancer Organoids by Restoring Apoptotic Response. Cell Death Dis. 2022, 13, 303. [Google Scholar] [CrossRef]
- Usui, T.; Sakurai, M.; Umata, K.; Elbadawy, M.; Ohama, T.; Yamawaki, H.; Hazama, S.; Takenouchi, H.; Nakajima, M.; Tsunedomi, R.; et al. Hedgehog Signals Mediate Anti-Cancer Drug Resistance in Three-Dimensional Primary Colorectal Cancer Organoid Culture. Int. J. Mol. Sci. 2018, 19, 1098. [Google Scholar] [CrossRef] [PubMed]
- Khalifa, A.; Guijarro, A.; Ravera, S.; Bertola, N.; Adorni, M.P.; Papotti, B.; Raffaghello, L.; Benelli, R.; Becherini, P.; Namatalla, A.; et al. Cyclic Fasting Bolsters Cholesterol Biosynthesis Inhibitors’ Anticancer Activity. Nat. Commun. 2023, 14, 6951. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Kwon, J.; Shin, H.-J.; Moon, S.M.; Kim, S.B.; Shin, U.S.; Han, Y.-H.; Kim, Y. Butyrate Enhances the Efficacy of Radiotherapy via FOXO3A in Colorectal Cancer Patient-derived Organoids. Int. J. Oncol. 2020, 57, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, T.; Goldhardt, T.; Edelmann, M.; Rogge, T.; Rauch, K.; Kyuchukov, N.D.; Menck, K.; Bleckmann, A.; Kalucka, J.; Khan, S.; et al. Effects of the Novel PFKFB3 Inhibitor KAN0438757 on Colorectal Cancer Cells and Its Systemic Toxicity Evaluation In Vivo. Cancers 2021, 13, 1011. [Google Scholar] [CrossRef]
- Hsu, K.-S.; Adileh, M.; Martin, M.L.; Makarov, V.; Chen, J.; Wu, C.; Bodo, S.; Klingler, S.; Sauvé, C.-E.G.; Szeglin, B.C.; et al. Colorectal Cancer Develops Inherent Radiosensitivity That Can Be Predicted Using Patient-Derived Organoids. Cancer Res. 2022, 82, 2298–2312. [Google Scholar] [CrossRef]
- Janakiraman, H.; Zhu, Y.; Becker, S.A.; Wang, C.; Cross, A.; Curl, E.; Lewin, D.; Hoffman, B.J.; Warren, G.W.; Hill, E.G.; et al. Modeling Rectal Cancer to Advance Neoadjuvant Precision Therapy. Int. J. Cancer 2020, 147, 1405–1418. [Google Scholar] [CrossRef]
- Narasimhan, V.; Wright, J.A.; Churchill, M.; Wang, T.; Rosati, R.; Lannagan, T.R.M.; Vrbanac, L.; Richardson, A.B.; Kobayashi, H.; Price, T.; et al. Medium-Throughput Drug Screening of Patient-Derived Organoids from Colorectal Peritoneal Metastases to Direct Personalized Therapy. Clin. Cancer Res. 2020, 26, 3662–3670. [Google Scholar] [CrossRef]
- Mo, S.; Tang, P.; Luo, W.; Zhang, L.; Li, Y.; Hu, X.; Ma, X.; Chen, Y.; Bao, Y.; He, X.; et al. Patient-Derived Organoids from Colorectal Cancer with Paired Liver Metastasis Reveal Tumor Heterogeneity and Predict Response to Chemotherapy. Adv. Sci. 2022, 9, e2204097. [Google Scholar] [CrossRef]
- Geevimaan, K.; Guo, J.-Y.; Shen, C.-N.; Jiang, J.-K.; Fann, C.S.J.; Hwang, M.-J.; Shui, J.-W.; Lin, H.-T.; Wang, M.-J.; Shih, H.-C.; et al. Patient-Derived Organoid Serves as a Platform for Personalized Chemotherapy in Advanced Colorectal Cancer Patients. Front. Oncol. 2022, 12, 883437. [Google Scholar] [CrossRef]
- Ooft, S.N.; Weeber, F.; Schipper, L.; Dijkstra, K.K.; McLean, C.M.; Kaing, S.; van de Haar, J.; Prevoo, W.; van Werkhoven, E.; Snaebjornsson, P.; et al. Prospective Experimental Treatment of Colorectal Cancer Patients Based on Organoid Drug Responses. ESMO Open 2021, 6, 100103. [Google Scholar] [CrossRef]
- Cho, Y.-W.; Min, D.-W.; Kim, H.-P.; An, Y.; Kim, S.; Youk, J.; Chun, J.; Im, J.P.; Song, S.-H.; Ju, Y.S.; et al. Patient-Derived Organoids as a Preclinical Platform for Precision Medicine in Colorectal Cancer. Mol. Oncol. 2022, 16, 2396–2412. [Google Scholar] [CrossRef] [PubMed]
- Ooft, S.N.; Weeber, F.; Dijkstra, K.K.; McLean, C.M.; Kaing, S.; van Werkhoven, E.; Schipper, L.; Hoes, L.; Vis, D.J.; van de Haar, J.; et al. Patient-Derived Organoids Can Predict Response to Chemotherapy in Metastatic Colorectal Cancer Patients. Sci. Transl. Med. 2019, 11, eaay2574. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Pan, W.; Zheng, H.; Zheng, H.; Wang, Z.; Li, J.J.; Deng, C.; Yan, J. Accuracy of Using a Patient-Derived Tumor Organoid Culture Model to Predict the Response to Chemotherapy Regimens In Stage IV Colorectal Cancer: A Blinded Study. Dis. Colon Rectum 2021, 64, 833–850. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Zao, X.-L.; Pan, X.-F.; Zhang, H.-G.; Wang, F.-J.; Qiao, P.-F. Application of Tumoroids Derived from Advanced Colorectal Cancer Patients to Predict Individual Response to Chemotherapy. J. Chemother. 2023, 35, 104–116. [Google Scholar] [CrossRef]
