
Oxidative Stress and Redox Imbalance: Common Mechanisms in Cancer Stem Cells and Neurodegenerative Diseases

 ,
,
Abstract
:1. Introduction
2. Oxidative Stress and Redox Imbalance: An Overview
2.1. Mechanisms of Redox Regulation
- NF-κB pathway: A master regulator of inflammatory responses that is highly sensitive to redox changes. It forms specific signaling complexes that regulate target gene expression during acute inflammation [25].
- MAPK pathway: Redox-sensitive and involved in cellular responses to OS [26].
- Nrf2-Keap1 pathway: Central to cellular defense against oxidative and electrophilic insults [27]. Nrf2 binds to the Antioxidant Response Element (ARE) to induce the expression of antioxidant and detoxifying enzymes [27]. Keap1 negatively regulates Nrf2 by targeting it for degradation. Disrupting the Keap1-Nrf2 interaction can enhance the antioxidant capacity of the brain and protect against OS and neuroinflammation [28].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Cancer Stem Cells (CSCs) | Neurons |
|---|---|---|
| MAPK Pathway (Mitogen-Activated Protein Kinase) | ||
| NF-κB Pathway (Nuclear Factor Kappa B) | ||
| Nrf2 Pathway (Nuclear Factor Erythroid 2-Related Factor 2) |
2.2. Consequences of OS and Redox Imbalance
2.3. Dual Roles of OS
2.4. Consequences of Oxidative Stress: Cancer Stem Cells and Neurodegeneration
3. Oxidative Stress in Cancer Stem Cells
- Metabolic reprogramming (Warburg effect);
- Autophagy;
- Quiescence.
- Inhibition of antioxidant systems: Drugs targeting glutathione (GSH) synthesis, superoxide dismutase (SOD) activity, or thioredoxin pathways can selectively impair CSC survival [84].
- ROS-inducing agents: Pro-oxidant therapies that elevate ROS levels beyond the tolerance threshold of CSCs can induce oxidative stress and apoptosis [125].
- Metabolic modulators: Agents that disrupt metabolic pathways involved in NADPH production, such as pentose phosphate pathway (PPP) inhibitors, can compromise CSC antioxidant defenses [126].
- Limited understanding of CSC antioxidant responses: The precise mechanisms through which CSCs modulate their antioxidant responses under different microenvironmental conditions remain poorly understood, hindering the development of targeted therapies.
- Therapeutic specificity: Developing agents that selectively target CSCs without affecting normal stem cells is critical to minimizing side effects.
- CSC heterogeneity: The diverse phenotypes and metabolic states of CSCs across different tumor types complicate therapeutic targeting.
- Resistance development: CSCs may develop resistance to redox-targeted therapies through compensatory pathways.
4. Oxidative Stress in Neurodegenerative Diseases
Oxidative Stress-Driven Mechanisms in NDDs
- Kavalactones attenuate amyloid beta-peptide toxicity by inducing Nrf2-mediated protective gene expression in vitro [176].
- Tert-butylhydroquinone treatment and adenoviral Nrf2 gene transfer protect against amyloid beta toxicity in Alzheimer’s disease models [177].
- Food-derived Nrf2/ARE pathway inducers, such as l-sulforaphane from broccoli and isoliquiritigenin from licorice, protect mitochondrial function in oxidative stress and neurodegenerative disease models [174]. The Nrf2-ARE pathway not only addresses OS but also modulates mitochondrial function, reduces neuroinflammation, and promotes neuroprotection.
- Apocynin, an NADPH oxidase inhibitor, attenuates microglial activation, oxidative stress damage, and induction of Alzheimer’s disease proteins in traumatic brain injury models [178].
- Inhibiting NADPH oxidases may reduce ROS-mediated damage to retinal ganglion cells and glial dysfunction in glaucoma [179].
5. Common Mechanisms in CSCs and Neurodegenerative Diseases
5.1. Oxidative Stress: Overlap Between Mechanisms in Cancer Stem Cells and Neurodegeneration
5.1.1. Oxidative Stress and Mitochondrial Dysfunction
5.1.2. Redox Signaling and Cellular Survival
5.1.3. Oxidative Stress and Chronic Inflammation
5.1.4. Oxidative Stress: Lipid Peroxidation and Ferroptosis
Iron Metabolism and Lipid Peroxidation: Contrasting Roles in CSCs and Neurons
Polyunsaturated Fatty Acids (PUFAs) and Ferroptosis Susceptibility
5.1.5. DNA Damage and Genomic Instability
5.1.6. Aging-Related Oxidative Damage in CSCs and NDDs
5.1.7. Epigenetic Consequences of Oxidative Stress
5.1.8. Autophagy and Oxidative Stress
5.1.9. Oxidative Stress and Nrf2
5.1.10. Oxidative Stress and Metabolic Reprogramming
5.1.11. Oxidative Stress: Inverse Relationships in Cancer and NDDs
- PARK2 (Parkin) and PARK5 (UCHL1): Associated with Parkinson’s disease (PD) and have roles in cancer and oxidative stress. Parkin is involved in mitochondrial quality control and protects against oxidative stress-induced neurodegeneration [307]. In cancer, PARK2 acts as a tumor suppressor. UCHL1, part of the ubiquitin-proteasome system, is downregulated in some cancers but upregulated in others [308].
- DJ-1: A redox-responsive cytoprotective protein involved in regulating oxidative stress and linked to Parkinson’s disease (PD). It acts as a transcriptional regulator of antioxidative genes and controls oxidative stress in ischemia, neuroinflammation, and age-related neurodegenerative processes [313]. DJ-1 is also connected to Nrf2, a master regulator of antioxidant gene expression. DJ-1 is involved in hepatocellular carcinoma (HCC) development, with a significant inverse correlation between DJ-1 expression and overall survival in HCC patients [314]. DJ-1 knockout mice displayed reduced tumorigenesis and cell proliferation, accompanied by decreased hepatic inflammation and IL-6/STAT3 activation in a DEN-induced murine HCC model [314].
- ABCA7: An ATP-binding cassette transporter identified as a susceptibility factor for late-onset Alzheimer’s disease (AD) [315]. It plays a role in amyloid precursor protein (APP) processing and amyloid-β (Aβ) generation. Loss of ABCA7 function results in increased β-secretase cleavage and elevated Aβ levels [316]. ABCA7 also mediates phagocytosis and affects membrane trafficking. In retinoblastoma, the Y79 cell line demonstrates high gene expression of ABCA7 along with several other ABC transporters [317]. This elevated expression suggests that ABCA7 might be a potential target for medical treatment of retinoblastoma.
- MAPT (Tau): A microtubule-associated protein implicated in various neurodegenerative diseases. Recent evidence suggests its involvement in DNA repair and p53 regulation, indicating a potential role in cancer. MAPT expression is associated with key cancer hallmarks and clinical outcomes in a context-specific manner [318]. The involvement of these genes in both cancer and NDDs, often with opposing effects, underscores the complex relationship between oxidative stress, cell survival, and disease pathogenesis. An understanding of these shared pathways may lead to novel therapeutic approaches for both cancer and neurodegenerative disorders.
| Biomarker | Description | Relevance to Cancer | Relevance to NDDs |
|---|---|---|---|
| F2-Isoprostanes | Lipid peroxidation products formed by free radical attack on arachidonic acid. | Elevated in various cancers due to increased oxidative stress and inflammation [319]. | Biomarker for lipid peroxidation in Alzheimer’s and Parkinson’s disease; associated with cognitive decline [320]. |
| 8-Oxo-2′-deoxyguanosine (8-oxo-dG) | Oxidative DNA damage marker resulting from reactive oxygen species (ROS) attack on guanine bases. | High levels found in tumor tissues, indicating genomic instability and cancer progression [321]. | Increased in Alzheimer’s, Parkinson’s, and ALS; contributes to neuronal DNA damage and apoptosis [322]. |
| Glutathione (GSH)/Glutathione Disulfide (GSSG) Ratio | Indicator of cellular redox status; higher GSH/GSSG suggests better antioxidant defense. | Reduced in tumor tissues, indicating oxidative stress and impaired detoxification [323]. | Low GSH/GSSG ratio in neurodegenerative diseases suggests oxidative stress-induced neuronal damage [324]. |
| Malondialdehyde (MDA) | Byproduct of lipid peroxidation; marker of oxidative damage to membranes. | Elevated in cancer patients, linked to tumor growth and progression [325]. | increased in AD and PD; contributes to neuronal degeneration [326]. |
| Protein Carbonyls | Markers of protein oxidation and dysfunction. | Increased levels found in various cancers, reflecting oxidative damage to proteins [327]. | Elevated in Alzheimer’s, Parkinson’s, and Huntington’s disease; associated with neurotoxicity [328]. |
| SOD, CAT, and Glutathione Peroxidase (GPx) | Enzymatic antioxidants that neutralize ROS. | Altered expression in cancer; reduced activity may promote tumor progression [329]. | Reduced levels contribute to oxidative stress in neurodegenerative diseases [330]. |
| Nitrotyrosine | Marker of peroxynitrite-mediated nitrosative stress. | High levels linked to cancer development and inflammation [331]. | Increased in Alzheimer’s and Parkinson’s; associated with mitochondrial dysfunction [332]. |
6. Future Directions, Challenges, and Gaps in Current Knowledge
- Molecular mechanisms: Unraveling the specific pathways through which reactive oxygen species (ROS) and reactive nitrogen species (RNS) contribute to the pathogenesis of neurodegenerative diseases and cancer.
- Mitochondrial dysfunction: Examining the relationship between mitochondrial impairment, oxidative stress, and their impact on cellular processes in both conditions.
- Biomarker development: Creating more sensitive and specific indicators of oxidative stress to enhance early detection and disease progression monitoring.
- Targeted antioxidant therapies: Exploring interventions that can selectively modulate ROS levels in affected tissues without disrupting physiological redox signaling.
- Cellular process interactions: Investigating the interplay between oxidative stress and other cellular mechanisms, such as inflammation, autophagy, and apoptosis, in the context of neurodegenerative diseases and cancer.
- Epigenetic modifications: Studying the role of oxidative stress-induced epigenetic changes in disease progression and potential therapeutic interventions.
- Lifestyle factors: Examining the impact of diet, exercise, and other lifestyle elements on redox balance and their potential as complementary approaches to manage oxidative stress-related diseases.
- Combination therapies: Investigating treatments that target multiple aspects of redox imbalance and oxidative stress-induced damage.
- Advanced drug delivery: Exploring nanotechnology and innovative drug delivery systems in order to enhance the efficacy and specificity of antioxidant therapies.
- Clinical trials: Conducting comprehensive, long-term studies to evaluate the effectiveness of novel antioxidant strategies in preventing or treating neurodegenerative diseases and cancer.
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| OS | oxidative stress |
| NDD | neurodegenerative diseases |
| ROS | reactive oxygen species |
| CSC | cancer stem cell |
| RNS | reactive nitrogen species |
| PUFA | polyunsaturated fatty acid |
References
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Aramini, B.; Masciale, V.; Grisendi, G.; Bertolini, F.; Maur, M.; Guaitoli, G.; Chrystel, I.; Morandi, U.; Stella, F.; Dominici, M.; et al. Dissecting Tumor Growth: The Role of Cancer Stem Cells in Drug Resistance and Recurrence. Cancers 2022, 14, 976. [Google Scholar] [CrossRef] [PubMed]
- Vera-Ramirez, L.; Sanchez-Rovira, P.; Ramirez-Tortosa, M.C.; Ramirez-Tortosa, C.L.; Granados-Principal, S.; Lorente, J.A.; Quiles, J.L. Free radicals in breast carcinogenesis, breast cancer progression and cancer stem cells. Biological bases to develop oxidative-based therapies. Crit. Rev. Oncol./Hematol. 2011, 80, 347–368. [Google Scholar] [CrossRef] [PubMed]
- García-Blanco, A.; Baquero, M.; Vento, M.; Gil, E.; Bataller, L.; Cháfer-Pericás, C. Potential oxidative stress biomarkers of mild cognitive impairment due to Alzheimer disease. J. Neurol. Sci. 2017, 373, 295–302. [Google Scholar] [CrossRef]
- Zabel, M.; Nackenoff, A.; Kirsch, W.M.; Harrison, F.E.; Perry, G.; Schrag, M. Markers of oxidative damage to lipids, nucleic acids and proteins and antioxidant enzymes activities in Alzheimer’s disease brain: A meta-analysis in human pathological specimens. Free. Radic. Biol. Med. 2018, 115, 351–360. [Google Scholar] [CrossRef]
- Dakubo, G.D. The Role of Mitochondrial Reactive Oxygen Species in Cancer. In Mitochondrial Genetics and Cancer; Springer: Berlin/Heidelberg, Germany, 2010; pp. 237–256. [Google Scholar] [CrossRef]
- Broto, G.E.; da Silva, J.C.; de Oliveira, S.T.; Garbim, M.R.; Oliveira, M.O.; Panis, C. Oxidative Stress-Related Mechanisms That Mediate Chemo-resistance in Cancer Stem Cells. In Handbook of Oxidative Stress in Cancer: Therapeutic Aspects; Springer: Berlin/Heidelberg, Germany, 2022; Available online: https://link.springer.com/content/pdf/10.1007/978-981-16-1247-3_101-1.pdf (accessed on 15 February 2025).
- Abolhassani, N.; Leon, J.; Sheng, Z.; Oka, S.; Hamasaki, H.; Iwaki, T.; Nakabeppu, Y. Molecular pathophysiology of impaired glucose metabolism, mitochondrial dysfunction, and oxidative DNA damage in Alzheimer’s disease brain. Mech. Ageing Dev. 2017, 161, 95–104. [Google Scholar] [CrossRef]
- Yun, H.R.; Jo, Y.H.; Kim, J.; Shin, Y.; Kim, S.S.; Choi, T.G. Roles of Autophagy in Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 3289. [Google Scholar] [CrossRef]
- Grieco, J.P.; Schmelz, E.M.; Smyth, J.W.; Craige, S.M.; Pickrell, A.M. Mitochondrial Quality Control Adaptations Support Malignant Progression of Serous Ovarian Cancer Cells and Spheroids; Virginia Tech: Blacksburg, VA, USA, 2022; Available online: https://vtechworks.lib.vt.edu/items/ea8cacb8-7e6e-47fc-be28-2e918eb9de84 (accessed on 15 February 2025).
