
Prostate-Specific Membrane Antigen-Targeted Antibody–Drug Conjugates: A Promising Approach for Metastatic Castration-Resistant Prostate Cancer



Abstract
:1. Introduction
2. Prostate-Specific Membrane Antigens
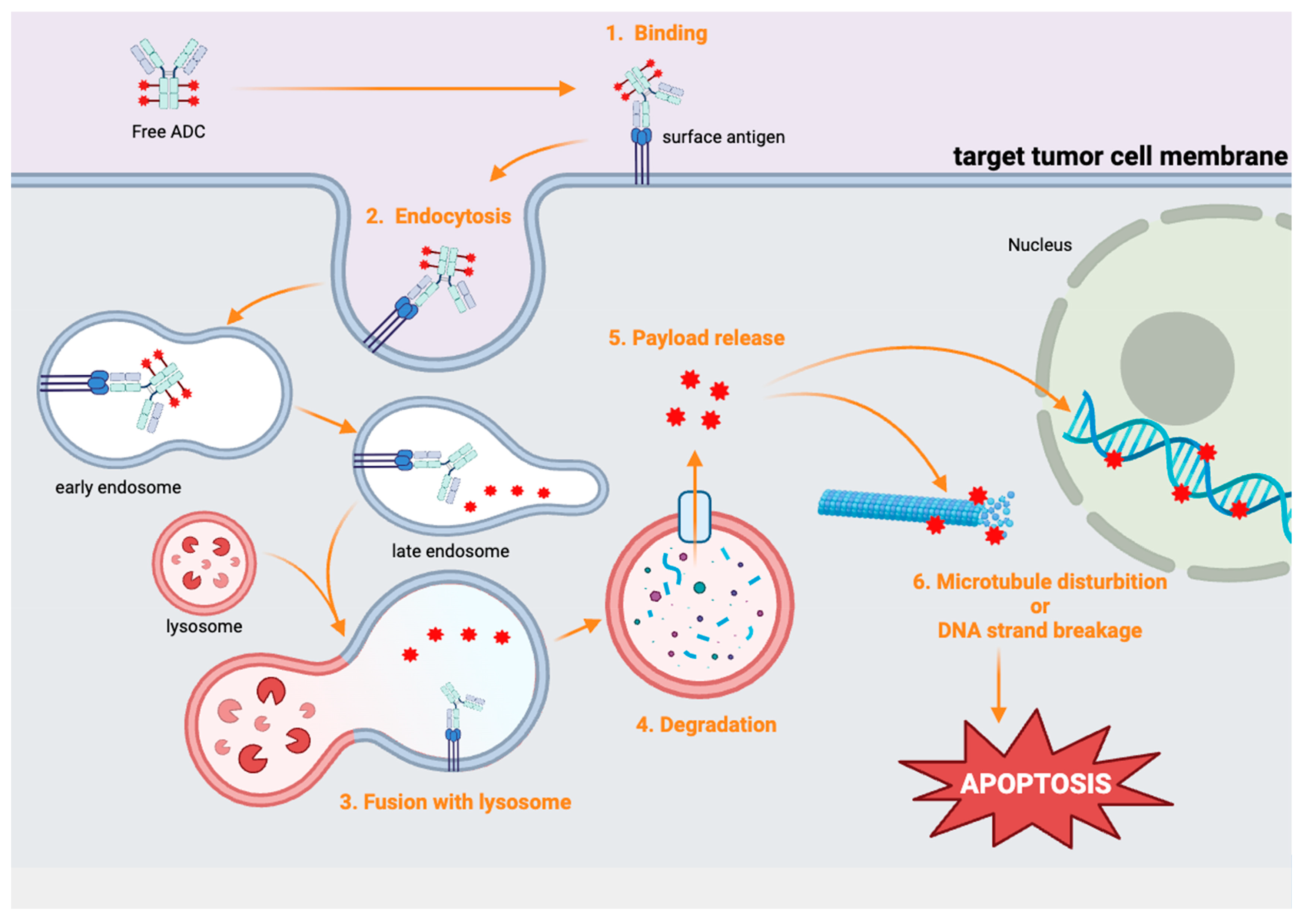
3. Antibody–Drug Conjugates (ADCs)
4. Selective Cytotoxicity of ADCs
5. Anti-PSMA Monoclonal Antibodies
6. 7E11
7. J591
8. 5D3
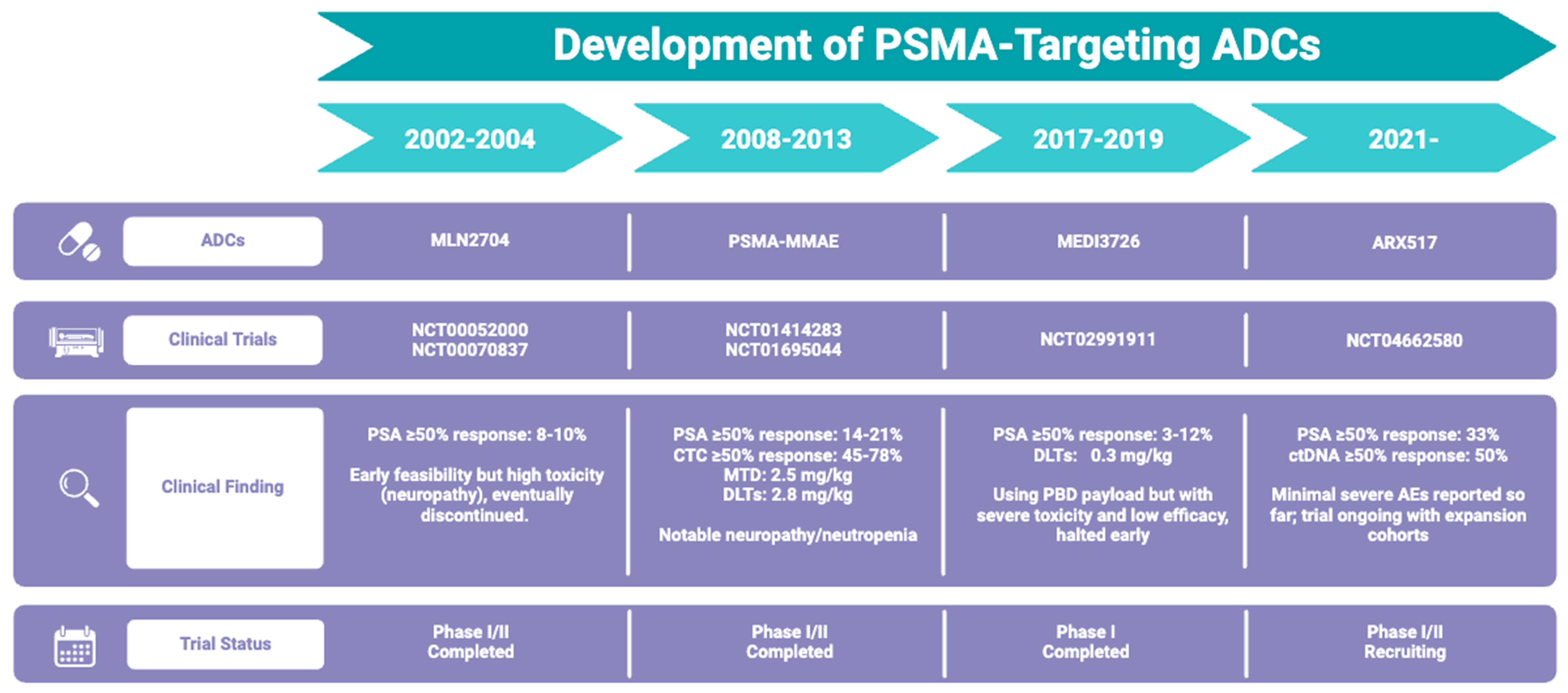
9. ADCs Targeting PSMA in PCa
9.1. PSMA-MMAE
9.1.1. NCT01414283
9.1.2. NCT01695044
9.2. ARX517
NCT04662580
9.3. MLN2704
9.4. MEDI3726
NCT02991911
10. Optimizing PSMA-Targeted ADCs: Balancing Efficacy and Safety
11. Comparison Between PSMA-Targeting ADCs and 177Lu-PSMA Radiotherapy
12. Future Perspectives: Optimizing PSMA-Targeting ADCs for mCRPC
13. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Rebello, R.J.; Oing, C.; Knudsen, K.E.; Loeb, S.; Johnson, D.C.; Reiter, R.E.; Gillessen, S.; Van der Kwast, T.; Bristow, R.G. Prostate cancer. Nat. Rev. Dis. Primers 2021, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Henríquez, I.; Roach, M., 3rd; Morgan, T.M.; Bossi, A.; Gómez, J.A.; Abuchaibe, O.; Couñago, F. Current and Emerging Therapies for Metastatic Castration-Resistant Prostate Cancer (mCRPC). Biomedicines 2021, 9, 1247. [Google Scholar] [CrossRef] [PubMed]
- Francini, E.; Gray, K.P.; Shaw, G.K.; Evan, C.P.; Hamid, A.A.; Perry, C.E.; Kantoff, P.W.; Taplin, M.E.; Sweeney, C.J. Impact of new systemic therapies on overall survival of patients with metastatic castration-resistant prostate cancer in a hospital-based registry. Prostate Cancer Prostatic Dis. 2019, 22, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.S. Overview of prostate-specific membrane antigen. Rev. Urol. 2004, 6 (Suppl. S10), S13–S18. [Google Scholar]
- Perner, S.; Hofer, M.D.; Kim, R.; Shah, R.B.; Li, H.; Möller, P.; Hautmann, R.E.; Gschwend, J.E.; Kuefer, R.; Rubin, M.A. Prostate-specific membrane antigen expression as a predictor of prostate cancer progression. Hum. Pathol. 2007, 38, 696–701. [Google Scholar] [CrossRef]
- Drake, P.M.; Rabuka, D. Recent Developments in ADC Technology: Preclinical Studies Signal Future Clinical Trends. BioDrugs 2017, 31, 521–531. [Google Scholar] [CrossRef]