- Strauss, J.; Figg, W.D. Epigenetic Approaches to Overcoming Chemotherapy Resistance. Lancet Oncol. 2015, 16, 1013–1015. [Google Scholar] [CrossRef]
- El Amrani, M.; Corfiotti, F.; Corvaisier, M.; Vasseur, R.; Fulbert, M.; Skrzypczyk, C.; Deshorgues, A.-C.; Gnemmi, V.; Tulasne, D.; Lahdaoui, F.; et al. Gemcitabine-Induced Epithelial-Mesenchymal Transition-like Changes Sustain Chemoresistance of Pancreatic Cancer Cells of Mesenchymal-like Phenotype. Mol. Carcinog. 2019, 58, 1985–1997. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Yan, S.; Wang, W.; Feng, Z.; Xue, J.; Liang, W.; Wu, X.; Tan, Z.; Zhang, X.; Zhang, S.; Li, X.; et al. Immune Checkpoint Inhibitors in Colorectal Cancer: Limitation and Challenges. Front. Immunol. 2024, 15, 1403533. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, X.; Zhou, H.; Jiang, M.; Wang, J.; Sui, B. Molecular Complexity of Colorectal Cancer: Pathways, Biomarkers, and Therapeutic Strategies. Cancer Manag. Res. 2024, 16, 1389–1403. [Google Scholar] [CrossRef]
- Hoffmann, O.I.; Ilmberger, C.; Magosch, S.; Joka, M.; Jauch, K.-W.; Mayer, B. Impact of the Spheroid Model Complexity on Drug Response. J. Biotechnol. 2015, 205, 14–23. [Google Scholar] [CrossRef]
- Cattaneo, C.M.; Dijkstra, K.K.; Fanchi, L.F.; Kelderman, S.; Kaing, S.; van Rooij, N.; van den Brink, S.; Schumacher, T.N.; Voest, E.E. Tumor Organoid–T-Cell Coculture Systems. Nat. Protoc. 2020, 15, 15–39. [Google Scholar] [CrossRef] [PubMed]
- Küçükköse, E.; Heesters, B.A.; Villaudy, J.; Verheem, A.; Cercel, M.; van Hal, S.; Boj, S.F.; Borel Rinkes, I.H.M.; Punt, C.J.A.; Roodhart, J.M.L.; et al. Modeling Resistance of Colorectal Peritoneal Metastases to Immune Checkpoint Blockade in Humanized Mice. J. Immunother. Cancer 2022, 10, e005345. [Google Scholar] [CrossRef] [PubMed]
- Esposito, A.; Agostini, A.; Quero, G.; Piro, G.; Priori, L.; Caggiano, A.; Scaglione, G.; Battaglia, A.; Calegari, M.A.; Salvatore, L.; et al. Colorectal Cancer Patients-Derived Immunity-Organoid Platform Unveils Cancer-Specific Tissue Markers Associated with Immunotherapy Resistance. Cell Death Dis. 2024, 15, 878. [Google Scholar] [CrossRef]
- Bar-Ephraim, Y.E.; Kretzschmar, K.; Clevers, H. Organoids in Immunological Research. Nat. Rev. Immunol. 2020, 20, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Cheung, A.F.; Chodon, T.; Koya, R.C.; Wu, Z.; Ng, C.; Avramis, E.; Cochran, A.J.; Witte, O.N.; Baltimore, D.; et al. Multifunctional T-Cell Analyses to Study Response and Progression in Adoptive Cell Transfer Immunotherapy. Cancer Discov. 2013, 3, 418–429. [Google Scholar] [CrossRef]
- Sconocchia, G.; Eppenberger, S.; Spagnoli, G.C.; Tornillo, L.; Droeser, R.; Caratelli, S.; Ferrelli, F.; Coppola, A.; Arriga, R.; Lauro, D.; et al. NK Cells and T Cells Cooperate during the Clinical Course of Colorectal Cancer. OncoImmunology 2014, 3, e952197. [Google Scholar] [CrossRef]
- Rastin, F.; Javid, H.; Oryani, M.A.; Rezagholinejad, N.; Afshari, A.-R.; Karimi-Shahri, M. Immunotherapy for Colorectal Cancer: Rational Strategies and Novel Therapeutic Progress. Int. Immunopharmacol. 2024, 126, 111055. [Google Scholar] [CrossRef]
- Liu, X.; Lai, W.; Li, Y.; Chen, S.; Liu, B.; Zhang, N.; Mo, J.; Lyu, C.; Zheng, J.; Du, Y.-R.; et al. N6-Methyladenine Is Incorporated into Mammalian Genome by DNA Polymerase. Cell Res. 2021, 31, 94–97. [Google Scholar] [CrossRef]
- Reis, B.S.; Darcy, P.W.; Khan, I.Z.; Moon, C.S.; Kornberg, A.E.; Schneider, V.S.; Alvarez, Y.; Eleso, O.; Zhu, C.; Schernthanner, M.; et al. TCR-Vγδ Usage Distinguishes Protumor from Antitumor Intestinal Γδ T Cell Subsets. Science 2022, 377, 276–284. [Google Scholar] [CrossRef]
