
Stem Cells in Cancer: From Mechanisms to Therapeutic Strategies


Abstract
:1. Introduction
2. Cancer Stem Cells (CSCs)
2.1. Origin of CSCs
2.1.1. Mutation in Normal Stem Cells
2.1.2. Dedifferentiation of Progenitor and Somatic Cells
2.1.3. Epithelial–Mesenchymal Transition (EMT)
2.2. Identification and Isolation of CSCs
2.3. Signaling Pathways in CSCs
2.3.1. The Wnt/β-Catenin Signaling Pathway
2.3.2. The Hedgehog Signaling Pathway
2.3.3. The Notch Signaling Pathway
2.3.4. The PI3K/AKT/mTOR Signaling Pathway
2.4. Cancer Stem Cell Niche
2.4.1. Hypoxia and Acidity
2.4.2. Immune Evasion
2.4.3. Extracellular Matrix Interactions
2.5. Role of CSCs in Tumorigenesis
2.5.1. Self-Renewal and Differentiation
2.5.2. Tumor Heterogeneity
2.6. Role of CSCs in Metastasis
2.6.1. Epithelial–Mesenchymal Transition (EMT)
2.6.2. Circulating Tumor Cells (CTCs)
2.7. Therapeutic Strategies Targeting CSCs
2.7.1. Targeting CSC-Specific Surface Markers
2.7.2. Inhibiting CSC Signaling Pathways
- (1)
- Targeting the Wnt/β-Catenin Signaling Pathways
- (2)
- Targeting the Hedgehog Signaling Pathways
- (3)
- Targeting the Notch Signaling Pathways
- (4)
- Targeting the PI3K Signaling Pathways
2.7.3. Inducing CSC Differentiation
2.7.4. Strategies to Overcome CSC Immune Evasion
2.7.5. Strategies to Overcome ABC Transporter-Mediated Resistance in CSCs
2.7.6. Targeting DNA Repair in CSCs for Therapy
2.7.7. Targeting Quiescent CSCs for Therapy
3. Normal Stem Cells in Cancer Therapy
3.1. Tissue Regeneration
3.2. Delivery Vehicles for Anticancer Agents
3.3. Stem Cells in Immunotherapy
4. Challenges and Future Directions
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACC | Adenoid cystic carcinoma |
| ALDH | Aldehyde dehydrogenase |
| AML | Acute myeloid leukemia |
| aSMA | actin smooth muscle |
| BER | Base excision repair |
| CAFs | Cancer-associated fibroblasts |
| CAR-T | Chimeric antigen receptor T cells |
| CICs | Cancer-initiating cells |
| CRCSCs | Colorectal cancer stem cells |
| CSCs | Cancer stem cells |
| CTCs | Circulating tumor cells |
| DSBs | Double-strand breaks |
| ECM | Extracellular matrix |
| EMT | Epithelial–mesenchymal transition |
| FAK | Focal adhesion kinase |
| FAP | Fibroblast activation protein |
| FSP1 | Fibroblast specific protein 1 |
| GSCs | Glioma stem cells |
| hESC | Human embryonic stem cell |
| HR | Homologous recombination |
| HSPCs | Hematopoietic stem and progenitor cells |
| iPSCs | Induced pluripotent stem cells |
| LSCs | Leukemia stem cells |
| MACS | Magnetic-activated cell sorting |
| MDSCs | Myeloid-derived suppressor cells |
| MMR | Mismatch repair |
| MSCs | Mesenchymal stem cells |
| NHEJ | Non-homologous end joining |
| NK | Natural Killer |
| NSCLC | Non-small cell lung cancer |
| NSCs | Neural stem cells |
| PDAC | Pancreatic ductal adenocarcinoma |
| RNAi | RNA interference |
| TAMs | Tumor-associated macrophages |
| TME | Tumor microenvironment |
| TNBC | Triple-negative breast cancer |
| VM | Vasculogenic mimicry |
References
- Quintana, E.; Shackleton, M.; Sabel, M.S.; Fullen, D.R.; Johnson, T.M.; Morrison, S.J. Efficient tumour formation by single human melanoma cells. Nature 2008, 456, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P.N.; Dakic, A.; Adams, J.M.; Nutt, S.L.; Strasser, A. Tumor growth need not be driven by rare cancer stem cells. Science 2007, 317, 337. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef] [PubMed]
- Hope, K.J.; Jin, L.; Dick, J.E. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat. Immunol. 2004, 5, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Loh, J.J.; Ma, S. Hallmarks of cancer stemness. Cell Stem Cell 2024, 31, 617–639. [Google Scholar] [CrossRef] [PubMed]