- Gogia, P.; Ashraf, H.; Bhasin, S.; Xu, Y. Antibody-Drug Conjugates: A Review of Approved Drugs and Their Clinical Level of Evidence. Cancers 2023, 15, 3886. [Google Scholar] [CrossRef]
- Shih, C.H.; Lin, Y.H.; Luo, H.L.; Sung, W.W. Antibody-drug conjugates targeting HER2 for the treatment of urothelial carcinoma: Potential therapies for HER2-positive urothelial carcinoma. Front. Pharmacol. 2024, 15, 1326296. [Google Scholar] [CrossRef]
- Nagayama, A.; Ellisen, L.W.; Chabner, B.; Bardia, A. Antibody-Drug Conjugates for the Treatment of Solid Tumors: Clinical Experience and Latest Developments. Target. Oncol. 2017, 12, 719–739. [Google Scholar] [CrossRef]
- Jain, N.; Smith, S.W.; Ghone, S.; Tomczuk, B. Current ADC Linker Chemistry. Pharm. Res. 2015, 32, 3526–3540. [Google Scholar] [CrossRef] [PubMed]
- Waight, A.B.; Bargsten, K.; Doronina, S.; Steinmetz, M.O.; Sussman, D.; Prota, A.E. Structural Basis of Microtubule Destabilization by Potent Auristatin Anti-Mitotics. PLoS ONE 2016, 11, e0160890. [Google Scholar] [CrossRef]
- Voigt, W.; Matsui, S.; Yin, M.B.; Burhans, W.C.; Minderman, H.; Rustum, Y.M. Topoisomerase-I inhibitor SN-38 can induce DNA damage and chromosomal aberrations independent from DNA synthesis. Anticancer. Res. 1998, 18, 3499–3505. [Google Scholar]
- Leyton, J.V. The endosomal-lysosomal system in ADC design and cancer therapy. Expert. Opin. Biol. Ther. 2023, 23, 1067–1076. [Google Scholar] [CrossRef]
- Giugliano, F.; Corti, C.; Tarantino, P.; Michelini, F.; Curigliano, G. Bystander effect of antibody-drug conjugates: Fact or fiction? Curr. Oncol. Rep. 2022, 24, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Miyahira, A.K.; Pienta, K.J.; Morris, M.J.; Bander, N.H.; Baum, R.P.; Fendler, W.P.; Goeckeler, W.; Gorin, M.A.; Hennekes, H.; Pomper, M.G.; et al. Meeting report from the Prostate Cancer Foundation PSMA-directed radionuclide scientific working group. Prostate 2018, 78, 775–789. [Google Scholar] [CrossRef]
- Wittrup, K.D.; Thurber, G.M.; Schmidt, M.M.; Rhoden, J.J. Practical theoretic guidance for the design of tumor-targeting agents. Methods Enzymol. 2012, 503, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M.; Tannock, I.F. The distribution of the therapeutic monoclonal antibodies cetuximab and trastuzumab within solid tumors. BMC Cancer 2010, 10, 255. [Google Scholar] [CrossRef]