- Calkins, M.J.; Johnson, D.A.; Townsend, J.A.; Vargas, M.R.; Dowell, J.A.; Williamson, T.P.; Kraft, A.D.; Lee, J.-M.; Li, J.; Johnson, J.A. The Nrf2/ARE Pathway as a Potential Therapeutic Target in Neurodegenerative Disease. Antioxid. Redox Signal. 2009, 11, 497–508. [Google Scholar] [CrossRef]
- Xu, J.; Du, W.; Zhao, Y.; Lim, K.; Lu, L.; Zhang, C.; Li, L. Mitochondria targeting drugs for neurodegenerative diseases—Design, mechanism and application. Acta Pharm. Sin. B 2022, 12, 2778–2789. [Google Scholar] [CrossRef]
- Gao, L.; Meng, F.; Yang, Z.; Lafuente-Merchan, M.; Fernández, L.M.; Cao, Y.; Kusamori, K.; Nishikawa, M.; Itakura, S.; Chen, J. Nano-drug delivery system for the treatment of multi-drug-resistant breast cancer: Current status and future perspectives. Biomed. Pharmacother. 2024, 179, 117327. [Google Scholar] [CrossRef] [PubMed]
- Barmaki, H.; Nourazarian, A.; Khaki-Khatibi, F. Proteostasis and neurodegeneration: A closer look at autophagy in Alzheimer’s disease. Front. Aging Neurosci. 2023, 15, 1281338. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, E.M.; Sotgia, F.; Lisanti, M.P. Cancer stem cells (CSCs): Metabolic strategies for their identification and eradication. Biochem. J. 2018, 475, 1611–1634. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Galley, M.; Brockett, C.; Spithourakis, G.P.; Gao, J.; Dolan, B. A persona-based neural conversation model. In Proceedings of the 54th Annual Meeting of the Association for Computational Linguistics, ACL 2016—Long Papers, Berlin, Germany, 7–12 August 2016; Association for Computational Linguistics: Kerrville, TX, USA, 2016; Volume 2, pp. 994–1003. [Google Scholar] [CrossRef]
- Milatovic, D.; Montine, T.J.; Aschner, M. Prostanoid signaling: Dual role for prostaglandin E2 in neurotoxicity. NeuroToxicology 2011, 32, 312–319. [Google Scholar] [CrossRef]
- Briyal, S.; Ranjan, A.K.; Gulati, A. Oxidative stress: A target to treat Alzheimer’s disease and stroke. Neurochem. Int. 2023, 165, 105509. [Google Scholar] [CrossRef]
- Rhodes, L.V.; Short, S.P.; Neel, N.F.; Salvo, V.A.; Zhu, Y.; Elliott, S.; Wei, Y.; Yu, D.; Sun, M.; Muir, S.E.; et al. Cytokine receptor CXCR4 mediates estro-gen-independent tumorigenesis, metastasis, and resistance to endocrine therapy in human breast cancer. Cancer Res. 2011, 71, 603–613. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Mukwevho, E.; Ferreira, Z.; Ayeleso, A. Potential Role of Sulfur-Containing Antioxidant Systems in Highly Oxidative Environments. Molecules 2014, 19, 19376–19389. [Google Scholar] [CrossRef]
- Paulsen, C.E.; Carroll, K.S. Cysteine-Mediated Redox Signaling: Chemistry, Biology, and Tools for Discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef]
- Allen, E.M.; Mieyal, J.J. Protein-Thiol Oxidation and Cell Death: Regulatory Role of Glutaredoxins. Antioxid. Redox Signal. 2012, 17, 1748–1763. [Google Scholar] [CrossRef]
- Ghezzi, P.; Bonetto, V. Redox proteomics: Identification of oxidatively modified proteins. Proteomics 2003, 3, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S. PGC-1α, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxid. Med. Cell. Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Zhang, Y.; Dusting, G.J. NADPH oxidase-mediated redox signaling: Roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol. Rev. 2011, 63, 218–242. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Tong, K.I.; Yamamoto, M. Molecular mechanism activating Nrf2–Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic. Biol. Med. 2004, 36, 1208–1213. [Google Scholar] [CrossRef]
- Bono, S.; Feligioni, M.; Corbo, M. Impaired antioxidant KEAP1-NRF2 system in amyotrophic lateral sclerosis: NRF2 activation as a potential therapeutic strategy. Mol. Neurodegener. 2021, 16, 71. [Google Scholar] [CrossRef]
- Chang, C.-H.; Pauklin, S. ROS and TGFβ: From pancreatic tumour growth to metastasis. J. Exp. Clin. Cancer Res. 2021, 40, 152. [Google Scholar] [CrossRef]
- Zhou, D.; Shao, L.; Spitz, D.R. Reactive oxygen species in normal and tumor stem cells. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherlands, 2014; Available online: https://www.sciencedirect.com/science/article/pii/B9780124201170000013 (accessed on 15 February 2025).
- He, J.; Zhong, W.; Zhang, M.; Zhang, R.; Hu, W. P38 mitogen-activated protein kinase and Parkinson’s disease. Transl. Neurosci. 2018, 9, 147–153. [Google Scholar] [CrossRef]
- Sahu, R.; Upadhayay, S.; Mehan, S. Inhibition of extracellular regulated kinase (ERK)-1/2 signaling pathway in the prevention of ALS: Target inhibitors and influences on neurological dysfunctions. Eur. J. Cell Biol. 2021, 100, 151179. [Google Scholar] [CrossRef]
- Corrêa, S.A.L.; Eales, K.L. The Role of p38 MAPK and Its Substrates in Neuronal Plasticity and Neurodegenerative Disease. J. Signal Transduct. 2012, 2012, 649079. [Google Scholar] [CrossRef]
- Guo, Q.; Jin, Y.; Chen, X.; Ye, X.; Shen, X.; Lin, M.; Zeng, C.; Zhou, T.; Zhang, J. NF-κB in biology and targeted therapy: New insights and translational implications. Signal Transduct. Target. Ther. 2024, 9, 53. [Google Scholar] [CrossRef]
- Verzella, D.; Pescatore, A.; Capece, D.; Vecchiotti, D.; Ursini, M.V.; Franzoso, G.; Alesse, E.; Zazzeroni, F. Life, death, and autophagy in cancer: NF-κB turns up everywhere. Cell Death Dis. 2020, 11, 210. [Google Scholar] [CrossRef] [PubMed]
- Maniati, E.; Bossard, M.; Cook, N.; Candido, J.B.; Emami-Shahri, N.; Nedospasov, S.A.; Balkwill, F.R.; Tuveson, D.A.; Hagemann, T. Crosstalk between the canonical NF-κB and Notch signaling pathways inhibits Pparγ expression and promotes pancreatic cancer progression in mice. J. Clin. Investig. 2011, 121, 4685–4699. [Google Scholar] [CrossRef] [PubMed]
- Shih, R.-H.; Wang, C.-Y.; Yang, C.-M. NF-kappaB Signaling Pathways in Neurological Inflammation: A Mini Review. Front. Mol. Neurosci. 2015, 8, 77. [Google Scholar] [CrossRef]
- Dresselhaus, E.C.; Meffert, M.K. Cellular Specificity of NF-κB Function in the Nervous System. Front. Immunol. 2019, 10, 1043. [Google Scholar] [CrossRef]
- Ryoo, I.-G.; Lee, S.-H.; Kwak, M.-K. Redox Modulating NRF2: A Potential Mediator of Cancer Stem Cell Resistance. Oxidative Med. Cell. Longev. 2016, 2016, 2428153. [Google Scholar] [CrossRef]
- Xue, D.; Zhou, X.; Qiu, J. Emerging role of NRF2 in ROS-mediated tumor chemo-resistance. Biomed. Pharmacother. 2020, 131, 110676. [Google Scholar] [CrossRef]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef]
- Schmidlin, C.J.; Dodson, M.B.; Madhavan, L.; Zhang, D.D. Redox regulation by NRF2 in aging and disease. Free. Radic. Biol. Med. 2019, 134, 702–707. [Google Scholar] [CrossRef]
- Mayer, C.; Riera-Ponsati, L.; Kauppinen, S.; Klitgaard, H.; Erler, J.T.; Hansen, S.N. Targeting the NRF2 pathway for disease modification in neurodegenerative diseases: Mechanisms and therapeutic implications. Front. Pharmacol. 2024, 15, 1437939. [Google Scholar] [CrossRef]
- Zweig, J.A.; Caruso, M.; Brandes, M.S.; Gray, N.E. Loss of NRF2 leads to impaired mitochondrial function, decreased synaptic density and exacerbated age-related cognitive deficits. Exp. Gerontol. 2020, 131, 110767. [Google Scholar] [CrossRef]
- Chiurchiù, V.; Maccarrone, M. Chronic Inflammatory Disorders and Their Redox Control: From Molecular Mechanisms to Therapeutic Opportunities. Antioxid. Redox Signal. 2011, 15, 2605–2641. [Google Scholar] [CrossRef]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef] [PubMed]
- Therond, P. Oxidative stress and damages to biomolecules (lipids, proteins, DNA). In Annales Pharmaceutiques Francaises; Elsevier: Amsterdam, The Netherlands, 2006; Available online: https://www.researchgate.net/publication/6679488_Oxidative_stress_and_damages_to_biomolecules_lipids_proteins_DNA (accessed on 15 February 2025).
- Kandola, K.; Bowman, A.; Birch-Machin, M.A. Oxidative stress—A key emerging impact factor in health, ageing, lifestyle and aesthetics. Int. J. Cosmet. Sci. 2015, 37, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Dobmeyer, T.S.; Findhammer, S.; Dobmeyer, J.M.; A Klein, S.; Raffel, B.; Hoelzer, D.; Helm, E.B.; Kabelitz, D.; Rossol, R. Ex Vivo Induction of Apoptosis in Lymphocytes Is Mediated by Oxidative Stress: Role for Lymphocyte Loss in HIV Infection. Free Radic. Biol. Med. 1997, 22, 775–785. [Google Scholar] [CrossRef]
- Buttke, T.M.; Sandstrom, P.A. Redox Regulation of Programmed Cell Death in Lymphocytes. Free. Radic. Res. 1995, 22, 389–397. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, Y.; Wang, G.; Ren, J. Molecular Mechanisms and Therapeutic Targeting of Ferroptosis in Doxorubicin-Induced Cardiotoxicity. JACC Basic Transl. Sci. 2024, 9, 811–826. [Google Scholar] [CrossRef]
- Lee, K.M.; Kim, T.H.; Noh, E.-J.; Han, J.W.; Kim, J.-S.; Lee, S.K. 25-Hydroxycholesterol induces oxidative stress, leading to apoptosis and ferroptosis in extravillous trophoblasts. Chem. Interact. 2024, 403, 111214. [Google Scholar] [CrossRef]
- Perluigi, M.; Butterfield, D.A. Oxidative Stress and Down Syndrome: A Route toward Alzheimer-Like Dementia. Curr. Gerontol. Geriatr. Res. 2011, 2012, 724904. [Google Scholar] [CrossRef]
- Ferrari, M.; Stagi, S. Oxidative Stress in Down and Williams-Beuren Syndromes: An Overview. Molecules 2021, 26, 3139. [Google Scholar] [CrossRef]
- Oliveira, B.F.; Nogueira-Machado, J.A.; Chaves, M.M. The Role of Oxidative Stress in the Aging Process. Sci. World J. 2010, 10, 1121–1128. [Google Scholar] [CrossRef]
- Ristow, M.; Schmeisser, K. Mitohormesis: Promoting Health and Lifespan by Increased Levels of Reactive Oxygen Species (ROS). Dose-Response 2014, 12, 288–341. [Google Scholar] [CrossRef]
- Li, Z.; Xu, X.; Leng, X.; He, M.; Wang, J.; Cheng, S.; Wu, H. Roles of reactive oxygen species in cell signaling pathways and immune responses to viral infections. Arch. Virol. 2017, 162, 603–610. [Google Scholar] [CrossRef]
- Di Meo, S.; Napolitano, G.; Venditti, P. Mediators of Physical Activity Protection against ROS-Linked Skeletal Muscle Damage. Int. J. Mol. Sci. 2019, 20, 3024. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.; Janmeda, P.; Docea, A.O.; Yeskaliyeva, B.; Razis, A.F.A.; Modu, B.; Calina, D.; Sharifi-Rad, J. Oxidative stress, free radicals and antioxidants: Potential crosstalk in the pathophysiology of human dis-eases. Front. Chem. 2023, 11, 1158198. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Zhang, Y.; Zheng, J.; Pan, J. Reactive Oxygen Species in Cancer Stem Cells. Antioxid. Redox Signal. 2012, 16, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Yu, W.; Liu, J.; Tang, D.; Yang, L.; Chen, X. Oxidative cell death in cancer: Mechanisms and therapeutic opportunities. Cell Death Dis. 2024, 15, 556. [Google Scholar] [CrossRef]