- Parikh, A.Y.; Masi, R.; Gasmi, B.; Hanada, K.-I.; Parkhurst, M.; Gartner, J.; Sindiri, S.; Prickett, T.; Robbins, P.; Zacharakis, N.; et al. Using Patient-Derived Tumor Organoids from Common Epithelial Cancers to Analyze Personalized T-Cell Responses to Neoantigens. Cancer Immunol. Immunother. 2023, 72, 3149–3162. [Google Scholar] [CrossRef]
- Westcott, P.M.K.; Sacks, N.J.; Schenkel, J.M.; Ely, Z.A.; Smith, O.; Hauck, H.; Jaeger, A.M.; Zhang, D.; Backlund, C.M.; Beytagh, M.C.; et al. Low Neoantigen Expression and Poor T-Cell Priming Underlie Early Immune Escape in Colorectal Cancer. Nat. Cancer 2021, 2, 1071–1085. [Google Scholar] [CrossRef] [PubMed]
- Schell, M.J.; Yang, M.; Teer, J.K.; Lo, F.Y.; Madan, A.; Coppola, D.; Monteiro, A.N.A.; Nebozhyn, M.V.; Yue, B.; Loboda, A.; et al. A Multigene Mutation Classification of 468 Colorectal Cancers Reveals a Prognostic Role for APC. Nat. Commun. 2016, 7, 11743. [Google Scholar] [CrossRef] [PubMed]
- Ara, A.; Ahmed, K.A.; Xiang, J. Multiple Effects of CD40-CD40L Axis in Immunity against Infection and Cancer. Immunotargets Ther. 2018, 7, 55–61. [Google Scholar] [CrossRef]
- Hargadon, K.M. Murine and Human Model Systems for the Study of Dendritic Cell Immunobiology. Int. Rev. Immunol. 2016, 35, 85–115. [Google Scholar] [CrossRef]
- Lanier, L.L. Five Decades of Natural Killer Cell Discovery. J. Exp. Med. 2024, 221, e20231222. [Google Scholar] [CrossRef]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.-H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988.e16. [Google Scholar] [CrossRef]
- Bonnereau, J.; Courau, T.; Asesio, N.; Salfati, D.; Bouhidel, F.; Corte, H.; Hamoudi, S.; Hammoudi, N.; Lavolé, J.; Vivier-Chicoteau, J.; et al. Autologous T Cell Responses to Primary Human Colorectal Cancer Spheroids Are Enhanced by Ectonucleotidase Inhibition. Gut 2023, 72, 699–709. [Google Scholar] [CrossRef]
- Moesta, A.K.; Li, X.-Y.; Smyth, M.J. Targeting CD39 in Cancer—PubMed. Nat. Rev. Immunol. 2020, 20, 739–755. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Z.; Zhao, X.; Xiao, J.; Bi, J.; Li, X.-Y.; Chen, G.; Lu, L. Review Immune Response of Targeting CD39 in Cancer. Biomark. Res. 2023, 11, 63. [Google Scholar] [CrossRef]
- Courau, T.; Bonnereau, J.; Chicoteau, J.; Bottois, H.; Remark, R.; Assante Miranda, L.; Toubert, A.; Blery, M.; Aparicio, T.; Allez, M.; et al. Cocultures of Human Colorectal Tumor Spheroids with Immune Cells Reveal the Therapeutic Potential of MICA/B and NKG2A Targeting for Cancer Treatment. J. Immunother. Cancer 2019, 7, 74. [Google Scholar] [CrossRef]
- Tsai, Y.-Y.; Nair, K.G.; Barot, S.V.; Xiang, S.; Kamath, S.; Melas, M.; Walker, C.P.; Srivastava, R.M.; Osborne, N.; Chan, T.A.; et al. Differences in Tumor-Associated T-Cell Receptor Repertoires between Early-Onset and Average-Onset Colorectal Cancer. J. Natl. Cancer Inst. 2024, 116, 1645–1653. [Google Scholar] [CrossRef] [PubMed]
- Tran, K.; Kumari, A.N.; Raghu, D.; Cox, D.R.A.; Goh, S.K.; Perini, M.V.; Muralidharan, V.; Tebbutt, N.C.; Behren, A.; Mariadason, J.; et al. T Cell Factor 1 (TCF-1) Defines T Cell Differentiation in Colorectal Cancer. iScience 2024, 27, 110754. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Zhou, Y.; Kuang, Y.; Wang, C.; Wang, W.; Hu, X.; Chen, X. Progress of Research on Γδ T Cells in Colorectal Cancer (Review). Oncol. Rep. 2024, 52, 160. [Google Scholar] [CrossRef]
- Zhu, D.; Ren, X.; Xie, W.; Chen, J.; Liang, S.; Jiang, M.; Wang, J.; Zheng, Z. Potential of Gamma/Delta T Cells for Solid Tumor Immunotherapy. Front. Immunol. 2024, 15, 1466266. [Google Scholar] [CrossRef]
- Di Mascolo, D.; Varesano, S.; Benelli, R.; Mollica, H.; Salis, A.; Zocchi, M.R.; Decuzzi, P.; Poggi, A. Nanoformulated Zoledronic Acid Boosts the Vδ2 T Cell Immunotherapeutic Potential in Colorectal Cancer. Cancers 2020, 12, 104. [Google Scholar] [CrossRef]