- Troyer, J.K.; Feng, Q.; Beckett, M.L.; Wright, G.L., Jr. Biochemical characterization and mapping of the 7E11-C5.3 epitope of the prostate-specific membrane antigen. Urol. Oncol. 1995, 1, 29–37. [Google Scholar] [CrossRef]
- Barren, R.J., 3rd; Holmes, E.H.; Boynton, A.L.; Misrock, S.L.; Murphy, G.P. Monoclonal antibody 7E11.C5 staining of viable LNCaP cells. Prostate 1997, 30, 65–68. [Google Scholar] [CrossRef]
- Aloysius, H.; Hu, L. Targeted prodrug approaches for hormone refractory prostate cancer. Med. Res. Rev. 2015, 35, 554–585. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, S.T.; Thomas, C.; Sartor, A.O.; Sun, M.; Stangl-Kremser, J.; Bissassar, M.; Vallabhajosula, S.; Huicochea Castellanos, S.; Nauseef, J.T.; Sternberg, C.N.; et al. Prostate-Specific Membrane Antigen-Targeting Alpha Emitter via Antibody Delivery for Metastatic Castration-Resistant Prostate Cancer: A Phase I Dose-Escalation Study of (225)Ac-J591. J. Clin. Oncol. 2024, 42, 842–851. [Google Scholar] [CrossRef]
- Holland, J.P.; Divilov, V.; Bander, N.H.; Smith-Jones, P.M.; Larson, S.M.; Lewis, J.S. 89Zr-DFO-J591 for immunoPET of prostate-specific membrane antigen expression in vivo. J. Nucl. Med. 2010, 51, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Nováková, Z.; Foss, C.A.; Copeland, B.T.; Morath, V.; Baranová, P.; Havlínová, B.; Skerra, A.; Pomper, M.G.; Barinka, C. Novel Monoclonal Antibodies Recognizing Human Prostate-Specific Membrane Antigen (PSMA) as Research and Theranostic Tools. Prostate 2017, 77, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.R.; Kumar, V.; Lisok, A.; Plyku, D.; Nováková, Z.; Brummet, M.; Wharram, B.; Barinka, C.; Hobbs, R.; Pomper, M.G. Evaluation of (111)In-DOTA-5D3, a Surrogate SPECT Imaging Agent for Radioimmunotherapy of Prostate-Specific Membrane Antigen. J. Nucl. Med. 2019, 60, 400–406. [Google Scholar] [CrossRef]
- Petrylak, D.P.; Kantoff, P.; Vogelzang, N.J.; Mega, A.; Fleming, M.T.; Stephenson, J.J., Jr.; Frank, R.; Shore, N.D.; Dreicer, R.; McClay, E.F.; et al. Phase 1 study of PSMA ADC, an antibody-drug conjugate targeting prostate-specific membrane antigen, in chemotherapy-refractory prostate cancer. Prostate 2019, 79, 604–613. [Google Scholar] [CrossRef]