- Pardal, R.; Clarke, M.F.; Morrison, S.J. Applying the principles of stem-cell biology to cancer. Nat. Rev. Cancer 2003, 3, 895–902. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Hancock, J.F. Ras proteins: Different signals from different locations. Nat. Rev. Mol. Cell Biol. 2003, 4, 373–384. [Google Scholar] [CrossRef]
- Voice, J.K.; Klemke, R.L.; Le, A.; Jackson, J.H. Four human ras homologs differ in their abilities to activate Raf-1, induce transformation, and stimulate cell motility. J. Biol. Chem. 1999, 274, 17164–17170. [Google Scholar] [CrossRef] [PubMed]
- Sinn, E.; Muller, W.; Pattengale, P.; Tepler, I.; Wallace, R.; Leder, P. Coexpression of MMTV/v-Ha-ras and MMTV/c-myc genes in transgenic mice: Synergistic action of oncogenes in vivo. Cell 1987, 49, 465–475. [Google Scholar] [CrossRef]
- Li, S.; Balmain, A.; Counter, C.M. A model for RAS mutation patterns in cancers: Finding the sweet spot. Nat. Rev. Cancer 2018, 18, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Ligresti, G.; Militello, L.; Steelman, L.S.; Cavallaro, A.; Basile, F.; Nicoletti, F.; Stivala, F.; McCubrey, J.A.; Libra, M. PIK3CA mutations in human solid tumors: Role in sensitivity to various therapeutic approaches. Cell Cycle 2009, 8, 1352–1358. [Google Scholar] [CrossRef] [PubMed]
- Vasan, N.; Razavi, P.; Johnson, J.L.; Shao, H.; Shah, H.; Antoine, A.; Ladewig, E.; Gorelick, A.; Lin, T.Y.; Toska, E.; et al. Double PIK3CA mutations in cis increase oncogenicity and sensitivity to PI3Kα inhibitors. Science 2019, 366, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Van Keymeulen, A.; Lee, M.Y.; Ousset, M.; Brohee, S.; Rorive, S.; Giraddi, R.R.; Wuidart, A.; Bouvencourt, G.; Dubois, C.; Salmon, I.; et al. Reactivation of multipotency by oncogenic PIK3CA induces breast tumour heterogeneity. Nature 2015, 525, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Madsen, R.R.; Knox, R.G.; Pearce, W.; Lopez, S.; Mahler-Araujo, B.; McGranahan, N.; Vanhaesebroeck, B.; Semple, R.K. Oncogenic PIK3CA promotes cellular stemness in an allele dose-dependent manner. Proc. Natl. Acad. Sci. USA 2019, 116, 8380–8389. [Google Scholar] [CrossRef]
- Little, C.D.; Nau, M.M.; Carney, D.N.; Gazdar, A.F.; Minna, J.D. Amplification and expression of the c-myc oncogene in human lung cancer cell lines. Nature 1983, 306, 194–196. [Google Scholar] [CrossRef]
- Lee, K.M.; Giltnane, J.M.; Balko, J.M.; Schwarz, L.J.; Guerrero-Zotano, A.L.; Hutchinson, K.E.; Nixon, M.J.; Estrada, M.V.; Sanchez, V.; Sanders, M.E.; et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab. 2017, 26, 633–647.E7. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, T.; Su, W.; Dou, Z.; Zhao, D.; Jin, X.; Lei, H.; Wang, J.; Xie, X.; Cheng, B.; et al. Mutant p53 in cancer: From molecular mechanism to therapeutic modulation. Cell Death Dis. 2022, 13, 974. [Google Scholar] [CrossRef]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Su, Z.; Tavana, O.; Gu, W. Understanding the complexity of p53 in a new era of tumor suppression. Cancer Cell 2024, 42, 946–967. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef]
- Banin, S.; Moyal, L.; Shieh, S.; Taya, Y.; Anderson, C.W.; Chessa, L.; Smorodinsky, N.I.; Prives, C.; Reiss, Y.; Shiloh, Y.; et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 1998, 281, 1674–1677. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef] [PubMed]
- Baslan, T.; Morris, J.P.T.; Zhao, Z.; Reyes, J.; Ho, Y.J.; Tsanov, K.M.; Bermeo, J.; Tian, S.; Zhang, S.; Askan, G.; et al. Ordered and deterministic cancer genome evolution after p53 loss. Nature 2022, 608, 795–802. [Google Scholar] [CrossRef]