- Fan, M.; Shi, Y.; Zhao, J.; Li, L. Cancer stem cell fate determination: Mito-nuclear communication. Cell Commun. Signal. 2023, 21, 159. [Google Scholar] [CrossRef]
- Wang, C.; Shao, L.; Pan, C.; Ye, J.; Ding, Z.; Wu, J.; Du, Q.; Ren, Y.; Zhu, C. Elevated level of mitochondrial reactive oxygen species via fatty acid β-oxidation in cancer stem cells promotes cancer metastasis by inducing epithelial–mesenchymal transition. Stem Cell Res. Ther. 2019, 10, 175. [Google Scholar] [CrossRef]
- Babajani, A.; Eftekharinasab, A.; Bekeschus, S.; Mehdian, H.; Vakhshiteh, F.; Madjd, Z. Reactive oxygen species from non-thermal gas plasma (CAP): Implication for targeting cancer stem cells. Cancer Cell Int. 2024, 24, 344. [Google Scholar] [CrossRef]
- Rudrapal, M.; Khairnar, S.J.; Khan, J.; Dukhyil, A.B.; Ansari, M.A.; Alomary, M.N.; Alshabrmi, F.M.; Palai, S.; Deb, P.K.; Devi, R. Dietary Polyphenols and Their Role in Oxidative Stress-Induced Human Diseases: Insights Into Protective Effects, Antioxidant Potentials and Mechanism(s) of Action. Front. Pharmacol. 2022, 13, 806470. [Google Scholar] [CrossRef]
- Castelli, V.; Giordano, A.; Benedetti, E.; Giansanti, F.; Quintiliani, M.; Cimini, A.; D’angelo, M. The Great Escape: The Power of Cancer Stem Cells to Evade Programmed Cell Death. Cancers 2021, 13, 328. [Google Scholar] [CrossRef]
- Kobayashi, C.I.; Suda, T. Regulation of reactive oxygen species in stem cells and cancer stem cells. J. Cell. Physiol. 2012, 227, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Simonian, N.; Getz, R.; Leveque, J.; Konradi, C.; Coyle, J. Kainic acid induces apoptosis in neurons. Neuroscience 1996, 75, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Gella, A.; Durany, N. Oxidative stress in Alzheimer disease. Cell Adhes. Migr. 2009, 3, 88–93. [Google Scholar] [CrossRef]
- Leuner, K.; Hauptmann, S.; Abdel-Kader, R.; Scherping, I.; Keil, U.; Strosznajder, J.B.; Eckert, A.; Müller, W.E. Mitochondrial Dysfunction: The First Domino in Brain Aging and Alzheimer’s Disease? Antioxid. Redox Signal. 2007, 9, 1659–1676. [Google Scholar] [CrossRef]
- Hashimoto, M.; Takeda, A.; Hsu, L.J.; Takenouchi, T.; Masliah, E. Role of Cytochrome c as a Stimulator of α-Synuclein Aggregation in Lewy Body Disease. J. Biol. Chem. 1999, 274, 28849–28852. [Google Scholar] [CrossRef]
- Chin-Chan, M.; Navarro-Yepes, J.; Quintanilla-Vega, B. Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front. Cell. Neurosci. 2015, 9, 124. [Google Scholar] [CrossRef]
- Akanji, M.A.; Rotimi, D.E.; Elebiyo, T.C.; Awakan, O.J.; Adeyemi, O.S. Redox Homeostasis and Prospects for Therapeutic Targeting in Neurodegenerative Disorders. Oxidative Med. Cell. Longev. 2021, 2021, 9971885. [Google Scholar] [CrossRef]
- Paloczi, J.; Varga, Z.V.; Hasko, G.; Pacher, P. Neuroprotection in Oxidative Stress-Related Neurodegenerative Diseases: Role of Endocannabinoid System Modulation. Antioxid. Redox Signal. 2018, 29, 75–108. [Google Scholar] [CrossRef]
- Haddad, J.J. Antioxidant and prooxidant mechanisms in the regulation of redox (y)-sensitive tran-scription factors. Cell Signal. 2002, 14, 879–897. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Sun, Y. Tumor microenvironment and cancer therapy resistance. Cancer Lett. 2016, 380, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Huang, Y.-H.; Chen, J.-L. Understanding and targeting cancer stem cells: Therapeutic implications and challenges. Acta Pharmacol. Sin. 2013, 34, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Turdo, A.; Veschi, V.; Gaggianesi, M.; Chinnici, A.; Bianca, P.; Todaro, M.; Stassi, G. Meeting the Challenge of Targeting Cancer Stem Cells. Front. Cell Dev. Biol. 2019, 7, 16. [Google Scholar] [CrossRef]
- Yadav, A.K.; Desai, N.S. Cancer Stem Cells: Acquisition, Characteristics, Therapeutic Implications, Targeting Strategies and Future Prospects. Stem Cell Rev. Rep. 2019, 15, 331–355. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells: Current status and evolving complexities. Cell Stem Cell 2012, 10, 717–728. [Google Scholar] [CrossRef]
- Sugihara, E.; Saya, H. Complexity of cancer stem cells. Int. J. Cancer 2013, 132, 1249–1259. [Google Scholar] [CrossRef]
- Kuşoğlu, A.; Avcı, Ç.B. Cancer stem cells: A brief review of the current status. Gene 2019, 681, 80–85. [Google Scholar] [CrossRef]
- Luo, M.; Wicha, M.S. Targeting Cancer Stem Cell Redox Metabolism to Enhance Therapy Responses. Semin. Radiat. Oncol. 2019, 29, 42–54. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, S.; Li, Y.; Liu, Z.; Mi, L.; Cai, Y.; Wang, X.; Chen, L.; Ran, H.; Xiao, D.; et al. Suppression of mitochondrial ROS by prohibitin drives glioblastoma progression and therapeutic resistance. Nat. Commun. 2021, 12, 3720. [Google Scholar] [CrossRef]
- Deshmukh, A.; Deshpande, K.; Arfuso, F.; Newsholme, P.; Dharmarajan, A. Cancer stem cell metabolism: A potential target for cancer therapy. Mol. Cancer 2016, 15, 69. [Google Scholar] [CrossRef] [PubMed]
- Ismail, T.; Kim, Y.; Lee, H.; Lee, D.-S.; Lee, H.-S. Interplay Between Mitochondrial Peroxiredoxins and ROS in Cancer Development and Progression. Int. J. Mol. Sci. 2019, 20, 4407. [Google Scholar] [CrossRef] [PubMed]
- Sonn, S.K.; Song, E.J.; Seo, S.; Kim, Y.Y.; Um, J.-H.; Yeo, F.J.; Lee, D.S.; Jeon, S.; Lee, M.-N.; Jin, J.; et al. Peroxiredoxin 3 deficiency induces cardiac hypertrophy and dysfunction by impaired mitochondrial quality control. Redox Biol. 2022, 51, 102275. [Google Scholar] [CrossRef]
- Song, I.-S.; Kim, H.-K.; Jeong, S.-H.; Lee, S.-R.; Kim, N.; Rhee, B.D.; Ko, K.S.; Han, J. Mitochondrial Peroxiredoxin III is a Potential Target for Cancer Therapy. Int. J. Mol. Sci. 2011, 12, 7163–7185. [Google Scholar] [CrossRef]
- Cox, A.G.; Winterbourn, C.C.; Hampton, M.B. Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. Biochem. J. 2010, 425, 313–325. [Google Scholar] [CrossRef]
- Sahoo, B.M.; Banik, B.K.; Borah, P.; Jain, A. Reactive Oxygen Species (ROS): Key Components in Cancer Therapies. Anti-Cancer Agents Med. Chem. 2022, 22, 215–222. [Google Scholar] [CrossRef]
- Ralph, S.J.; Rodríguez-Enríquez, S.; Neuzil, J.; Saavedra, E.; Moreno-Sánchez, R. The causes of cancer revisited: “mitochondrial malignancy” and ROS-induced oncogenic transformation–why mitochondria are targets for cancer therapy. Mol. Asp. Med. 2010, 31, 145–170. [Google Scholar] [CrossRef]
- Maya-Mendoza, A.; Ostrakova, J.; Kosar, M.; Hall, A.; Duskova, P.; Mistrik, M.; Merchut-Maya, J.M.; Hodny, Z.; Bartkova, J.; Christensen, C.; et al. Myc and Ras oncogenes engage different energy metabolism programs and evoke distinct patterns of oxidative and DNA replication stress. Mol. Oncol. 2014, 9, 601–616. [Google Scholar] [CrossRef]
- Tarrado-Castellarnau, M.; de Atauri, P.; Cascante, M. Oncogenic regulation of tumor metabolic reprogramming. Oncotarget 2016, 7, 62726. [Google Scholar] [CrossRef]
- Li, B.; Simon, M.C. Molecular pathways: Targeting MYC-induced metabolic reprogramming and oncogenic stress in cancer. Clin. Cancer Res. 2013, 19, 5835–5841. [Google Scholar] [CrossRef]
- Casacuberta-Serra, S.; González-Larreategui, Í.; Capitán-Leo, D.; Soucek, L. MYC and KRAS cooperation: From historical challenges to therapeutic opportunities in cancer. Signal Transduct. Target. Ther. 2024, 9, 205. [Google Scholar] [CrossRef] [PubMed]
- Bansal, M.P. Redox Regulation and Therapeutic Approaches in Cancer; Springer Nature: Dordrecht, The Netherlands, 2023. [Google Scholar]
- Khan, S.U.; Rayees, S.; Sharma, P.; Malik, F. Targeting redox regulation and autophagy systems in cancer stem cells. Clin. Exp. Med. 2023, 23, 1405–1423. [Google Scholar] [CrossRef] [PubMed]
- Tuy, K.; Rickenbacker, L.; Hjelmeland, A.B. Reactive oxygen species produced by altered tumor metabolism impacts cancer stem cell maintenance. Redox Biol. 2021, 44, 101953. [Google Scholar] [CrossRef] [PubMed]
- Quiles, J.L.; Sánchez-González, C.; Vera-Ramírez, L.; Giampieri, F.; Navarro-Hortal, M.D.; Xiao, J.; Llopis, J.; Battino, M.; Varela-López, A. Reductive Stress, Bioactive Compounds, Redox-Active Metals, and Dormant Tumor Cell Biology to Develop Redox-Based Tools for the Treatment of Cancer. Antioxid. Redox Signal. 2020, 33, 860–881. [Google Scholar] [CrossRef]
- Movahed, Z.G.; Yarani, R.; Mohammadi, P.; Mansouri, K. Sustained oxidative stress instigates differentiation of cancer stem cells into tumor endothelial cells: Pentose phosphate pathway, reactive oxygen species and autophagy crosstalk. Biomed. Pharmacother. 2021, 139, 111643. [Google Scholar] [CrossRef]
- Kciuk, M.; Gielecińska, A.; Budzinska, A.; Mojzych, M.; Kontek, R. Metastasis and MAPK Pathways. Int. J. Mol. Sci. 2022, 23, 3847. [Google Scholar] [CrossRef]
- Nazio, F.; Bordi, M.; Cianfanelli, V.; Locatelli, F.; Cecconi, F. Autophagy and cancer stem cells: Molecular mechanisms and therapeutic applications. Cell Death Differ. 2019, 26, 690–702. [Google Scholar] [CrossRef]
- Kahroba, H.; Shirmohamadi, M.; Hejazi, M.S.; Samadi, N. The Role of Nrf2 signaling in cancer stem cells: From stemness and self-renewal to tumorigenesis and chemoresistance. Life Sci. 2019, 239, 116986. [Google Scholar] [CrossRef]
- Ezhilarasan, D.; Elumalai, P.; Anandan, B.; Muralidharan, A. Role of Stem Cells and Reactive Oxygen Species in Cancer: An Insight into Nrf2 Signaling. In Handbook of Oxidative Stress in Cancer: Therapeutic Aspects; Springer: Berlin/Heidelberg, Germany, 2022. [Google Scholar]
- Luo, M.; Bao, L.; Xue, Y.; Zhu, M.; Kumar, A.; Xing, C.; Wang, J.E.; Wang, Y.; Luo, W. ZMYND8 protects breast cancer stem cells against oxidative stress and ferroptosis through activation of NRF2. J. Clin. Investig. 2024, 134, e171166. [Google Scholar] [CrossRef]
- Yang, W.; Shen, Y.; Wei, J.; Liu, F. MicroRNA-153/Nrf-2/GPx1 pathway regulates radiosensitivity and stemness of glioma stem cells via reactive oxygen species. Oncotarget 2015, 6, 22006. [Google Scholar] [CrossRef]
- Moubarak, M. Study of the Role of NRF2 in the Progression and Metabolic Adaptation of Glioblastoma. Ph.D. Thesis, Université de Bordeaux, Bordeaux, France, 2024. Available online: https://theses.hal.science/tel-04788234/ (accessed on 15 February 2025).
- Cesário, R.S.R. The Effects of Ionizing and UVA Radiation Induced Oxidative Stress in Glioblastoma Stem Cells Silenced for NRF2. Master’s Thesis, Universidade do Porto, Porto, Portugal, 2021. Available online: https://repositorio-aberto.up.pt/bitstream/10216/138583/2/520911.pdf (accessed on 15 February 2025).