- Petrylak, D.P.; Vogelzang, N.J.; Chatta, K.; Fleming, M.T.; Smith, D.C.; Appleman, L.J.; Hussain, A.; Modiano, M.; Singh, P.; Tagawa, S.T.; et al. PSMA ADC monotherapy in patients with progressive metastatic castration-resistant prostate cancer following abiraterone and/or enzalutamide: Efficacy and safety in open-label single-arm phase 2 study. Prostate 2020, 80, 99–108. [Google Scholar] [CrossRef]
- Skidmore, L.K.; Mills, D.; Kim, J.Y.; Knudsen, N.A.; Nelson, J.D.; Pal, M.; Wang, J.; Gc, K.; Gray, M.J.; Barkho, W.; et al. Preclinical Characterization of ARX517, a Site-Specific Stable PSMA-Targeted Antibody-Drug Conjugate for the Treatment of Metastatic Castration-Resistant Prostate Cancer. Mol. Cancer Ther. 2024, 23, 1842–1853. [Google Scholar] [CrossRef]
- Shen, J.; Pachynski, R.; Nordquist, L.T.; Adra, N.; Bilen, M.A.; Aggarwal, R.; Reichert, Z.; Schweizer, M.; Iravani, A.; Aung, S.; et al. 1804P APEX-01: First-in-human phase I/II study of ARX517 an anti- prostate-specific membrane antigen (PSMA) antibody-drug conjugate (ADC) in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC). Ann. Oncol. 2023, 34, S974–S975. [Google Scholar] [CrossRef]
- Doehn, C.; Jocham, D. Technology evaluation: MLN-591, Cornell University/BZL Biologics/ImmunoGen/Millennium. Curr. Opin. Mol. Ther. 2002, 4, 606–613. [Google Scholar]
- Milowsky, M.I.; Galsky, M.D.; Morris, M.J.; Crona, D.J.; George, D.J.; Dreicer, R.; Tse, K.; Petruck, J.; Webb, I.J.; Bander, N.H.; et al. Phase 1/2 multiple ascending dose trial of the prostate-specific membrane antigen-targeted antibody drug conjugate MLN2704 in metastatic castration-resistant prostate cancer. Urol. Oncol. 2016, 34, 530.e515–530.e521. [Google Scholar] [CrossRef]
- Cho, S.; Zammarchi, F.; Williams, D.G.; Havenith, C.E.G.; Monks, N.R.; Tyrer, P.; D’Hooge, F.; Fleming, R.; Vashisht, K.; Dimasi, N.; et al. Antitumor Activity of MEDI3726 (ADCT-401), a Pyrrolobenzodiazepine Antibody-Drug Conjugate Targeting PSMA, in Preclinical Models of Prostate Cancer. Mol. Cancer Ther. 2018, 17, 2176–2186. [Google Scholar] [CrossRef]