- Mikulak, J.; Oriolo, F.; Bruni, E.; Roberto, A.; Colombo, F.S.; Villa, A.; Bosticardo, M.; Bortolomai, I.; Lo Presti, E.; Meraviglia, S.; et al. NKp46-Expressing Human Gut-Resident Intraepithelial Vδ1 T Cell Subpopulation Exhibits High Antitumor Activity against Colorectal Cancer. JCI Insight 2025, 4, e125884. [Google Scholar] [CrossRef]
- Stary, V.; Pandey, R.V.; List, J.; Kleissl, L.; Deckert, F.; Kabiljo, J.; Laengle, J.; Gerakopoulos, V.; Oehler, R.; Watzke, L.; et al. Dysfunctional Tumor-Infiltrating Vδ1 + T Lymphocytes in Microsatellite-Stable Colorectal Cancer. Nat. Commun. 2024, 15, 6949. [Google Scholar] [CrossRef]
- Benelli, R.; Costa, D.; Salvini, L.; Tardito, S.; Tosetti, F.; Villa, F.; Zocchi, M.R.; Poggi, A. Targeting of Colorectal Cancer Organoids with Zoledronic Acid Conjugated to the Anti-EGFR Antibody Cetuximab. J. Immunother. Cancer 2022, 10, e005660. [Google Scholar] [CrossRef]
- Herrmann, T.; Karunakaran, M.M. Phosphoantigen Recognition by Vγ9Vδ2 T Cells. Eur. J. Immunol. 2024, 54, e2451068. [Google Scholar] [CrossRef]
- Fulford, T.S.; Soliman, C.; Castle, R.G.; Rigau, M.; Ruan, Z.; Dolezal, O.; Seneviratna, R.; Brown, H.G.; Hanssen, E.; Hammet, A.; et al. Vγ9Vδ2 T Cells Recognize Butyrophilin 2A1 and 3A1 Heteromers. Nat. Immunol. 2024, 25, 1355–1366. [Google Scholar] [CrossRef]
- Rigau, M.; Ostrouska, S.; Fulford, T.S.; Johnson, D.N.; Woods, K.; Ruan, Z.; McWilliam, H.E.G.; Hudson, C.; Tutuka, C.; Wheatley, A.K.; et al. Butyrophilin 2A1 Is Essential for Phosphoantigen Reactivity by Γδ T Cells. Science 2020, 367, eaay5516. [Google Scholar] [CrossRef] [PubMed]
- Vavassori, S.; Kumar, A.; Wan, G.S.; Ramanjaneyulu, G.S.; Cavallari, M.; El Daker, S.; Beddoe, T.; Theodossis, A.; Williams, N.K.; Gostick, E.; et al. Butyrophilin 3A1 Binds Phosphorylated Antigens and Stimulates Human Γδ T Cells. Nat. Immunol. 2013, 14, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Mehra, N.; Voortman, J.; Wise, D.R.; Calvo, E.; Piulats, J.M.; Gonzalez-Billalabeitia, E.; Ortego, I.; Swami, U.; Picus, J.; Karlik, M.; et al. Phase I/II Trial of LAVA-1207, a Novel Bispecific Gamma-Delta T-Cell Engager Alone, or with Low Dose IL-2 or Pembrolizumab, in Metastatic Castration Resistant Prostate Cancer (MCRPC). J. Clin. Oncol. 2024, 42, TPS2672. [Google Scholar] [CrossRef]
- Radi, H.; Ferdosi-Shahandashti, E.; Kardar, G.A.; Hafezi, N. An Updated Review of Interleukin-2 Therapy in Cancer and Autoimmune Diseases. J. Interferon Cytokine Res. 2024, 44, 143–157. [Google Scholar] [CrossRef]
- Kedmi, R.; Littman, D.R. Antigen-Presenting Cells as Specialized Drivers of Intestinal T Cell Functions. Immunity 2024, 57, 2269–2279. [Google Scholar] [CrossRef]
- Ielpo, S.; Barberini, F.; Dabbagh Moghaddam, F.; Pesce, S.; Cencioni, C.; Spallotta, F.; De Ninno, A.; Businaro, L.; Marcenaro, E.; Bei, R.; et al. Crosstalk and Communication of Cancer-Associated Fibroblasts with Natural Killer and Dendritic Cells: New Frontiers and Unveiled Opportunities for Cancer Immunotherapy. Cancer Treat. Rev. 2024, 131, 102843. [Google Scholar] [CrossRef]
- Chen, M.Y.; Zhang, F.; Goedegebuure, S.P.; Gillanders, W.E. Dendritic Cell Subsets and Implications for Cancer Immunotherapy. Front. Immunol. 2024, 15, 1393451. [Google Scholar] [CrossRef]
- Hato, L.; Vizcay, A.; Eguren, I.; Pérez-Gracia, J.L.; Rodríguez, J.; Gállego Pérez-Larraya, J.; Sarobe, P.; Inogés, S.; Díaz de Cerio, A.L.; Santisteban, M. Dendritic Cells in Cancer Immunology and Immunotherapy. Cancers 2024, 16, 981. [Google Scholar] [CrossRef]
- Alcántara-Hernández, M.; Leylek, R.; Wagar, L.E.; Engleman, E.G.; Keler, T.; Marinkovich, M.P.; Davis, M.M.; Nolan, G.P.; Idoyaga, J. High-Dimensional Phenotypic Mapping of Human Dendritic Cells Reveals Interindividual Variation and Tissue Specialization. Immunity 2017, 47, 1037–1050.e6. [Google Scholar] [CrossRef]