- Zhu, J.-M.; Chen, C.; Kong, M.; Zhu, L.; Li, Y.-L.; Zhang, J.-F.; Yu, Z.-P.; Xu, S.-S.; Kong, L.-Y.; Luo, J.-G. Discovery and optimization of indirubin derivatives as novel ferroptosis inducers for the treatment of colon cancer. Eur. J. Med. Chem. 2023, 261, 115829. [Google Scholar] [CrossRef] [PubMed]
- Ramisetti, S.V.; Patra, T.; Munirathnam, V.; Sainath, J.V.; Veeraiyan, D.; Namani, A. NRF2 Signaling Pathway in Chemo/Radio/Immuno-Therapy Resistance of Lung Cancer: Looking Beyond the Tip of the Iceberg. Arch. Bronc. 2024, 60, S59–S66. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Farhood, B.; Mortezaee, K.; Kharazinejad, E.; Majidpoor, J.; Ahadi, R. Hypoxia in solid tumors: A key promoter of cancer stem cell (CSC) resistance. J. Cancer Res. Clin. Oncol. 2020, 146, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Farhood, B.; Mortezaee, K. Cancer stem cells (CSCs) in cancer progression and therapy. J. Cell. Physiol. 2019, 234, 8381–8395. [Google Scholar] [CrossRef]
- Götting, I.; Jendrossek, V.; Matschke, J. A New Twist in Protein Kinase B/Akt Signaling: Role of Altered Cancer Cell Metabolism in Akt-Mediated Therapy Resistance. Int. J. Mol. Sci. 2020, 21, 8563. [Google Scholar] [CrossRef]
- Deng, H.; Chen, Y.; Wang, L.; Zhang, Y.; Hang, Q.; Li, P.; Zhang, P.; Ji, J.; Song, H.; Chen, M.; et al. PI3K/mTOR inhibitors promote G6PD autophagic degradation and exacerbate oxidative stress damage to radiosensitize small cell lung cancer. Cell Death Dis. 2023, 14, 652. [Google Scholar] [CrossRef]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.-G.; Lee, S.-H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef]
- Marengo, B.; Nitti, M.; Furfaro, A.L.; Colla, R.; De Ciucis, C.; Marinari, U.M.; Pronzato, M.A.; Traverso, N.; Domenicotti, C. Redox Homeostasis and Cellular Antioxidant Systems: Crucial Players in Cancer Growth and Therapy. Oxid. Med. Cell. Longev. 2016, 2016, 6235641. [Google Scholar] [CrossRef]
- Gu, X.; Mu, C.; Zheng, R.; Zhang, Z.; Zhang, Q.; Liang, T. The Cancer Antioxidant Regulation System in Therapeutic Resistance. Antioxidants 2024, 13, 778. [Google Scholar] [CrossRef]
- Stouras, I.; Vasileiou, M.; Kanatas, P.F.; Tziona, E.; Tsianava, C.; Theocharis, S. Metabolic Profiles of Cancer Stem Cells and Normal Stem Cells and Their Therapeutic Significance. Cells 2023, 12, 2686. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Kuo, W.-T.; Huang, Y.-C.; Lee, T.-C.; Yu, L.C.H. Resistance to hypoxia-induced necroptosis is conferred by glycolytic pyruvate scavenging of mitochondrial superoxide in colorectal cancer cells. Cell Death Dis. 2013, 4, e622. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Yan, Y.; Niu, F.; Wang, Y.; Chen, X.; Su, G.; Liu, Y.; Zhao, X.; Qian, L.; Liu, P.; et al. Ferroptosis: A cell death connecting oxidative stress, inflammation and cardiovascular diseases. Cell Death Discov. 2021, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Paskeh, M.D.A.; Asadi, S.; Zabolian, A.; Saleki, H.; Khoshbakht, M.A.; Sabet, S.; Naghdi, M.J.; Hashemi, M.; Hushmandi, K.; Ashrafizadeh, M.; et al. Targeting Cancer Stem Cells by Dietary Agents: An Important Therapeutic Strategy against Human Malignancies. Int. J. Mol. Sci. 2021, 22, 11669. [Google Scholar] [CrossRef]
- Abdullah, L.N.; Chow, E.K.-H. Mechanisms of chemoresistance in cancer stem cells. Clin. Transl. Med. 2013, 2, 3. [Google Scholar] [CrossRef]
- Yoshida, G.J.; Saya, H. Therapeutic strategies targeting cancer stem cells. Cancer Sci. 2016, 107, 5–11. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, H.S.; Seo, Y.R. Understanding of ROS-Inducing Strategy in Anticancer Therapy. Oxid. Med. Cell. Longev. 2019, 2019, 5381692. [Google Scholar] [CrossRef]
- Sun, F.; Li, W.; Du, R.; Liu, M.; Cheng, Y.; Ma, J.; Yan, S. Impact of glycolysis enzymes and metabolites in regulating DNA damage repair in tumorigenesis and therapy. Cell Commun. Signal. 2025, 23, 44. [Google Scholar] [CrossRef]
- Fu, Y.; Chang, H.; Peng, X.; Bai, Q.; Yi, L.; Zhou, Y.; Zhu, J.; Mi, M. Resveratrol Inhibits Breast Cancer Stem-Like Cells and Induces Autophagy via Suppressing Wnt/β-Catenin Signaling Pathway. PLoS ONE 2014, 9, e102535. [Google Scholar] [CrossRef]
- Yuan, Y.; Xue, X.; Guo, R.; Sun, X.; Hu, G. Resveratrol Enhances the Antitumor Effects of Temozolomide in Glioblastoma via ROS-dependent AMPK-TSC-mTOR Signaling Pathway. CNS Neurosci. Ther. 2012, 18, 536–546. [Google Scholar] [CrossRef]
- Zhong, G.; Qin, S.; Townsend, D.; Schulte, B.A.; Tew, K.D.; Wang, G.Y. Oxidative stress induces senescence in breast cancer stem cells. Biochem. Biophys. Res. Commun. 2019, 514, 1204–1209. [Google Scholar] [CrossRef]
- Yan, M.; Wang, H.; Wei, R.; Li, W. Arsenic trioxide: Applications, mechanisms of action, toxicity and rescue strategies to date. Arch. Pharmacal Res. 2024, 47, 249–271. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, G.J.; Saya, H. Molecular pathology underlying the robustness of cancer stem cells. Regen. Ther. 2021, 17, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef]
- Ebrahimi, S.; Hashemy, S.I. MicroRNA-mediated redox regulation modulates therapy resistance in cancer cells: Clinical perspectives. Cell. Oncol. 2019, 42, 131–141. [Google Scholar] [CrossRef]
- Parihar, A.; Sharma, P.; Choudhary, N.K.; Khan, R. Neurobiosensors: Novel approaches towards early diagnostics of neurodegenerative disorders. In Smart Diagnostics for Neurodegenerative Disorders; Academic Press: Cambridge, MA, USA, 2024. [Google Scholar]
- Ciurea, A.V.; Mohan, A.G.; Covache-Busuioc, R.-A.; Costin, H.-P.; Glavan, L.-A.; Corlatescu, A.-D.; Saceleanu, V.M. Unraveling Molecular and Genetic Insights into Neurodegenerative Diseases: Advances in Understanding Alzheimer’s, Parkinson’s, and Huntington’s Diseases and Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2023, 24, 10809. [Google Scholar] [CrossRef] [PubMed]
- Pathak, N.; Vimal, S.K.; Tandon, I.; Agrawal, L.; Hongyi, C.; Bhattacharyya, S. Neurodegenerative Disorders of Alzheimer, Parkinsonism, Amyotrophic Lateral Sclerosis and Multiple Sclerosis: An Early Diagnostic Approach for Precision Treatment. Metab. Brain Dis. 2022, 37, 67–104. [Google Scholar] [CrossRef]
- Elfawy, H.A.; Das, B. Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: Etiologies and therapeutic strategies. Life Sci. 2019, 218, 165–184. [Google Scholar] [CrossRef]
- Kowalczyk, P.; Sulejczak, D.; Kleczkowska, P.; Bukowska-Ośko, I.; Kucia, M.; Popiel, M.; Wietrak, E.; Kramkowski, K.; Wrzosek, K.; Kaczyńska, K. Mitochondrial Oxidative Stress—A Causative Factor and Therapeutic Target in Many Diseases. Int. J. Mol. Sci. 2021, 22, 13384. [Google Scholar] [CrossRef]
- Radi, E.; Formichi, P.; Battisti, C.; Federico, A. Apoptosis and Oxidative Stress in Neurodegenerative Diseases. J. Alzheimer’s Dis. 2014, 42 (Suppl. S3), S125–S152. [Google Scholar] [CrossRef]
- Islam, M.T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative Stress in Neurodegenerative Diseases: From a Mitochondrial Point of View. Oxidative Med. Cell. Longev. 2019, 2019, 2105607. [Google Scholar] [CrossRef] [PubMed]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative Stress in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef] [PubMed]
- Storkebaum, E.; Quaegebeur, A.; Vikkula, M.; Carmeliet, P. Cerebrovascular disorders: Molecular insights and therapeutic opportunities. Nat. Neurosci. 2011, 14, 1390–1397. [Google Scholar] [CrossRef]
- Freeman, L.R.; Keller, J.N. Oxidative stress and cerebral endothelial cells: Regulation of the blood–brain-barrier and antioxidant based interventions. Biochim. Biophys. Acta-Mol. Basis Dis. 2012, 1822, 822–829. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, M.; Liang, X. The role of mitochondria in iron overload-induced damage. J. Transl. Med. 2024, 22, 1057. [Google Scholar] [CrossRef]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Cassano, T.; Pace, L.; Bedse, G.; Lavecchia, A.M.; De Marco, F.; Gaetani, S.; Serviddio, G. Glutamate and Mitochondria: Two Prominent Players in the Oxidative Stress-Induced Neurodegeneration. Curr. Alzheimer Res. 2016, 13, 185–197. [Google Scholar] [CrossRef]
- Armada-Moreira, A.; Gomes, J.I.; Pina, C.C.; Savchak, O.K.; Gonçalves-Ribeiro, J.; Rei, N.; Pinto, S.; Morais, T.P.; Martins, R.S.; Ribeiro, F.F.; et al. Going the Extra (Synaptic) Mile: Excitotoxicity as the Road Toward Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14, 90. [Google Scholar] [CrossRef]
- Yadav, V.K.; Choudhary, N.; Gacem, A.; Verma, R.K.; Hasan, M.A.; Imam, M.T.; Almalki, Z.S.; Yadav, K.K.; Park, H.-K.; Ghosh, T.; et al. Deeper insight into ferroptosis: Association with Alzheimer’s, Parkinson’s disease, and brain tumors and their possible treatment by nanomaterials induced ferroptosis. Redox Rep. 2023, 28, 2269331. [Google Scholar] [CrossRef]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef] [PubMed]
- Viktorinova, A.; Durfinova, M. Mini-review: Is iron-mediated cell death (ferroptosis) an identical factor contributing to the pathogenesis of some neurodegenerative diseases? Neurosci. Lett. 2021, 745, 135627. [Google Scholar] [CrossRef] [PubMed]
- Mahoney-Sánchez, L.; Bouchaoui, H.; Ayton, S.; Devos, D.; Duce, J.A.; Devedjian, J.-C. Ferroptosis and its potential role in the physiopathology of Parkinson’s Disease. Prog. Neurobiol. 2021, 196, 101890. [Google Scholar] [CrossRef]
- Masaldan, S.; Bush, A.I.; Devos, D.; Rolland, A.S.; Moreau, C. Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. Free. Radic. Biol. Med. 2019, 133, 221–233. [Google Scholar] [CrossRef]
- DeGregorio-Rocasolano, N.; Martí-Sistac, O.; Gasull, T. Deciphering the iron side of stroke: Neurodegeneration at the crossroads between iron dyshomeostasis, excitotoxicity, and ferroptosis. Front. Neurosci. 2019, 13, 85. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar]
- Giorgi, C.; Marchi, S.; Simoes, I.C.; Ren, Z.; Morciano, G.; Perrone, M.; Patalas-Krawczyk, P.; Borchard, S.; Jędrak, P.; Pierzynowska, K.; et al. Mitochondria and reactive oxygen species in aging and age-related diseases. Int. Rev. Cell Mol. Biol. 2018, 340, 209–344. [Google Scholar] [CrossRef]
- Sun, K.; De Pablo, Y.; Vincent, F.; Shah, K. Deregulated Cdk5 promotes oxidative stress and mitochondrial dysfunction. J. Neurochem. 2008, 107, 265–278. [Google Scholar] [CrossRef]
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 2015, 74, 101–110. [Google Scholar] [CrossRef]
- Taso, O.V.; Philippou, A.; Moustogiannis, A.; Zevolis, E.; Koutsilieris, M. Lipid peroxidation products and their role in neurodegenerative diseases. Ann. Res. Hosp. 2019, 3, 2. [Google Scholar] [CrossRef]