- Adair, J.R.; Howard, P.W.; Hartley, J.A.; Williams, D.G.; Chester, K.A. Antibody-drug conjugates—A perfect synergy. Expert Opin. Biol. Ther. 2012, 12, 1191–1206. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.S.; Fleming, M.T.; Wang, J.S.; Cathomas, R.; Miralles, M.S.; Bothos, J.; Hinrichs, M.J.; Zhang, Q.; He, P.; Williams, M.; et al. Phase I Study of MEDI3726: A Prostate-Specific Membrane Antigen-Targeted Antibody-Drug Conjugate, in Patients with mCRPC after Failure of Abiraterone or Enzalutamide. Clin. Cancer Res. 2021, 27, 3602–3609. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.S.; Scher, H.I.; Montgomery, R.B.; Parker, C.; Miller, M.C.; Tissing, H.; Doyle, G.V.; Terstappen, L.W.; Pienta, K.J.; Raghavan, D. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin. Cancer Res. 2008, 14, 6302–6309. [Google Scholar] [CrossRef]
- Zhang, D.; Yu, S.F.; Khojasteh, S.C.; Ma, Y.; Pillow, T.H.; Sadowsky, J.D.; Su, D.; Kozak, K.R.; Xu, K.; Polson, A.G.; et al. Intratumoral Payload Concentration Correlates with the Activity of Antibody-Drug Conjugates. Mol. Cancer Ther. 2018, 17, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Zhang, D. Linker Design Impacts Antibody-Drug Conjugate Pharmacokinetics and Efficacy via Modulating the Stability and Payload Release Efficiency. Front. Pharmacol. 2021, 12, 687926. [Google Scholar] [CrossRef]
- Johann, F.; Wöll, S.; Gieseler, H. “Negative” Impact: The Role of Payload Charge in the Physicochemical Stability of Auristatin Antibody–Drug Conjugates. J. Pharm. Sci. 2024, 113, 2433–2442. [Google Scholar] [CrossRef]
- Wang, Z.; Li, H.; Gou, L.; Li, W.; Wang, Y. Antibody-drug conjugates: Recent advances in payloads. Acta Pharm. Sin. B 2023, 13, 4025–4059. [Google Scholar] [CrossRef]
- de Bono, J.S.; Logothetis, C.J.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B., Jr.; Saad, F.; et al. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 2011, 364, 1995–2005. [Google Scholar] [CrossRef]
- Berthold, D.R.; Pond, G.R.; Soban, F.; de Wit, R.; Eisenberger, M.; Tannock, I.F. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer: Updated survival in the TAX 327 study. J. Clin. Oncol. 2008, 26, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Sartor, O.; de Bono, J.; Chi, K.N.; Fizazi, K.; Herrmann, K.; Rahbar, K.; Tagawa, S.T.; Nordquist, L.T.; Vaishampayan, N.; El-Haddad, G.; et al. Lutetium-177-PSMA-617 for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2021, 385, 1091–1103. [Google Scholar] [CrossRef]
- Alati, S.; Singh, R.; Pomper, M.G.; Rowe, S.P.; Banerjee, S.R. Preclinical Development in Radiopharmaceutical Therapy for Prostate Cancer. Semin. Nucl. Med. 2023, 53, 663–686. [Google Scholar] [CrossRef]