- van der Meijs, N.L.; Travecedo, M.A.; Marcelo, F.; van Vliet, S.J. The Pleiotropic CLEC10A: Implications for Harnessing This Receptor in the Tumor Microenvironment. Expert Opin. Ther. Targets 2024, 28, 601–612. [Google Scholar] [CrossRef]
- Subtil, B.; Iyer, K.K.; Poel, D.; Bakkerus, L.; Gorris, M.A.J.; Escalona, J.C.; van den Dries, K.; Cambi, A.; Verheul, H.M.W.; de Vries, I.J.M.; et al. Dendritic Cell Phenotype and Function in a 3D Co-Culture Model of Patient-Derived Metastatic Colorectal Cancer Organoids. Front. Immunol. 2023, 14, 1105244. [Google Scholar] [CrossRef]
- Collin, M.; Bigley, V. Human Dendritic Cell Subsets: An Update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Subtil, B.; van der Hoorn, I.A.E.; Cuenca-Escalona, J.; Becker, A.M.D.; Alvarez-Begue, M.; Iyer, K.K.; Janssen, J.; van Oorschot, T.; Poel, D.; Gorris, M.A.J.; et al. CDC2 Plasticity and Acquisition of a DC3-like Phenotype Mediated by IL-6 and PGE2 in a Patient-Derived Colorectal Cancer Organoids Model. Eur. J. Immunol. 2024, 54, e2350891. [Google Scholar] [CrossRef] [PubMed]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic Cells in Cancer Immunology and Immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Di Blasio, S.; van Wigcheren, G.F.; Becker, A.; van Duffelen, A.; Gorris, M.; Verrijp, K.; Stefanini, I.; Bakker, G.J.; Bloemendal, M.; Halilovic, A.; et al. The Tumour Microenvironment Shapes Dendritic Cell Plasticity in a Human Organotypic Melanoma Culture. Nat. Commun. 2020, 11, 2749. [Google Scholar] [CrossRef]
- Kabiljo, J.; Theophil, A.; Homola, J.; Renner, A.F.; Stürzenbecher, N.; Ammon, D.; Zirnbauer, R.; Stang, S.; Tran, L.; Laengle, J.; et al. Cancer-Associated Fibroblasts Shape Early Myeloid Cell Response to Chemotherapy-Induced Immunogenic Signals in next Generation Tumor Organoid Cultures. J. Immunother. Cancer 2024, 12, e009494. [Google Scholar] [CrossRef]
- Rayner, A.A.; Grimm, E.A.; Lotze, M.T.; Wilson, D.J.; Rosenberg, S.A. Lymphokine-Activated Killer (LAK) Cell Phenomenon. IV. Lysis by LAK Cell Clones of Fresh Human Tumor Cells from Autologous and Multiple Allogeneic Tumors. J. Natl. Cancer Inst. 1985, 75, 67–75. [Google Scholar]
- Linn, Y.C.; Hui, K.M. Cytokine-Induced Killer Cells: NK-like T Cells with Cytotolytic Specificity against Leukemia. Leuk. Lymphoma 2003, 44, 1457–1462. [Google Scholar] [CrossRef]
- Gutting, T.; Hauber, V.; Pahl, J.; Klapproth, K.; Wu, W.; Dobrota, I.; Herweck, F.; Reichling, J.; Helm, L.; Schroeder, T.; et al. PPARγ Induces PD-L1 Expression in MSS+ Colorectal Cancer Cells. Oncoimmunology 2021, 10, 1906500. [Google Scholar] [CrossRef]
- Hollenberg, A.N. Metabolic Health and Nuclear-Receptor Sensitivity. N. Engl. J. Med. 2012, 366, 1345–1347. [Google Scholar] [CrossRef]
- Schnalzger, T.E.; de Groot, M.H.; Zhang, C.; Mosa, M.H.; Michels, B.E.; Röder, J.; Darvishi, T.; Wels, W.S.; Farin, H.F. 3D Model for CAR-Mediated Cytotoxicity Using Patient-Derived Colorectal Cancer Organoids. EMBO J. 2019, 38, e100928. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.H.; Maki, G.; Klingemann, H.G. Characterization of a Human Cell Line (NK-92) with Phenotypical and Functional Characteristics of Activated Natural Killer Cells. Leukemia 1994, 8, 652–658. [Google Scholar] [PubMed]
- Annaratone, L.; De Palma, G.; Bonizzi, G.; Sapino, A.; Botti, G.; Berrino, E.; Mannelli, C.; Arcella, P.; Di Martino, S.; Steffan, A.; et al. Basic Principles of Biobanking: From Biological Samples to Precision Medicine for Patients. Virchows Arch. 2021, 479, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Goossens, N.; Nakagawa, S.; Sun, X.; Hoshida, Y. Cancer Biomarker Discovery and Validation. Transl. Cancer Res. 2015, 4, 256–269. [Google Scholar] [CrossRef]
- Riegman, P.H.J.; Becker, K.F.; Zatloukal, K.; Pazzagli, M.; Schröder, U.; Oelmuller, U. How Standardization of the Pre-Analytical Phase of Both Research and Diagnostic Biomaterials Can Increase Reproducibility of Biomedical Research and Diagnostics. New Biotechnol. 2019, 53, 35–40. [Google Scholar] [CrossRef]