- Stachowicz, K. The role of polyunsaturated fatty acids in neuronal signaling in depression and cognitive processes. Arch. Biochem. Biophys. 2023, 737, 109555. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Zhang, Z.; Saha, A.; Sarkar, C.; Zhao, Z.; Xu, Y.; Mukherjee, A.B. Omega-3 and omega-6 fatty acids suppress ER- and oxidative stress in cultured neurons and neuronal progenitor cells from mice lacking PPT1. Neurosci. Lett. 2010, 479, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Michael-Titus, A. Omega-3 fatty acids: Their neuroprotective and regenerative potential in traumatic neurological injury. Clin. Lipidol. 2009, 4, 343–353. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Potapova, E.V.; Dremin, V.V.; Dunaev, A.V. Interaction of Oxidative Stress and Misfolded Proteins in the Mechanism of Neurodegeneration. Life 2020, 10, 101. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Zaltieri, M.; Longhena, F.; Pizzi, M.; Missale, C.; Spano, P.; Bellucci, A. Mitochondrial Dysfunction andα-Synuclein Synaptic Pathology in Parkinson’s Disease: Who’s on First? Park. Dis. 2015, 2015, 108029. [Google Scholar] [CrossRef]
- Alqahtani, T.; Deore, S.L.; Kide, A.A.; Shende, B.A.; Sharma, R.; Chakole, R.D.; Nemade, L.S.; Kale, N.K.; Borah, S.; Deokar, S.S.; et al. Mitochondrial dysfunction and oxidative stress in Alzheimer’s disease, and Parkinson’s disease, Huntington’s disease and amyotrophic lateral sclerosis-an updated review. Mitochondrion 2023, 71, 83–92. [Google Scholar] [CrossRef]
- Uversky, V.N. Looking at the recent advances in understanding α-synuclein and its aggregation through the proteoform prism. F1000Research 2017, 6, 525. [Google Scholar] [CrossRef]
- Deas, E.; Cremades, N.; Angelova, P.R.; Ludtmann, M.H.; Yao, Z.; Chen, S.; Horrocks, M.H.; Banushi, B.; Little, D.; Devine, M.J.; et al. Alpha-Synuclein Oligomers Interact with Metal Ions to Induce Oxidative Stress and Neuronal Death in Parkinson’s Disease. Antioxid. Redox Signal. 2016, 24, 376–391. [Google Scholar] [CrossRef]
- ALKHALIFA, A.; Alkhalifa, O.; Durdanovic, I.; Ibrahim, D. Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease: Insights into Pathophysiology and Treatment. Preprints 2024, 2024081375. [Google Scholar] [CrossRef]
- Salkovic-Petrisic, M.; Osmanovic, J.; Grünblatt, E.; Riederer, P.; Hoyer, S. Modeling Sporadic Alzheimer’s Disease: The Insulin Resistant Brain State Generates Multiple Long-Term Morphobiological Abnormalities Including Hyperphosphorylated Tau Protein and Amyloid-β. J. Alzheimer’s Dis. 2009, 18, 729–750. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed]
- Fujii, J.; Katsuyama, M.; Matsuno, K.; Yabeenishimura, C. Physiological roles of NOX/NADPH oxidase, the superox-ide-generating enzyme. J. Clin. Biochem. Nutr. 2011, 50, 9–22. [Google Scholar] [CrossRef]
- Hannan, M.A.; Dash, R.; Sohag, A.A.M.; Haque, M.N.; Moon, I.S. Neuroprotection Against Oxidative Stress: Phytochemicals Targeting TrkB Signaling and the Nrf2-ARE Antioxidant System. Front. Mol. Neurosci. 2020, 13, 116. [Google Scholar] [CrossRef]
- Brandes, M.S.; Gray, N.E. NRF2 as a Therapeutic Target in Neurodegenerative Diseases. ASN Neuro 2020, 12, 1759091419899782. [Google Scholar] [CrossRef]
- Wruck, C.J.; Go, M.E.; Herdegen, T.; Varoga, D.; Brandenburg, L.-O.; Pufe, T. Kavalactones protect neural cells against amyloid β pep-tide-induced neurotoxicity via extracellular signal-regulated kinase 1/2-dependent nuclear factor erythroid 2. Mol. Pharmacol. 2008, 73, 1785–1795. [Google Scholar] [CrossRef]
- Kanninen, K.; Malm, T.M.; Jyrkkänen, H.-K.; Goldsteins, G.; Keksa-Goldsteine, V.; Tanila, H.; Yamamoto, M.; Ylä-Herttuala, S.; Levonen, A.-L.; Koistinaho, J. Nuclear factor erythroid 2-related factor 2 protects against beta amyloid. Mol. Cell. Neurosci. 2008, 39, 302–313. [Google Scholar] [CrossRef]
- Zhang, Q.-G.; Laird, M.D.; Han, D.; Nguyen, K.; Scott, E.; Dong, Y.; Dhandapani, K.M.; Brann, D.W. Critical Role of NADPH Oxidase in Neuronal Oxidative Damage and Microglia Activation following Traumatic Brain Injury. PLoS ONE 2012, 7, e34504. [Google Scholar] [CrossRef]
- Tezel, G. Oxidative stress in glaucomatous neurodegeneration: Mechanisms and consequences. Prog. Retin. Eye Res. 2006, 25, 490–513. [Google Scholar] [CrossRef]
- Altenhöfer, S.; Radermacher, K.A.; Kleikers, P.W.; Wingler, K.; Schmidt, H.H. Evolution of NADPH Oxidase Inhibitors: Selectivity and Mechanisms for Target Engagement. Antioxid. Redox Signal. 2015, 23, 406–427. [Google Scholar] [CrossRef]
- Barbosa, M.C.; Grosso, R.A.; Fader, C.M. Hallmarks of Aging: An Autophagic Perspective. Front. Endocrinol. 2019, 9, 790. [Google Scholar] [CrossRef] [PubMed]
- Houck, A.L.; Seddighi, S.; Driver, J.A. At the Crossroads Between Neurodegeneration and Cancer: A Review of Overlapping Biology and Its Implications. Curr. Aging Sci. 2018, 11, 77–89. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Tian, W.; Ning, J.; Xiao, G.; Zhou, Y.; Wang, Z.; Zhai, Z.; Tanzhu, G.; Yang, J.; Zhou, R. Cancer stem cells: Advances in knowledge and implications for cancer therapy. Signal Transduct. Target. Ther. 2024, 9, 170. [Google Scholar] [CrossRef]
- Wilson, D.M.; Cookson, M.R.; Bosch, L.V.D.; Zetterberg, H.; Holtzman, D.M.; Dewachter, I. Hallmarks of neurodegenerative diseases. Cell 2023, 186, 693–714. [Google Scholar] [CrossRef]
- Ding, S.; Li, C.; Cheng, N.; Cui, X.; Xu, X.; Zhou, G. Redox Regulation in Cancer Stem Cells. Oxidative Med. Cell. Longev. 2015, 2015, 750798. [Google Scholar] [CrossRef]
- Bailo, P.S.; Martín, E.L.; Calmarza, P.; Breva, S.M.; Gómez, A.B.; Giráldez, A.P.; Callau, J.J.S.-P.; Santamaría, J.M.V.; Khialani, A.D.; Micó, C.C.; et al. The role of oxidative stress in neurodegenerative diseases and potential antioxidant therapies. Adv. Lab. Med./Av. En Med. De Lab. 2022, 3, 342–350. [Google Scholar] [CrossRef]
- Tanabe, A.; Sahara, H. The metabolic heterogeneity and flexibility of cancer stem cells. Cancers 2020, 12, 2780. [Google Scholar] [CrossRef]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef]
- Suzen, S.; Tucci, P.; Profumo, E.; Buttari, B.; Saso, L. A Pivotal Role of Nrf2 in Neurodegenerative Disorders: A New Way for Therapeutic Strategies. Pharmaceuticals 2022, 15, 692. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Sanchez, M.J.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Dueñas, C.; Romero-Camarero, I.; Cobaleda, C.; Sánchez-García, I. Function of oncogenes in cancer development: A changing paradigm. EMBO J. 2013, 32, 1502–1513. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Parkinson’s disease-associated protein Parkin: An unusual player in cancer. Cancer Commun. 2018, 38, 1–8. [Google Scholar] [CrossRef]
- Wu, B.; Shi, X.; Jiang, M.; Liu, H. Cross-talk between cancer stem cells and immune cells: Potential therapeutic targets in the tumor immune microenvironment. Mol. Cancer 2023, 22, 38. [Google Scholar] [CrossRef]
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H. Role of neuroinflammation in neurodegeneration development. Signal Transduct. Target. Ther. 2023, 8, 267. [Google Scholar] [CrossRef]
- Toader, C.; Tataru, C.P.; Munteanu, O.; Serban, M.; Covache-Busuioc, R.-A.; Ciurea, A.V.; Enyedi, M. Decoding Neurodegeneration: A Review of Molecular Mechanisms and Therapeutic Advances in Alzheimer’s, Parkinson’s, and ALS. Int. J. Mol. Sci. 2024, 25, 12613. [Google Scholar] [CrossRef]
- Hassan, W.; Noreen, H.; Rehman, S.; Kamal, M.A.; da Rocha, J.B.T. Association of Oxidative Stress with Neurological Disorders. Curr. Neuropharmacol. 2022, 20, 1046–1072. [Google Scholar] [CrossRef]
- Kim, T.Y.; Leem, E.; Lee, J.M.; Kim, S.R. Control of Reactive Oxygen Species for the Prevention of Parkinson’s Disease: The Possible Application of Flavonoids. Antioxidants 2020, 9, 583. [Google Scholar] [CrossRef]
- Zhang, J.; Lei, W.; Chen, X.; Wang, S.; Qian, W. Oxidative stress response induced by chemotherapy in leukemia treatment. Mol. Clin. Oncol. 2018, 8, 391–399. [Google Scholar] [CrossRef]
- Kim, E.-K.; Jang, M.; Song, M.-J.; Kim, D.; Kim, Y.; Jang, H.H. Redox-Mediated Mechanism of Chemoresistance in Cancer Cells. Antioxidants 2019, 8, 471. [Google Scholar] [CrossRef]
- Boonpraman, N.; Yi, S.S. NADPH oxidase 4 (NOX4) as a biomarker and therapeutic target in neurodegenerative diseases. Neural Regen. Res. 2024, 19, 1961–1966. [Google Scholar] [CrossRef] [PubMed]
- Klaunig, J.E. Oxidative stress and cancer. Curr. Phar-Maceutical Des. 2018, 24, 4771–4778. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.-J.; Jhe, Y.-L.; Kim, J.; Lim, J.Y.; Lee, J.E.; Shin, M.-K.; Cheong, J.-H. FoxM1-dependent and fatty acid oxidation-mediated ROS modulation is a cell-intrinsic drug resistance mechanism in cancer stem-like cells. Redox Biol. 2020, 36, 101589. [Google Scholar] [CrossRef] [PubMed]
- Encarnação, C.C.; Faria, G.M.; Franco, V.A.; Botelho, L.G.X.; Moraes, J.A.; Renovato-Martins, M. Interconnections within the tumor microenvironment: Extra-cellular vesicles as critical players of metabolic reprogramming in tumor cells. J. Cancer Metastasis Treat. 2024, 10, 28. [Google Scholar] [CrossRef]
- Jurcău, M.C.; Andronie-Cioara, F.L.; Jurcău, A.; Marcu, F.; Ţiț, D.M.; Pașcalău, N.; Nistor-Cseppentö, D.C. The Link between Oxidative Stress, Mitochondrial Dysfunction and Neuroinflammation in the Pathophysiology of Alzheimer’s Disease: Therapeutic Implications and Future Perspectives. Antioxidants 2022, 11, 2167. [Google Scholar] [CrossRef]
- Liochev, S.I.; Fridovich, I. Mutant Cu, Zn superoxide dismutases and familial amyotrophic lateral sclerosis: Evaluation of oxidative hypotheses. Free Radic. Biol. Med. 2003, 34, 1383–1389. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Chen, X.; Qian, Y.; Wu, S. The Warburg effect: Evolving interpretations of an established concept. Free. Radic. Biol. Med. 2014, 79, 253–263. [Google Scholar] [CrossRef]
- Zhou, D.; Duan, Z.; Li, Z.; Ge, F.; Wei, R.; Kong, L. The significance of glycolysis in tumor progression and its relationship with the tumor microenvironment. Front. Pharmacol. 2022, 13, 1091779. [Google Scholar] [CrossRef]
- Tang, B.L. Glucose, glycolysis, and neurodegenerative diseases. J. Cell. Physiol. 2020, 235, 7653–7662. [Google Scholar] [CrossRef]
- Zhu, T.; Zheng, J.; Zhuo, W.; Pan, P.; Li, M.; Zhang, W.; Zhou, H.; Gao, Y.; Li, X.; Liu, Z. ETV4 promotes breast cancer cell stemness by activating glycolysis and CXCR4-mediated sonic Hedgehog signaling. Cell Death Discov. 2021, 7, 126. [Google Scholar] [CrossRef] [PubMed]
- Simpson, I.A.; Chundu, K.R.; Davies-Hill, T.; Honer, W.G.; Davies, P. Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer’s disease. Ann. Neurol. 1994, 35, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Tufail, M.; Jiang, C.-H.; Li, N. Altered metabolism in cancer: Insights into energy pathways and therapeutic targets. Mol. Cancer 2024, 23, 203. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, H.; Manfredi, G. Proteinopathies and OXPHOS dysfunction in neurodegenerative diseases. J. Cell Biol. 2017, 216, 3917–3929. [Google Scholar] [CrossRef]
- Winklhofer, K.F.; Haass, C. Mitochondrial dysfunction in Parkinson’s disease. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2010, 1802, 29–44. [Google Scholar] [CrossRef]