- Hennrich, U.; Eder, M. [177Lu]Lu-PSMA-617 (PluvictoTM): The First FDA-Approved Radiotherapeutical for Treatment of Prostate Cancer. Pharmaceuticals 2022, 15, 1292. [Google Scholar] [CrossRef]
- Hofman, M.S.; Violet, J.; Hicks, R.J.; Ferdinandus, J.; Thang, S.P.; Akhurst, T.; Iravani, A.; Kong, G.; Ravi Kumar, A.; Murphy, D.G.; et al. [177Lu]-PSMA-617 radionuclide treatment in patients with metastatic castration-resistant prostate cancer (LuPSMA trial): A single-centre, single-arm, phase 2 study. Lancet Oncol. 2018, 19, 825–833. [Google Scholar] [CrossRef]
- Heynickx, N.; Herrmann, K.; Vermeulen, K.; Baatout, S.; Aerts, A. The salivary glands as a dose limiting organ of PSMA- targeted radionuclide therapy: A review of the lessons learnt so far. Nucl. Med. Biol. 2021, 98–99, 30–39. [Google Scholar] [CrossRef]
- Chi, K.N.; Yip, S.M.; Bauman, G.; Probst, S.; Emmenegger, U.; Kollmannsberger, C.K.; Martineau, P.; Niazi, T.; Pouliot, F.; Rendon, R.; et al. 177Lu-PSMA-617 in Metastatic Castration-Resistant Prostate Cancer: A Review of the Evidence and Implications for Canadian Clinical Practice. Curr. Oncol. 2024, 31, 1400–1415. [Google Scholar] [CrossRef]
- Nambiar, D.K.; Mishra, D.; Singh, R.P. Targeting DNA repair for cancer treatment: Lessons from PARP inhibitor trials. Oncol. Res. 2023, 31, 405–421. [Google Scholar] [CrossRef] [PubMed]
- Salifu, I.; Singh, N.; Berraondo, M.; Remon, J.; Salifu, S.; Severson, E.; Quintana, A.; Peiró, S.; Ramkissoon, S.; Vidal, L.; et al. Antibody-drug conjugates, immune-checkpoint inhibitors, and their combination in advanced non-small cell lung cancer. Cancer Treat. Res. Commun. 2023, 36, 100713. [Google Scholar] [CrossRef]
- Dale, D.C. Advances in the treatment of neutropenia. Curr. Opin. Support. Palliat. Care 2009, 3, 207–212. [Google Scholar] [CrossRef]
- McCombs, J.R.; Owen, S.C. Antibody drug conjugates: Design and selection of linker, payload and conjugation chemistry. AAPS J. 2015, 17, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, D.A.; Rüschoff, J.H.; Muehlematter, U.J.; Kranzbühler, B.; Müller, J.; Messerli, M.; Husmann, L.; Hermanns, T.; Eberli, D.; Rupp, N.J.; et al. Immunohistochemical PSMA expression patterns of primary prostate cancer tissue are associated with the detection rate of biochemical recurrence with (68)Ga-PSMA-11-PET. Theranostics 2020, 10, 6082–6094. [Google Scholar] [CrossRef] [PubMed]