- Dagher, G. Quality Matters: International Standards for Biobanking. Cell Prolif. 2022, 55, e13282. [Google Scholar] [CrossRef]
- Grizzle, W.E.; Gunter, E.W.; Sexton, K.C.; Bell, W.C. Quality Management of Biorepositories. Biopreservation. Biobank. 2015, 13, 183–194. [Google Scholar] [CrossRef]
- Bledsoe, M.J. Ethical Legal and Social Issues of Biobanking: Past, Present, and Future. Biopreservation. Biobank. 2017, 15, 142–147. [Google Scholar] [CrossRef]
- Staunton, C.; Slokenberga, S.; Mascalzoni, D. The GDPR and the Research Exemption: Considerations on the Necessary Safeguards for Research Biobanks. Eur. J. Hum. Genet. 2019, 27, 1159–1167. [Google Scholar] [CrossRef]
- Halley, M.C.; Olson, N.W.; Ashley, E.A.; Goldenberg, A.J.; Tabor, H.K. A Just Genomics Needs an ELSI of Translation. Hastings Cent. Rep. 2024, 54, S126–S135. [Google Scholar] [CrossRef]
- Li, H.; Liu, H.; Chen, K. Living Biobank-Based Cancer Organoids: Prospects and Challenges in Cancer Research. Cancer Biol. Med. 2022, 19, 965–982. [Google Scholar] [CrossRef] [PubMed]
- Rabouille, C.; Deschamps, J. On the Shoulders of Hubrecht: From Embryos to Stem Cells. Dev. Biol. 2017, 428, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Perrone, F.; Zilbauer, M. Biobanking of Human Gut Organoids for Translational Research. Exp. Mol. Med. 2021, 53, 1451–1458. [Google Scholar] [CrossRef]
- Xie, X.; Li, X.; Song, W. Tumor Organoid Biobank-New Platform for Medical Research. Sci. Rep. 2023, 13, 1819. [Google Scholar] [CrossRef]
- Yu, Y.; Zhu, Y.; Xiao, Z.; Chen, Y.; Chang, X.; Liu, Y.; Tang, Q.; Zhang, H. The Pivotal Application of Patient-Derived Organoid Biobanks for Personalized Treatment of Gastrointestinal Cancers. Biomark. Res. 2022, 10, 73. [Google Scholar] [CrossRef]
- Kim, J.; Koo, B.-K.; Knoblich, J.A. Human Organoids: Model Systems for Human Biology and Medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef]
- Sorrentino, G.; Rezakhani, S.; Yildiz, E.; Nuciforo, S.; Heim, M.H.; Lutolf, M.P.; Schoonjans, K. Mechano-Modulatory Synthetic Niches for Liver Organoid Derivation. Nat. Commun. 2020, 11, 3416. [Google Scholar] [CrossRef]
- Lee, H.; Yang, S.; Lee, K.J.; Kim, S.-N.; Jeong, J.-S.; Kim, K.Y.; Jung, C.-R.; Jeon, S.; Kwon, D.; Lee, S.; et al. Standardization and Quality Assessment for Human Intestinal Organoids. Front. Cell Dev. Biol. 2024, 12, 1383893. [Google Scholar] [CrossRef]
- Kataoka, M.; Gyngell, C.; Savulescu, J.; Sawai, T. The Donation of Human Biological Material for Brain Organoid Research: The Problems of Consciousness and Consent. Sci. Eng. Ethics 2024, 30, 3. [Google Scholar] [CrossRef]
- Bredenoord, A.L.; Clevers, H.; Knoblich, J.A. Human Tissues in a Dish: The Research and Ethical Implications of Organoid Technology. Science 2017, 355, eaaf9414. [Google Scholar] [CrossRef]
- Lensink, M.A.; Boers, S.N.; Gulmans, V.A.M.; Jongsma, K.R.; Bredenoord, A.L. Mini-Gut Feelings: Perspectives of People with Cystic Fibrosis on the Ethics and Governance of Organoid Biobanking. Pers. Med. 2021, 18, 241–254. [Google Scholar] [CrossRef]
- Lensink, M.A.; Boers, S.N.; Jongsma, K.R.; Carter, S.E.; van der Ent, C.K.; Bredenoord, A.L. Organoids for Personalized Treatment of Cystic Fibrosis: Professional Perspectives on the Ethics and Governance of Organoid Biobanking. J. Cyst. Fibros. 2021, 20, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Lensink, M.A.; Jongsma, K.R.; Boers, S.N.; Noordhoek, J.J.; Beekman, J.M.; Bredenoord, A.L. Responsible Use of Organoids in Precision Medicine: The Need for Active Participant Involvement. Development 2020, 147, dev177972. [Google Scholar] [CrossRef] [PubMed]
- de Jongh, D.; Massey, E.K.; Berishvili, E.; Fonseca, L.M.; Lebreton, F.; Bellofatto, K.; Bignard, J.; Seissler, J.; Buerck, L.W.; Honarpisheh, M.; et al. Organoids: A Systematic Review of Ethical Issues. Stem Cell Res. Ther. 2022, 13, 337. [Google Scholar] [CrossRef]