- Lushchak, V.I.; Duszenko, M.; Gospodaryov, D.V.; Garaschuk, O. Oxidative Stress and Energy Metabolism in the Brain: Midlife as a Turning Point. Antioxidants 2021, 10, 1715. [Google Scholar] [CrossRef]
- Yang, H.-C.; Wu, Y.-H.; Yen, W.-C.; Liu, H.-Y.; Hwang, T.-L.; Stern, A.; Chiu, D.T.-Y. The Redox Role of G6PD in Cell Growth, Cell Death, and Cancer. Cells 2019, 8, 1055. [Google Scholar] [CrossRef]
- Tiwari, M. Glucose 6 phosphatase dehydrogenase (G6PD) and neurodegenerative disorders: Mapping diagnostic and therapeutic opportunities. Genes Dis. 2017, 4, 196–203. [Google Scholar] [CrossRef]
- Mascaraque, M.; Courtois, S.; Royo-García, A.; Barneda, D.; Stoian, A.M.; Villaoslada, I.; Espiau-Romera, P.; Bokil, A.; Cano-Galiano, A.; Jagust, P.; et al. Fatty acid oxidation is critical for the tumorigenic potential and chemoresistance of pancreatic cancer stem cells. J. Transl. Med. 2024, 22, 797. [Google Scholar] [CrossRef]
- Montine, T.J.; Morrow, J.D. Fatty Acid Oxidation in the Pathogenesis of Alzheimer’s Disease. Am. J. Pathol. 2005, 166, 1283–1289. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, L.; Chen, W.; Shen, L.; Jiang, J.; Sun, S.; Chen, Z. Role of Intra- and Extracellular Lipid Signals in Cancer Stemness and Potential Therapeutic Strategy. Front. Pharmacol. 2021, 12, 730751. [Google Scholar] [CrossRef] [PubMed]
- Block, R.C.; Dorsey, E.R.; Beck, C.A.; Brenna, J.T.; Shoulson, I. Altered cholesterol and fatty acid metabolism in Huntington disease. J. Clin. Lipidol. 2010, 4, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Thanan, R.; Oikawa, S.; Hiraku, Y.; Ohnishi, S.; Ma, N.; Pinlaor, S.; Yongvanit, P.; Kawanishi, S.; Murata, M. Oxidative Stress and Its Significant Roles in Neurodegenerative Diseases and Cancer. Int. J. Mol. Sci. 2015, 16, 193–217. [Google Scholar] [CrossRef]
- Smolková, K.; Mikó, E.; Kovács, T.; Leguina-Ruzzi, A.; Sipos, A.; Bai, P. Nuclear Factor Erythroid 2-Related Factor 2 in Regulating Cancer Metabolism. Antioxid. Redox Signal. 2020, 33, 966–997. [Google Scholar] [CrossRef] [PubMed]
- Sevastre-Berghian, A.C.; Ielciu, I.; Mitre, A.O.; Filip, G.A.; Oniga, I.; Vlase, L.; Benedec, D. Targeting Oxidative Stress Reduction and Inhibition of HDAC1, MECP2, and NF-kB Pathways in Rats With Experimentally Induced Hyperglycemia by Administration of Thymus marshallianus Willd. Extracts. Front. Pharmacol. 2020, 11, 581470. [Google Scholar] [CrossRef]
- Hallis, S.P.; Kim, J.M.; Kwak, M.-K. Emerging Role of NRF2 Signaling in Cancer Stem Cell Phenotype. Mol. Cells 2024, 46, 153–164. [Google Scholar] [CrossRef]
- Chen, B.; Cao, H.; Chen, L.; Yang, X.; Tian, X.; Li, R.; Cheng, O. Rifampicin Attenuated Global Cerebral Ischemia Injury via Activating the Nuclear Factor Erythroid 2-Related Factor Pathway. Front. Cell. Neurosci. 2016, 10, 273. [Google Scholar] [CrossRef]
- He, Z.; Li, X.; Han, S.; Ren, B.; Hu, X.; Li, N.; Du, X.; Ni, J.; Yang, X.; Liu, Q. Bis(ethylmaltolato) oxidovanadium (IV) attenuates amyloid-beta-mediated neuroinflammation by inhibiting NF-κB signaling pathway via a PPARγ-dependent mechanism. Metallomics 2021, 13, mfab036. [Google Scholar] [CrossRef]
- Meng, T.; Fu, S.; He, D.; Hu, G.; Gao, X.; Zhang, Y.; Huang, B.; Du, J.; Zhou, A.; Su, Y.; et al. Evodiamine Inhibits Lipopolysaccharide (LPS)-Induced Inflammation in BV-2 Cells via Regulating AKT/Nrf2-HO-1/NF-κB Signaling Axis. Cell. Mol. Neurobiol. 2021, 41, 115–127. [Google Scholar] [CrossRef]
- Teleanu, D.M.; Niculescu, A.-G.; Lungu, I.I.; Radu, C.I.; Vladâcenco, O.; Roza, E.; Costăchescu, B.; Grumezescu, A.M.; Teleanu, R.I. An Overview of Oxidative Stress, Neuroinflammation, and Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 5938. [Google Scholar] [CrossRef]
- Numakawa, T.; Matsumoto, T.; Numakawa, Y.; Richards, M.; Yamawaki, S.; Kunugi, H. Protective Action of Neurotrophic Factors and Estrogen against Oxidative Stress-Mediated Neurodegeneration. J. Toxicol. 2011, 2011, 405194. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.-D.; Zhao, X.; Li, Y.; Li, G.-R.; Liu, X.-L. Damage to dopaminergic neurons by oxidative stress in Parkinson’s disease (Review). Int. J. Mol. Med. 2018, 41, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Guo, J.; Ye, X.-Y.; Xie, Y.; Xie, T. Oxidative stress: The core pathogenesis and mechanism of Alzheimer’s disease. Ageing Res. Rev. 2022, 77, 101619. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Maier, O. Interrelation of Oxidative Stress and Inflammation in Neurodegenerative Disease: Role of TNF. Oxidative Med. Cell. Longev. 2015, 2015, 610813. [Google Scholar] [CrossRef]
- Korbecki, J.; Simińska, D.; Gąssowska-Dobrowolska, M.; Listos, J.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. Chronic and cycling hypoxia: Drivers of cancer chronic inflammation through HIF-1 and NF-κB activation: A review of the molecular mechanisms. Int. J. Mol. Sci. 2021, 22, 10701. [Google Scholar] [CrossRef]
- Sreesada, P.; Vandana; Krishnan, B.; Amrutha, R.; Chavan, Y.; Alfia, H.; Jyothis, A.; Venugopal, P.; Aradhya, R.; Suravajhala, P.; et al. Matrix metalloproteinases: Master regulators of tissue morphogenesis. Gene 2024, 933, 148990. [Google Scholar] [CrossRef]
- Kumar, G.B.; Nair, B.G.; Perry, J.J.P.; Martin, D.B. Recent insights into natural product inhibitors of matrix metalloproteinases. MedChemComm 2019, 10, 2024–2037. [Google Scholar] [CrossRef]
- Haorah, J.; Ramirez, S.H.; Schall, K.; Smith, D.; Pandya, R.; Persidsky, Y. Oxidative stress activates protein tyrosine kinase and matrix metalloproteinases leading to blood–brain barrier dysfunction. J. Neurochem. 2007, 101, 566–576. [Google Scholar] [CrossRef]
- Li, Z.; Takino, T.; Endo, Y.; Sato, H. Activation of MMP-9 by membrane type-1 MMP/MMP-2 axis stimulates tumor metastasis. Cancer Sci. 2017, 108, 347–353. [Google Scholar] [CrossRef]
- Ashok, A.; Andrabi, S.S.; Mansoor, S.; Kuang, Y.; Kwon, B.K.; Labhasetwar, V. Antioxidant Therapy in Oxidative Stress-Induced Neurodegenerative Diseases: Role of Nanoparticle-Based Drug Delivery Systems in Clinical Translation. Antioxidants 2022, 11, 408. [Google Scholar] [CrossRef]
- Wang, B.; Wang, Y.; Zhang, J.; Hu, C.; Jiang, J.; Li, Y.; Peng, Z. ROS-induced lipid peroxidation modulates cell death outcome: Mechanisms behind apoptosis, autophagy, and ferroptosis. Arch. Toxicol. 2023, 97, 1439–1451. [Google Scholar] [CrossRef] [PubMed]
- Farooqui, T.; Farooqui, A.A. Lipid-Mediated Oxidative Stress and Inflammation in the Pathogenesis of Parkinson’s Disease. Park. Dis. 2011, 2011, 247467. [Google Scholar] [CrossRef]
- La Rosa, P.; Petrillo, S.; Fiorenza, M.T.; Bertini, E.S.; Piemonte, F. Ferroptosis in Friedreich’s Ataxia: A Metal-Induced Neurodegenerative Disease. Biomolecules 2020, 10, 1551. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.-Y.; Liu, T.; Zhang, L.; Wang, M.-J.; Yang, Y.; Gao, J. Role of ferroptosis in neurological diseases. Neurosci. Lett. 2021, 747, 135614. [Google Scholar] [CrossRef]
- Wang, Y.; Wei, Z.; Pan, K.; Li, J.; Chen, Q. The function and mechanism of ferroptosis in cancer. Apoptosis 2020, 25, 786–798. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, R.; Ouyang, Y.; Shen, Y.; Hu, L.; Xu, C. Cancer stem cells: A target for overcoming therapeutic resistance and relapse. Cancer Biol. Med. 2024, 20, 985–1020. [Google Scholar] [CrossRef]
- Vitalakumar, D.; Sharma, A.; Flora, S.J.S. Ferroptosis: A potential therapeutic target for neurodegenerative diseases. J. Biochem. Mol. Toxicol. 2021, 35, e22830. [Google Scholar] [CrossRef]
- Li, Y.; Du, Y.; Zhou, Y.; Chen, Q.; Luo, Z.; Ren, Y.; Chen, X.; Chen, G. Iron and copper: Critical executioners of ferroptosis, cuproptosis and other forms of cell death. Cell Commun. Signal. 2023, 21, 327. [Google Scholar] [CrossRef]
- Ying, J.F.; Lu, Z.B.; Fu, L.Q.; Tong, Y.; Wang, Z.; Li, W.F.; Mou, X.Z. The role of iron homeostasis and iron-mediated ROS in cancer. Am. J. Cancer Res. 2021, 11, 1895. [Google Scholar]
- Ru, Q.; Li, Y.; Chen, L.; Wu, Y.; Min, J.; Wang, F. Iron homeostasis and ferroptosis in human diseases: Mechanisms and therapeutic prospects. Signal Transduct. Target. Ther. 2025, 9, 271. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, Z.; Song, L.; Gao, J.; Liu, Y. Lipid metabolism of cancer stem cells (Review). Oncol. Lett. 2022, 23, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, M.S.; Ruiz, J.; Watts, J.L. Polyunsaturated Fatty Acids Drive Lipid Peroxidation during Ferroptosis. Cells 2023, 12, 804. [Google Scholar] [CrossRef] [PubMed]
- Sokol, K.H.; Lee, C.J.; Rogers, T.J.; Waldhart, A.; Ellis, A.E.; Madireddy, S.; Daniels, S.R.; House, R.J.; Ye, X.; Olesnavich, M.; et al. Lipid availability influences ferroptosis sensitivity in cancer cells by regulating polyunsaturated fatty acid trafficking. Cell Chem. Biol. 2024, 21, 10. [Google Scholar] [CrossRef]
- Qiu, B.; Zandkarimi, F.; Bezjian, C.T.; Reznik, E.; Soni, R.K.; Gu, W.; Jiang, X.; Stockwell, B.R. Phospholipids with two polyunsaturated fatty acyl tails promote ferroptosis. Cell 2024, 187, 1177–1190.e18. [Google Scholar] [CrossRef]
- Singh, B.; Dutt, S. Role of cancer stem cells in maintenance of tumor heterogeneity in brain tumors. In Cancer Stem Cells and Signaling Pathways; Academic Press: Cambridge, MA, USA, 2024. [Google Scholar]
- Nieborowska-Skorska, M.; Kopinski, P.K.; Ray, R.; Hoser, G.; Ngaba, D.; Flis, S.; Cramer, K.; Reddy, M.M.; Koptyra, M.; Penserga, T.; et al. Rac2-MRC-cIII–generated ROS cause genomic insta-bility in chronic myeloid leukemia stem cells and primitive progenitors. Blood 2012, 119, 4253–4263. [Google Scholar] [CrossRef]
- Pawlowska, E.; Blasiak, J. DNA Repair—A Double-Edged Sword in the Genomic Stability of Cancer Cells—The Case of Chronic Myeloid Leukemia. Int. J. Mol. Sci. 2015, 16, 27535–27549. [Google Scholar] [CrossRef]
- Coppedè, F.; Migliore, L. DNA damage in neurodegenerative diseases. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2015, 776, 84–97. [Google Scholar] [CrossRef]
- Mao, P.; Reddy, P.H. Aging and amyloid beta-induced oxidative DNA damage and mitochondrial dysfunction in Alzheimer’s disease: Implications for early intervention and therapeutics. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2011, 1812, 1359–1370. [Google Scholar] [CrossRef]
- Guo, J.; Huang, X.; Dou, L.; Yan, M.; Shen, T.; Tang, W.; Li, J. Aging and aging-related diseases: From molecular mechanisms to interventions and treatments. Signal Transduct. Target. Ther. 2022, 7, 391. [Google Scholar] [CrossRef]
- Feng, J.; Zheng, Y.; Guo, M.; Ares, I.; Lopez-Torres, B.; Martínez-Larrañaga, M.-R.; Wang, X.; Anadón, A.; Martínez, M.-A. Oxidative stress, the blood–brain barrier and neurodegenerative diseases: The critical beneficial role of dietary antioxidants. Acta Pharm. Sin. B 2023, 13, 3988–4024. [Google Scholar] [CrossRef]
- Mileo, A.M.; Miccadei, S. Polyphenols as Modulator of Oxidative Stress in Cancer Disease: New Therapeutic Strategies. Oxidative Med. Cell. Longev. 2015, 2016, 6475624. [Google Scholar] [CrossRef] [PubMed]