- Sommer, U.; Siciliano, T.; Ebersbach, C.; Beier, A.K.; Stope, M.B.; Jöhrens, K.; Baretton, G.B.; Borkowetz, A.; Thomas, C.; Erb, H.H.H. Impact of Androgen Receptor Activity on Prostate-Specific Membrane Antigen Expression in Prostate Cancer Cells. Int. J. Mol. Sci. 2022, 23, 1046. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| ADC Name | Antibody | Linker Type | Drug Payload | Clinical Trial | Stage | Years | Inclusion Criteria | Enrollment | Status |
|---|---|---|---|---|---|---|---|---|---|
| PSMA-MMAE | Fully human anti-PSMA mAb | Valine–citrulline linker | MMAE | NCT01414283 | Phase 1 | 2008–2013 | Patients with mCRPC who have received prior chemotherapy and/or androgen receptor inhibitors; measurable disease required | 52 | Completed |
| NCT01695044 | Phase 2 | 2012–2015 | 119 | Completed | |||||
| ARX517 | Humanized anti-PSMA IgG1κ | Non-cleavable polyethylene glycol linker | AS269 | NCT04662580 | Phase 1/2 | 2021 till now | Patients with mCRPC who have received ≥ 2 prior lines of therapy, including AR inhibitors and chemotherapy | 352 (estimated) | Recruiting |
| MLN2704 | Humanized MLN591 | Disulfide linker | Maytansinoid-1 | NCT00052000 | Phase 1 | 2002–2004 | Patients with mCRPC with failed hormonal therapy | 29 | Completed |
| NCT00070837 | Phase 1/2 | 2003–2004 | 46 | Completed | |||||
| MEDI3726 | Humanized J591 | Cleavable linker | PBD dimer | NCT02991911 | Phase 1 | 2017–2019 | Patients with mCRPC after prior receipt of abiraterone and/or enzalutamide and taxane-based chemotherapy | 33 | Completed |
| Therapy | Mechanism | Reported Clinical Efficacy | Common Toxicities | Clinical Role |
|---|---|---|---|---|
| AR inhibitors (e.g., abiraterone and enzalutamide) | Inhibit androgen production or AR pathway signaling | ~4–5 mo OS benefit; high PSA response in early mCRPC [40] | Hypertension, fatigue, and liver enzyme elevations | First line in advanced PC; most of patients develop AR resistance |
| Chemotherapy (docetaxel and cabazitaxel) | Systemic cytotoxicity via microtubule disruption | ~2–3 mo OS benefit vs. control; ~30–45% PSA decline [41] | Neutropenia, neuropathy, and alopecia | Standard second line after AR inhibitors; broad cytotoxic approach |