- Boers, S.N.; Bredenoord, A.L. Consent for Governance in the Ethical Use of Organoids. Nat. Cell Biol. 2018, 20, 642–645. [Google Scholar] [CrossRef]
- Boers, S.N.; van Delden, J.J.; Clevers, H.; Bredenoord, A.L. Organoid Biobanking: Identifying the Ethics. EMBO Rep. 2016, 17, 938–941. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Omics Type | Sample Type | Technique Used | Key Findings | Reference |
|---|---|---|---|---|
| Genomics | CRC organoids | WGS | Comparison of mutational profiling between organoids and parental tumor Mutations and drug response prediction | [105] |
| Genomics | CRC organoids | WES | Comparison of mutational profiling between organoids and parental tumor | [101,106] |
| Genomics | CRC organoids | WES | Genomic characterization | [17] |
| Genomics | CRC organoids | Targeted sequencing | Genomic characterization | [107] |
| Genomics | CRC organoids | Targeted sequencing WES Comparative genomic hybridization/SNV microarray | Mutational profiling Copy number alterations | [16] |
| Genomics | CRC organoids | WGS | Origin of SBS44 mutation signature | [108] |
| Genomics | CRC organoids | WGS | SBS88 mutation signature induced by colibactin | [74] |
| Genomics | Healthy organoids | WGS | SBS17b mutational signature induced by chemotherapy | [109] |
| Genomics | CD organoids | Targeted sequencing | SNP identification | [110] |
| Genomics | CF organoids | WGS | Identification of common and rare mutations of CF | [111] |
| Epigenomics | CRC organoids | Bisulfite conversion microarray | Association between methylator phenotypes and drug sensitivity | [112] |
| Epigenomics | FAP organoids | Bisulfite conversion microarray | Identification of differentially methylated regions and association with CRC development | [113] |
| Epigenomics | LS organoids | Bisulfite conversion microarray | Identification of a hypermethylated region of MSH4 gene proposed as biomarker of LS | [114] |
| Epigenomics | CRC organoids | ChIP-seq | Histone modifications and increased chromatin accessibility of specific enhancers | [115] |
| Epigenomics | CD organoids | Bisulfite conversion microarray | Loss of DNA methylation in MHC-I and in its transcriptional transactivator NLRC5 | [116] |
| Epigenomics | Healthy organoids | Bisulfite conversion microarray | Dynamic changes in DNA methylation of different intestinal segments during development | [117] |
| Transcriptomics | CRC organoids | Microarray analysis | Gene expression profiling of different CRC subtypes | [16] |
| Transcriptomics | CRC organoids | Microarray analysis | CRC subtype classification based on transcriptomics profiling | [17] |
| Transcriptomics | CRC organoids | RNA-seq | Transcriptomics profiling and drug response signatures | [106] |
| Transcriptomics | CRC organoids | scRNA-seq | Comparison of transcriptomics profiling between organoids and parental tumor | [118] |
| Transcriptomics | CRC organoids | RNA-seq | Identification of drug targets by transcriptomics profiling | [119] |
| Transcriptomics | CRC organoids | RNA-seq | Expression of immune-related genes | [107] |
| Transcriptomics | CRC organoids | RNA-seq | Identification of biomarkers associated with anticancer drug resistance | [120] |
| Transcriptomics | CRC organoids | RNA-seq | Proteotranscriptomics profiling and drug response | [121] |
| Transcriptomics | CD organoids | RNA-seq | Identification of different clusters through gene expression profiling | [110] |
| Transcriptomics | CD organoids | scRNA-seq | Gene expression profiling depending on the presence or absence of inflammation | [122] |
| Proteomics | Healthy organoids | SILAC/MS | Protein expression profiles after drug treatment | [123] |
| Proteomics | CRC organoids | Nano-UHPLC/MS | Identification of personalized proteomics profiles | [124] |
| Proteomics | IBD organoids | LC/MS | Characterization of proteomics profiling | [125] |