- Shaban, S.; El-Husseny, M.W.A.; Abushouk, A.I.; Salem, A.M.A.; Mamdouh, M.; Abdel-Daim, M.M. Effects of Antioxidant Supplements on the Survival and Differentiation of Stem Cells. Oxidative Med. Cell. Longev. 2017, 2017, 5032102. [Google Scholar] [CrossRef] [PubMed]
- Di Carlo, E.; Sorrentino, C. Oxidative Stress and Age-Related Tumors. Antioxidants 2024, 13, 1109. [Google Scholar] [CrossRef] [PubMed]
- Wickens, A.P. Ageing and the free radical theory. Respir. Physiol. 2001, 128, 379–391. [Google Scholar] [CrossRef]
- Allemann, M.S.; Lee, P.; Beer, J.H.; Saravi, S.S.S. Targeting the redox system for cardiovascular regeneration in aging. Aging Cell 2023, 22, e14020. [Google Scholar] [CrossRef]
- Smith, J.A.; Park, S.; Krause, J.S.; Banik, N.L. Oxidative stress, DNA damage, and the telomeric complex as therapeutic targets in acute neurodegeneration. Neurochem. Int. 2013, 62, 764–775. [Google Scholar] [CrossRef]
- Konovalova, J.; Gerasymchuk, D.; Parkkinen, I.; Chmielarz, P.; Domanskyi, A. Interplay between MicroRNAs and Oxidative Stress in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 6055. [Google Scholar] [CrossRef]
- Niu, Y.; DesMarais, T.L.; Tong, Z.; Yao, Y.; Costa, M. Oxidative stress alters global histone modification and DNA methylation. Free. Radic. Biol. Med. 2015, 82, 22–28. [Google Scholar] [CrossRef]
- Mahalingaiah, P.K.S.; Ponnusamy, L.; Singh, K.P. Chronic Oxidative Stress Leads to Malignant Transformation Along with Acquisition of Stem Cell Characteristics, and Epithelial to Mesenchymal Transition in Human Renal Epithelial Cells. J. Cell. Physiol. 2015, 230, 1916–1928. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, H.; Gao, P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell 2022, 13, 877–919. [Google Scholar] [CrossRef]
- García-Giménez, J.L.; Romá-Mateo, C.; Pérez-Machado, G.; Peiró-Chova, L.; Pallardó, F.V. Role of glutathione in the regulation of epigenetic mechanisms in disease. Free. Radic. Biol. Med. 2017, 112, 36–48. [Google Scholar] [CrossRef]
- Bekdash, R.A. Methyl Donors, Epigenetic Alterations, and Brain Health: Understanding the Connection. Int. J. Mol. Sci. 2023, 24, 2346. [Google Scholar] [CrossRef] [PubMed]
- García-Guede, Á.; Vera, O.; Ibáñez-De-Caceres, I. When Oxidative Stress Meets Epigenetics: Implications in Cancer Development. Antioxidants 2020, 9, 468. [Google Scholar] [CrossRef] [PubMed]
- Fabrizio, F.P.; Sparaneo, A.; Muscarella, L.A. NRF2 Regulation by Noncoding RNAs in Cancers: The Present Knowledge and the Way Forward. Cancers 2020, 12, 3621. [Google Scholar] [CrossRef]
- Ionescu-Tucker, A.; Cotman, C.W. Emerging roles of oxidative stress in brain aging and Alzheimer’s disease. Neurobiol. Aging 2021, 107, 86–95. [Google Scholar] [CrossRef]
- Shen, L.; Song, C.-X.; He, C.; Zhang, Y. Mechanism and Function of Oxidative Reversal of DNA and RNA Methylation. Annu. Rev. Biochem. 2014, 83, 585–614. [Google Scholar] [CrossRef]
- Richa, R.; Sinha, R.P. Hydroxymethylation of DNA: An epigenetic marker. EXCLI J. 2014, 13, 592–610. [Google Scholar]
- Guillaumet-Adkins, A.; Yañez, Y.; Peris-Diaz, M.D.; Calabria, I.; Palanca-Ballester, C.; Sandoval, J. Epigenetics and Oxidative Stress in Aging. Oxidative Med. Cell. Longev. 2017, 2017, 9175806. [Google Scholar] [CrossRef]
- Snigdha, S.; Prieto, G.A.; Petrosyan, A.; Loertscher, B.M.; Dieskau, A.P.; Overman, L.E.; Cotman, C.W. H3K9me3 Inhibition Improves Memory, Promotes Spine Formation, and Increases BDNF Levels in the Aged Hippocampus. J. Neurosci. 2016, 36, 3611–3622. [Google Scholar] [CrossRef]
- Griñán-Ferré, C.; Marsal-García, L.; Bellver-Sanchis, A.; Kondengaden, S.M.; Turga, R.C.; Vázquez, S.; Pallàs, M. Pharmacological inhibition of G9a/GLP restores cognition and reduces oxidative stress, neuroinflammation and β-Amyloid plaques in an early-onset Alzheimer’s disease mouse model. Aging 2019, 11, 11591–11608. [Google Scholar] [CrossRef]
- Lei, Y.; Zhang, D.; Yu, J.; Dong, H.; Zhang, J.; Yang, S. Targeting autophagy in cancer stem cells as an anticancer therapy. Cancer Lett. 2017, 393, 33–39. [Google Scholar] [CrossRef]
- Dadgar, T.; Ebrahimi, N.; Gholipour, A.R.; Akbari, M.; Khani, L.; Ahmadi, A.; Hamblin, M.R. Targeting the metabolism of cancer stem cells by energy disruptor molecules. Crit. Rev. Oncol. 2022, 169, 103545. [Google Scholar] [CrossRef] [PubMed]
- Evangelia, K.; Roza, L.; Olga, T.; Kyriaki-Nefeli, P.; Constantina, S. Autophagy and neurodegenerative disorders. Neural Regen. Res. 2013, 8, 2275–2283. [Google Scholar]
- Yin, Y.; Sun, G.; Li, E.; Kiselyov, K.; Sun, D. ER stress and impaired autophagy flux in neuronal degeneration and brain injury. Ageing Res. Rev. 2017, 34, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Xilouri, M.; Stefanis, L. Autophagy in the central nervous system: Implications for neurodegenerative disorders. CNS Neurol. Disord.-Drug Targets 2010, 9, 701–719. [Google Scholar] [CrossRef]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef]
- Feng, J.; Read, O.J.; Dinkova-Kostova, A.T. Nrf2 in TIME: The Emerging Role of Nuclear Factor Erythroid 2-Related Factor 2 in the Tumor Immune Microenvironment. Mol. Cells 2023, 46, 142–152. [Google Scholar] [CrossRef]
- Ngo, V.; Duennwald, M.L. Nrf2 and Oxidative Stress: A General Overview of Mechanisms and Implications in Human Disease. Antioxidants 2022, 11, 2345. [Google Scholar] [CrossRef]
- Syu, J.-P.; Chi, J.-T.; Kung, H.-N. Nrf2 is the key to chemotherapy resistance in MCF7 breast cancer cells under hypoxia. Oncotarget 2016, 7, 14659–14672. [Google Scholar] [CrossRef]
- Godoy, P.R.D.V.; Pour Khavari, A.; Rizzo, M.; Sakamoto-Hojo, E.T.; Haghdoost, S. Targeting NRF2, Regulator of Antioxidant System, to Sensitize Glioblastoma Neurosphere Cells to Radiation-Induced Oxidative Stress. Oxidative Med. Cell. Longev. 2020, 2020, 2534643. [Google Scholar] [CrossRef]
- Qin, S.; He, X.; Lin, H.; Schulte, B.A.; Zhao, M.; Tew, K.D.; Wang, G.Y. Nrf2 inhibition sensitizes breast cancer stem cells to ionizing radiation via suppressing DNA repair. Free Radic. Biol. Med. 2021, 169, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Esteras, N.; Abramov, A.Y. Nrf2 as a regulator of mitochondrial function: Energy metabolism and beyond. Free Radic. Biol. Med. 2022, 189, 136–153. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Kazantsev, A.G. The role of Nrf2 signaling in counteracting neurodegenerative diseases. FEBS J. 2018, 285, 3576–3590. [Google Scholar] [CrossRef] [PubMed]
- Fragoulis, A.; Siegl, S.; Fendt, M.; Jansen, S.; Soppa, U.; Brandenburg, L.-O.; Pufe, T.; Weis, J.; Wruck, C.J. Oral administration of methysticin improves cognitive deficits in a mouse model of Alzheimer’s disease. Redox Biol. 2017, 12, 843–853. [Google Scholar] [CrossRef]
- Liu, M.; Liu, S.; Lin, Z.; Chen, X.; Jiao, Q.; Du, X.; Jiang, H. Targeting the Interplay Between Autophagy and the Nrf2 Pathway in Parkinson’s Disease with Potential Therapeutic Implications. Biomolecules 2025, 15, 149. [Google Scholar] [CrossRef]
- Han, H.; He, T.; Wu, Y.; He, T.; Zhou, W. Multidimensional analysis of tumor stem cells: From biological properties, metabolic adaptations to immune escape mechanisms. Front. Cell Dev. Biol. 2024, 12, 1441081. [Google Scholar] [CrossRef]
- Farhadi, P.; Yarani, R.; Dokaneheifard, S.; Mansouri, K. The emerging role of targeting cancer metabolism for cancer therapy. Tumor Biol. 2020, 42, 1441081. [Google Scholar] [CrossRef]
- Agrawal, K.; Asthana, S.; Kumar, D. Role of Oxidative Stress in Metabolic Reprogramming of Brain Cancer. Cancers 2023, 15, 4920. [Google Scholar] [CrossRef]
- Miao, J.; Chen, L.; Pan, X.; Li, L.; Zhao, B.; Lan, J. Microglial Metabolic Reprogramming: Emerging Insights and Therapeutic Strategies in Neurodegenerative Diseases. Cell. Mol. Neurobiol. 2023, 43, 3191–3210. [Google Scholar] [CrossRef]
- Hu, Y.; Lu, W.; Chen, G.; Wang, P.; Chen, Z.; Zhou, Y.; Ogasawara, M.; Trachootham, D.; Feng, L.; Pelicano, H.; et al. K-rasG12V transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. 2012, 22, 399–412. [Google Scholar] [CrossRef]
- Lee, Y.H.; Park, J.Y.; Lee, H.; Song, E.S.; Kuk, M.U.; Joo, J.; Oh, S.; Kwon, H.W.; Park, J.T.; Park, S.C. Targeting Mitochondrial Metabolism as a Strategy to Treat Senescence. Cells 2021, 10, 3003. [Google Scholar] [CrossRef] [PubMed]
- Cobb, C.A.; Cole, M.P. Oxidative and nitrative stress in neurodegeneration. Neurobiol. Dis. 2015, 84, 4–21. [Google Scholar] [CrossRef] [PubMed]
- Yeo, H.; A Lyssiotis, C.; Zhang, Y.; Ying, H.; Asara, J.M.; Cantley, L.C.; Paik, J.-H. FoxO3 coordinates metabolic pathways to maintain redox balance in neural stem cells. EMBO J. 2013, 32, 2589–2602. [Google Scholar] [CrossRef]
- Fernández-Checa, J.C.; Fernández, A.; Morales, A.; Marí, M.; García-Ruiz, C.; Colell, A. Oxidative stress and altered mito-chondrial function in neurodegenerative diseases: Lessons from mouse models. CNS Neurol. Disord.-Drug Targets 2010, 9, 439–454. [Google Scholar] [CrossRef]
- Bragina, E.Y.; Gomboeva, D.E.; Saik, O.V.; Ivanisenko, V.A.; Freidin, M.B.; Nazarenko, M.S.; Puzyrev, V.P. Apoptosis Genes as a Key to Identification of Inverse Comorbidity of Huntington’s Disease and Cancer. Int. J. Mol. Sci. 2023, 24, 9385. [Google Scholar] [CrossRef]
- Wilhelmus, M.M.; Nijland, P.G.; Drukarch, B.; de Vries, H.E.; van Horssen, J. Involvement and interplay of Parkin, PINK1, and DJ1 in neurodegenerative and neuroinflammatory disorders. Free Radic. Biol. Med. 2012, 53, 983–992. [Google Scholar] [CrossRef]
- Aliabadi, F.; Sohrabi, B.; Mostafavi, E.; Pazoki-Toroudi, H.; Webster, T.J. Ubiquitin–proteasome system and the role of its inhibitors in cancer therapy. Open Biol. 2021, 11, 200390. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Mattson, M.P. Apolipoprotein E and oxidative stress in brain with relevance to Alzheimer’s disease. Neurobiol. Dis. 2020, 138, 104795. [Google Scholar] [CrossRef]
- Zhang, Q.; Guo, S.; Zhang, X.; Tang, S.; Shao, W.; Han, X.; Wang, L.; Du, Y. Inverse relationship between cancer and Alzheimer’s disease: A systemic review meta-analysis. Neurol. Sci. 2015, 36, 1987–1994. [Google Scholar] [CrossRef]
- Vara, J.Á.F.; Casado, E.; de Castro, J.; Cejas, P.; Belda-Iniesta, C.; González-Barón, M. PI3K/Akt signalling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef]
- Yu, Q.; Zhang, R.; Li, T.; Yang, L.; Zhou, Z.; Hou, L.; Wu, W.; Zhao, R.; Chen, X.; Yao, Y.; et al. Mitochondrial Hydrogen Peroxide Activates PTEN and Inactivates Akt Leading to Autophagy Inhibition-Dependent Cell Death in Neuronal Models of Parkinson’s Disease. Mol. Neurobiol. 2023, 60, 3345–3364. [Google Scholar] [CrossRef] [PubMed]
- De Mónica, A. Oxidoreductase Protein Family Interaction with DJ-1 and Oxidative Stress-Induced Modulation of HADHA in-Teractome. Master’s Thesis, Universidade de Coimbra, Coimbra, Portugal, 2016. Available online: https://search.proquest.com/openview/41113324fb23f955e8e77230cc5403ea/1?pq-origsite=gscholar&cbl=2026366&diss=y (accessed on 15 February 2025).