| 177Lu-PSMA radioligand therapy | Beta-emitting isotope targeting PSMA-positive tumor cells | OS prolongation (phase III VISION trial: ~4 mo); 50–80% PSA decline [42] | Fatigue, dry mouth, and nausea | Approved for post-AR and post-chemo mCRPC; requires sufficient PSMA expression |
| PSMA-targeted ADC | Antibody-directed delivery of potent cytotoxic agents | PSA response rates ~15–50% in early trials | Myelosuppression, dryness, and fatigue | Potential salvage or combination strategy; promising new approach under study |
| PSMA ADC | Response Rate | Adverse Effects | Toxicity Profile |
|---|---|---|---|
| PSMA-MMAE | PSA ≥ 50% response: 14–21% CTC ≥ 50% response: 45–78% | Neutropenia (36%) Fatigue (34%) Decreased electrolytes (29%) Anemia (25%) | MTD: 2.5 mg/kg DLTs: 2.8 mg/kg |
| ARX517 | PSA ≥ 50% response: 33% ctDNA ≥ 50% response: 50% | Dry mouth (41.7%) Fatigue (33.3%) Diarrhea (20.8%) | N/A |
| MLN2704 | PSA ≥ 50% response: 8–10% | Peripheral neuropathy (71%) Nausea (61%) Fatigue (60%) | N/A |
| MEDI3726 | PSA ≥ 50% response: 3–12% | TRAE (90.9%) Grade 3/4 AEs (45.5%) Elevations in liver enzymes (30.3–36.4%) Fatigue (30.3%) | DLTs: 0.3 mg/kg |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shih, C.-H.; Hsieh, T.-Y.; Sung, W.-W. Prostate-Specific Membrane Antigen-Targeted Antibody–Drug Conjugates: A Promising Approach for Metastatic Castration-Resistant Prostate Cancer. Cells 2025, 14, 513. https://doi.org/10.3390/cells14070513
Shih C-H, Hsieh T-Y, Sung W-W. Prostate-Specific Membrane Antigen-Targeted Antibody–Drug Conjugates: A Promising Approach for Metastatic Castration-Resistant Prostate Cancer. Cells. 2025; 14(7):513. https://doi.org/10.3390/cells14070513
Chicago/Turabian StyleShih, Chia-Hsien, Tzuo-Yi Hsieh, and Wen-Wei Sung. 2025. "Prostate-Specific Membrane Antigen-Targeted Antibody–Drug Conjugates: A Promising Approach for Metastatic Castration-Resistant Prostate Cancer" Cells 14, no. 7: 513. https://doi.org/10.3390/cells14070513
APA StyleShih, C.-H., Hsieh, T.-Y., & Sung, W.-W. (2025). Prostate-Specific Membrane Antigen-Targeted Antibody–Drug Conjugates: A Promising Approach for Metastatic Castration-Resistant Prostate Cancer. Cells, 14(7), 513. https://doi.org/10.3390/cells14070513