| Metabolomics | CRC organoids | HR-MAS MRS | Metabolic alterations associated with tumor progression | [126] |
| Metabolomics | CRC organoids | LC-QTOF-MS | Characterization of metabolic drug response | [127] |
| Metabolomics | CRC organoids | GC/MS | Metabolic changes after drug treatment | [128] |
| NCT Number | Study Title | Study Status | Results | Conditions | Study Type | Locations |
|---|---|---|---|---|---|---|
| NCT05832398 | Precision Chemotherapy Based on Organoid Drug Sensitivity for Colorectal Cancer | RECRUITING | NO | CRC | INT | Guangzhou (CN) |
| NCT05384184 | Next Generation Pre-clinical Model for Colorectal Cancer Metastases and Hepatocellular Carcinomas (BORG) | COMPLETED | NO | CRCm; HCC | OBS | Marseille (FR) |
| NCT05304741 | The Culture of Advanced/Recurrent/Metastatic Colorectal Cancer Organoids and Drug Screening | RECRUITING | NO | CRC | OBS | Chongqing (CN) |
| NCT06100016 | A Clinical Study Aims to Assess the Consistency of Clinical Efficacy in Colorectal Cancer Treatment and Drug Susceptibility Outcomes Using a Novel Drug Susceptibility Testing Method | RECRUITING | NO | CRC | OBS | Shenyang (CN) |
| NCT05401318 | Tailoring Treatment in Colorectal Cancer | RECRUITING | NO | CRN | OBS | Viken (NO) |
| NCT05267912 | Prospective Multicenter Study Evaluating Feasibility and Efficacy of Tumor Organoid-based Precision Medicine in Patients With Advanced Refractory Cancers | ACTIVE, NOT RECRUITING | NO | APST | INT | Villejuif (FR) |
| NCT05725200 | Study to Investigate Outcome of Individualized Treatment in Patients With Metastatic Colorectal Cancer | RECRUITING | NO | CRCm | INT | Oslo (NO) |
| NCT05038358 | Tumor Immune Microenvironment Involvement in Colorectal Cancer Chemoresistance Mechanisms | RECRUITING | NO | CRC | OBS | Grenoble (FR) |
| NCT06136949 | The Theranostic Value of STARD3 in Colorectal Cancer: The STAR Study | RECRUITING | NO | CRC | OBS | Aviano (IT) |
| NCT04896684 | Chronic Intestinal Pathologies Analytical Cohort at TouLouse | RECRUITING | NO | IBD; CRC | OBS | Toulouse (FR) |
| NCT02732860 | Personalized Patient Derived Xenograft (pPDX) Modeling to Test Drug Response in Matching Host | RECRUITING | NO | CRN/CRC; BN/BC; ON/OC | OBS | Toronto (CA) |
| NCT06349590 | Manipulation of the Gut Microbiome by a Standardized Preoperative Diet to Prevent Colorectal Cancer Recurrence and Metastasis Following Surgery | RECRUITING | NO | CRC | INT | Chicago (US) |
| NCT04622423 | Advanced Therapies for Liver Metastases | RECRUITING | NO | PDAC CRC (LM) | OBS | Milan (IT) |
| NCT04587128 | Early-Line Anti-EGFR Therapy to Facilitate Retreatment for Select Patients With mCRC | RECRUITING | NO | CRCm | INT | Madison (US) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taglieri, M.; Di Gregorio, L.; Matis, S.; Uras, C.R.M.; Ardy, M.; Casati, S.; Marchese, M.; Poggi, A.; Raffaghello, L.; Benelli, R. Colorectal Organoids: Models, Imaging, Omics, Therapy, Immunology, and Ethics. Cells 2025, 14, 457. https://doi.org/10.3390/cells14060457
Taglieri M, Di Gregorio L, Matis S, Uras CRM, Ardy M, Casati S, Marchese M, Poggi A, Raffaghello L, Benelli R. Colorectal Organoids: Models, Imaging, Omics, Therapy, Immunology, and Ethics. Cells. 2025; 14(6):457. https://doi.org/10.3390/cells14060457
Chicago/Turabian StyleTaglieri, Martina, Linda Di Gregorio, Serena Matis, Chiara Rosa Maria Uras, Massimo Ardy, Sara Casati, Monica Marchese, Alessandro Poggi, Lizzia Raffaghello, and Roberto Benelli. 2025. "Colorectal Organoids: Models, Imaging, Omics, Therapy, Immunology, and Ethics" Cells 14, no. 6: 457. https://doi.org/10.3390/cells14060457
APA StyleTaglieri, M., Di Gregorio, L., Matis, S., Uras, C. R. M., Ardy, M., Casati, S., Marchese, M., Poggi, A., Raffaghello, L., & Benelli, R. (2025). Colorectal Organoids: Models, Imaging, Omics, Therapy, Immunology, and Ethics. Cells, 14(6), 457. https://doi.org/10.3390/cells14060457