- Qiu, B.; Wang, J.; Yu, Y.; Zhen, C.; Gu, J.; Liu, W.; Wen, Y.; Chen, L.; Gao, Y.; Xia, Q.; et al. DJ-1 promotes development of DEN-induced hepatocellular carcinoma and proliferation of liver cancer cells. Oncotarget 2016, 8, 8499–8511. [Google Scholar] [CrossRef]
- Satoh, K.; Abe-Dohmae, S.; Yokoyama, S.; George-Hyslop, P.S.; Fraser, P.E. ATP-binding Cassette Transporter A7 (ABCA7) Loss of Function Alters Alzheimer Amyloid Processing. J. Biol. Chem. 2015, 290, 24152–24165. [Google Scholar] [CrossRef]
- Zhao, Q.-F.; Yu, J.-T.; Tan, M.-S.; Tan, L. ABCA7 in Alzheimer’s Disease. Mol. Neurobiol. 2015, 51, 1008–1016. [Google Scholar] [CrossRef]
- Hendig, D.; Langmann, T.; Zarbock, R.; Schmitz, G.; Kleesiek, K.; Götting, C. Characterization of the ATP-binding cassette transporter gene expression profile in Y79: A retinoblastoma cell line. Mol. Cell. Biochem. 2009, 328, 85–92. [Google Scholar] [CrossRef]
- Callari, M.; Sola, M.; Magrin, C.; Rinaldi, A.; Bolis, M.; Paganetti, P.; Colnaghi, L.; Papin, S. Cancer-specific association between Tau (MAPT) and cellular pathways, clinical outcome, and drug response. Sci. Data 2023, 10, 637. [Google Scholar] [CrossRef]
- Milne, G.L. Classifying oxidative stress by F2-Isoprostane levels in human disease: The re-imagining of a biomarker. Redox Biol. 2017, 12, 897–898. [Google Scholar] [CrossRef]
- Milatovic, D.; Aschner, M. Measurement of Isoprostanes as Markers of Oxidative Stress in Neuronal Tissue. Curr. Protoc. Toxicol. 2009, 39, 12–14. [Google Scholar] [CrossRef]
- Roszkowski, K.; Jozwicki, W.; Blaszczyk, P.; Mucha-Malecka, A.; Siomek, A. Oxidative damage DNA: 8-oxoGua and 8-oxodG as molecular markers of cancer. Med. Sci. Monit. 2011, 17, CR329–CR333. [Google Scholar] [CrossRef]
- Nakabeppu, Y. Neurodegeneration caused by accumulation of an oxidized base lesion, 8-oxoguanine, in nuclear and mitochondrial dna: From animal models to human diseases. In The Base Excision Repair Pathway: Molecular Mechanisms and Role in Disease Development and Therapeutic Design; World Scientific: Singapore, 2017; pp. 523–556. [Google Scholar] [CrossRef]
- Zitka, O.; Skalickova, S.; Gumulec, J.; Masarik, M.; Adam, V.; Hubalek, J.; Trnkova, L.; Kruseova, J.; Eckschlager, T.; Kizek, R. Redox status expressed as GSH:GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol. Lett. 2012, 4, 1247–1253. [Google Scholar] [CrossRef]
- Schulz, J.B.; Lindenau, J.; Seyfried, J.; Dichgans, J. Glutathione, oxidative stress and neurodegeneration. Eur. J. Biochem. 2000, 267, 4904–4911. [Google Scholar] [CrossRef] [PubMed]
- Lepara, Z.; Lepara, O.; Fajkić, A.; Rebić, D.; Alić, J.; Spahović, H. Serum malondialdehyde (MDA) level as a potential biomarker of cancer progression for patients with bladder cancer. Rom. J. Intern. Med. 2020, 58, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Vondrakova, D.; Lawson, M.; Valko, M. Metals, oxidative stress and neurodegenerative disorders. Mol. Cell. Biochem. 2010, 345, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, S.; Cao, Y.; Tian, X.; Zeng, R.; Liao, D.-F.; Cao, D. Oxidative Stress and Carbonyl Lesions in Ulcerative Colitis and Associated Colorectal Cancer. Oxidative Med. Cell. Longev. 2015, 2016, 9875298. [Google Scholar] [CrossRef]
- Dasuri, K.; Zhang, L.; Keller, J.N. Oxidative stress, neurodegeneration, and the balance of protein degradation and protein synthesis. Free Radic. Biol. Med. 2013, 62, 170–185. [Google Scholar] [CrossRef]
- Gopčević, K.R.; Rovčanin, B.R.; Tatić, S.B.; Krivokapić, Z.V.; Gajić, M.M.; Dragutinović, V.V. Activity of Superoxide Dismutase, Catalase, Glutathione Peroxidase, and Glutathione Reductase in Different Stages of Colorectal Carcinoma. Dig. Dis. Sci. 2013, 58, 2646–2652. [Google Scholar] [CrossRef]
- Pong, K. Oxidative stress in neurodegenerative diseases: Therapeutic implications for superoxide dismutase mimetics. Expert Opin. Biol. Ther. 2003, 3, 127–139. [Google Scholar] [CrossRef]
- Zhan, X.; Huang, Y.; Qian, S. Protein Tyrosine Nitration in Lung Cancer: Current Research Status and Future Perspectives. Curr. Med. Chem. 2018, 25, 3435–3454. [Google Scholar] [CrossRef]
- Bandookwala, M.; Sengupta, P. 3-Nitrotyrosine: A versatile oxidative stress biomarker for major neurodegenerative diseases. Int. J. Neurosci. 2020, 130, 1047–1062. [Google Scholar] [CrossRef]
- Ratnam, D.V.; Ankola, D.; Bhardwaj, V.; Sahana, D.; Kumar, M.R. Role of antioxidants in prophylaxis and therapy: A pharmaceutical perspective. J. Control. Release 2006, 113, 189–207. [Google Scholar] [CrossRef]
- Babazadeh, A.; Vahed, F.M.; Jafari, S.M. Nanocarrier-mediated brain delivery of bioactives for treatment/prevention of neurodegenerative diseases. J. Control. Release 2020, 321, 211–221. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta-Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]


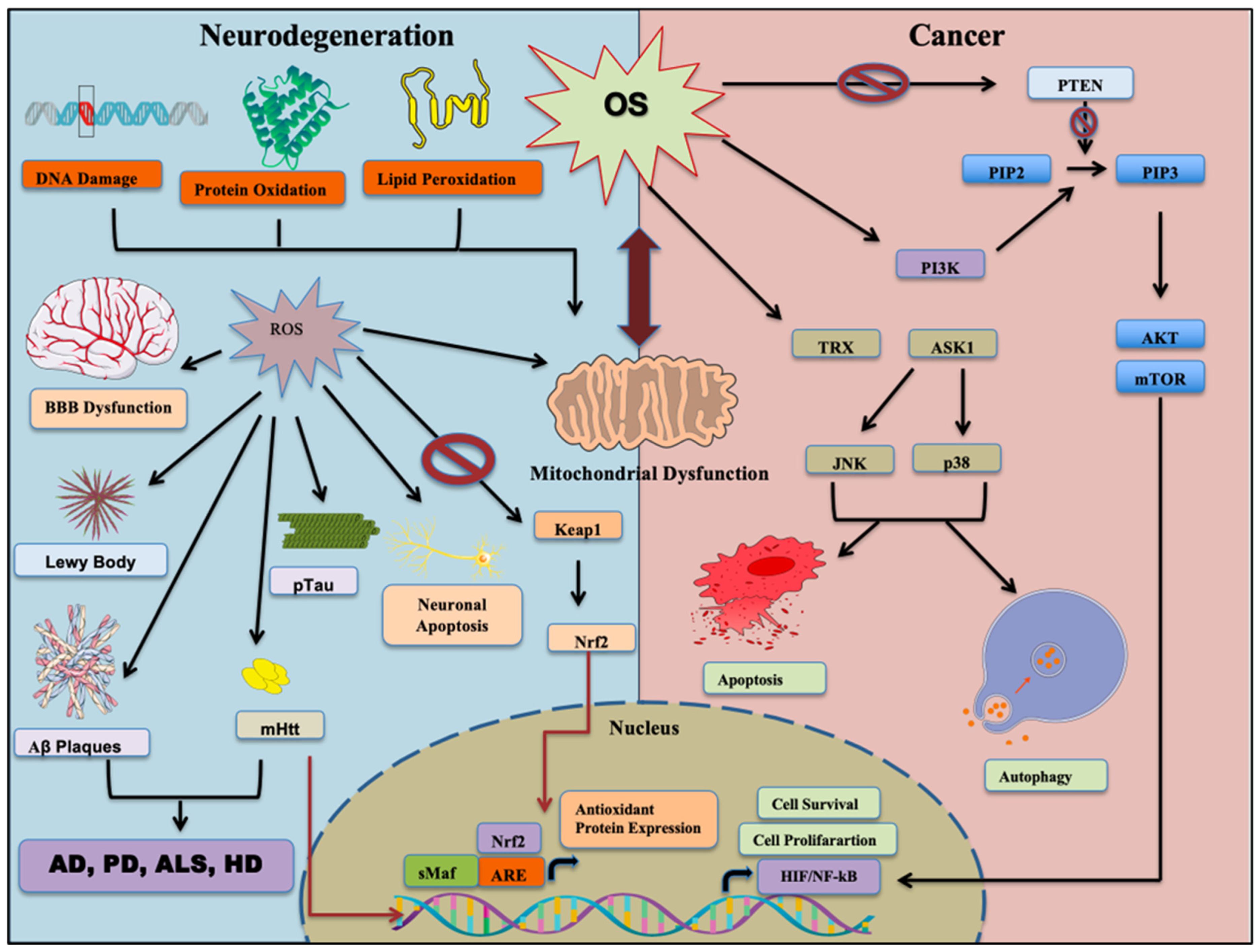
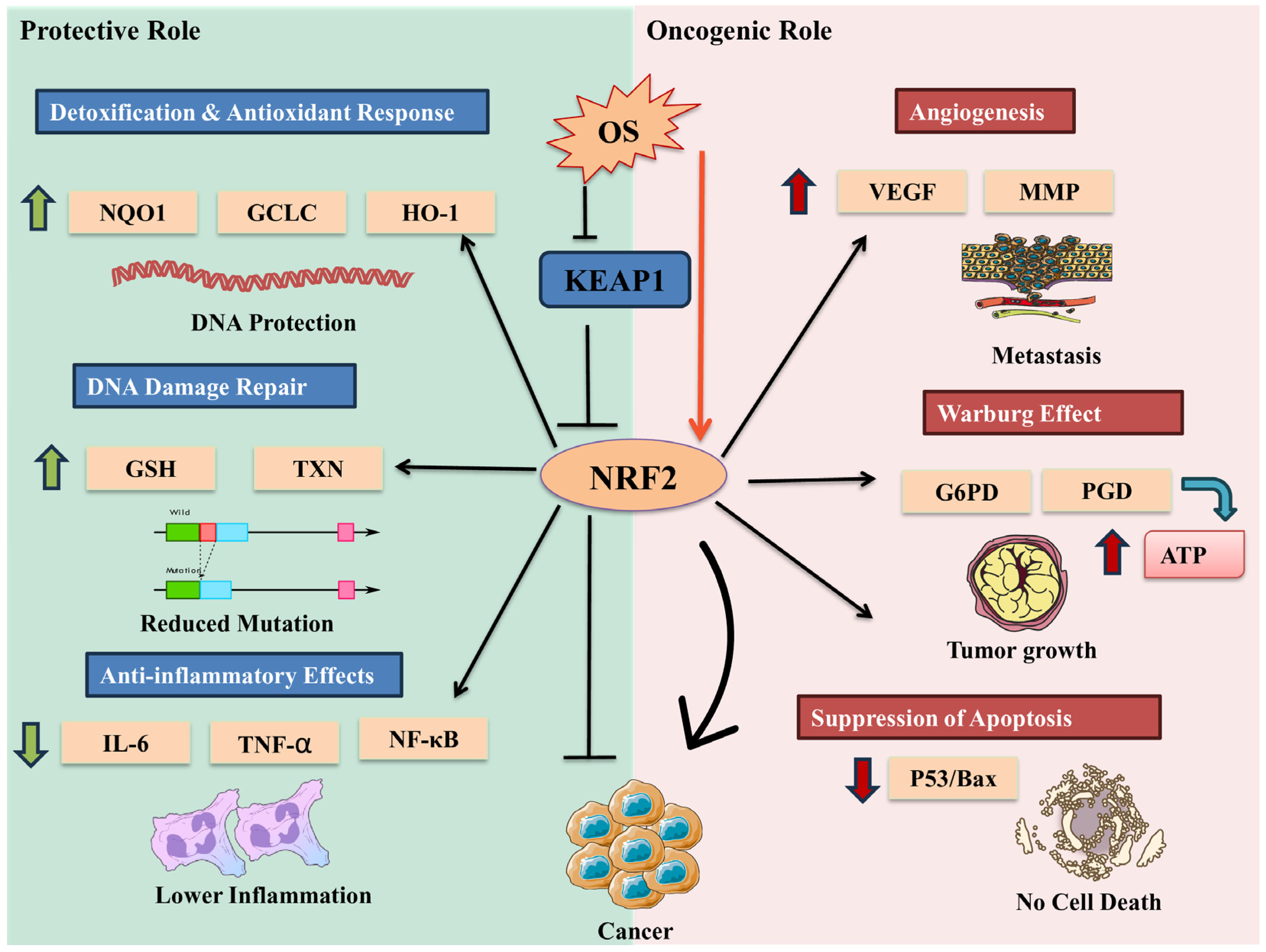
| Features | CSCs | NDDs |
|---|---|---|
| Cellular Behavior | Uncontrolled proliferation and tumorigenesis [184] | Progressive loss of neurons and synapses [185] |
| ROS levels and Antioxidant defenses | Generally maintain lower ROS levels compared to non-CSCs; Enhanced antioxidant systems (e.g., increased SOD2, GSH) [186] | Exhibit elevated ROS levels; Impaired antioxidant responses [187] |
| Mitochondrial function | Metabolic flexibility; can switch between OXPHOS and glycolysis [188] | Early mitochondrial dysfunction [189] |
| Nrf2 pathway | Dual role: promotes survival and chemoresistance and anticancer activity | Attenuation increases oxidative stress [190] |
| Autophagy | Can promote both stemness maintenance and loss [189] | Impairment contributes to protein aggregation [191] |
| Genetic influence | Oncogenes promote cell survival and growth (e.g., PARK7/DJ-1 antagonizing PTEN) [192] | Tumor suppressor-like genes involved in neuroprotection (e.g., PARK2/Parkin, PARK5) [193] |
| Inflammation | Immune evasion and immunosuppressive microenvironment [194] | Chronic neuroinflammation and microglial activation [195] |
| Therapeutic Focus | Targeting proliferative signaling, redox balance to eliminate CSCs and immune evasion (e.g., chemotherapy, immunotherapy) [84] | Enhancing neuronal survival and reducing misfolded proteins (e.g., anti-amyloid, neuroprotective agents), and boosting antioxidant defenses [196]. |
| Metabolic Process | CSC | NDDs | Examples |
|---|---|---|---|
| Glycolysis | |||
| Oxidative Phosphorylation (OXPHOS) |
|
| |
| Pentose Phosphate Pathway (PPP) |
|
| |
| Fatty Acid Oxidation (FAO) |
|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Selvaraj, N.R.; Nandan, D.; Nair, B.G.; Nair, V.A.; Venugopal, P.; Aradhya, R. Oxidative Stress and Redox Imbalance: Common Mechanisms in Cancer Stem Cells and Neurodegenerative Diseases. Cells 2025, 14, 511. https://doi.org/10.3390/cells14070511
Selvaraj NR, Nandan D, Nair BG, Nair VA, Venugopal P, Aradhya R. Oxidative Stress and Redox Imbalance: Common Mechanisms in Cancer Stem Cells and Neurodegenerative Diseases. Cells. 2025; 14(7):511. https://doi.org/10.3390/cells14070511
Chicago/Turabian StyleSelvaraj, Nikhil Raj, Durga Nandan, Bipin G. Nair, Vipin A. Nair, Parvathy Venugopal, and Rajaguru Aradhya. 2025. "Oxidative Stress and Redox Imbalance: Common Mechanisms in Cancer Stem Cells and Neurodegenerative Diseases" Cells 14, no. 7: 511. https://doi.org/10.3390/cells14070511
APA StyleSelvaraj, N. R., Nandan, D., Nair, B. G., Nair, V. A., Venugopal, P., & Aradhya, R. (2025). Oxidative Stress and Redox Imbalance: Common Mechanisms in Cancer Stem Cells and Neurodegenerative Diseases. Cells, 14(7), 511. https://doi.org/10.3390/cells14070511