- Sun, X.; Klingbeil, O.; Lu, B.; Wu, C.; Ballon, C.; Ouyang, M.; Wu, X.S.; Jin, Y.; Hwangbo, Y.; Huang, Y.H.; et al. BRD8 maintains glioblastoma by epigenetic reprogramming of the p53 network. Nature 2023, 613, 195–202. [Google Scholar] [CrossRef]
- Burkhart, D.L.; Sage, J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat. Rev. Cancer 2008, 8, 671–682. [Google Scholar] [CrossRef]
- Huang, H.J.; Yee, J.K.; Shew, J.Y.; Chen, P.L.; Bookstein, R.; Friedmann, T.; Lee, E.Y.; Lee, W.H. Suppression of the neoplastic phenotype by replacement of the RB gene in human cancer cells. Science 1988, 242, 1563–1566. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Kitajima, S.; Kohno, S.; Yoshida, A.; Tange, S.; Sasaki, S.; Okada, N.; Nishimoto, Y.; Muranaka, H.; Nagatani, N.; et al. Retinoblastoma Inactivation Induces a Protumoral Microenvironment via Enhanced CCL2 Secretion. Cancer Res. 2019, 79, 3903–3915. [Google Scholar] [CrossRef]
- Calo, E.; Quintero-Estades, J.A.; Danielian, P.S.; Nedelcu, S.; Berman, S.D.; Lees, J.A. Rb regulates fate choice and lineage commitment in vivo. Nature 2010, 466, 1110–1114. [Google Scholar] [CrossRef]
- Vidotto, T.; Melo, C.M.; Lautert-Dutra, W.; Chaves, L.P.; Reis, R.B.; Squire, J.A. Pan-cancer genomic analysis shows hemizygous PTEN loss tumors are associated with immune evasion and poor outcome. Sci. Rep. 2023, 13, 5049. [Google Scholar] [CrossRef]
- Lee, Y.R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Gao, J.; Lei, Q.; Rozengurt, N.; Pritchard, C.; Jiao, J.; Thomas, G.V.; Li, G.; Roy-Burman, P.; Nelson, P.S.; et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 2003, 4, 209–221. [Google Scholar] [CrossRef]
- Zhang, Y.; Kwok-Shing Ng, P.; Kucherlapati, M.; Chen, F.; Liu, Y.; Tsang, Y.H.; de Velasco, G.; Jeong, K.J.; Akbani, R.; Hadjipanayis, A.; et al. A Pan-Cancer Proteogenomic Atlas of PI3K/AKT/mTOR Pathway Alterations. Cancer Cell 2017, 31, 820–832.E3. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA methylation landscape of cancer. Trends Genet. 2021, 37, 1012–1027. [Google Scholar] [CrossRef]
- Herman, J.G.; Merlo, A.; Mao, L.; Lapidus, R.G.; Issa, J.P.; Davidson, N.E.; Sidransky, D.; Baylin, S.B. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res. 1995, 55, 4525–4530. [Google Scholar] [PubMed]
- Hansen, K.D.; Timp, W.; Bravo, H.C.; Sabunciyan, S.; Langmead, B.; McDonald, O.G.; Wen, B.; Wu, H.; Liu, Y.; Diep, D.; et al. Increased methylation variation in epigenetic domains across cancer types. Nat. Genet. 2011, 43, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Spencer, D.H.; Russler-Germain, D.A.; Ketkar, S.; Helton, N.M.; Lamprecht, T.L.; Fulton, R.S.; Fronick, C.C.; O’Laughlin, M.; Heath, S.E.; Shinawi, M.; et al. CpG Island Hypermethylation Mediated by DNMT3A Is a Consequence of AML Progression. Cell 2017, 168, 801–816.e13. [Google Scholar] [CrossRef] [PubMed]
- Marsolier, J.; Prompsy, P.; Durand, A.; Lyne, A.M.; Landragin, C.; Trouchet, A.; Bento, S.T.; Eisele, A.; Foulon, S.; Baudre, L.; et al. H3K27me3 conditions chemotolerance in triple-negative breast cancer. Nat. Genet. 2022, 54, 459–468. [Google Scholar] [CrossRef]
- Gollner, S.; Oellerich, T.; Agrawal-Singh, S.; Schenk, T.; Klein, H.U.; Rohde, C.; Pabst, C.; Sauer, T.; Lerdrup, M.; Tavor, S.; et al. Loss of the histone methyltransferase EZH2 induces resistance to multiple drugs in acute myeloid leukemia. Nat. Med. 2017, 23, 69–78. [Google Scholar] [CrossRef]
- Yamagishi, M.; Kuze, Y.; Kobayashi, S.; Nakashima, M.; Morishima, S.; Kawamata, T.; Makiyama, J.; Suzuki, K.; Seki, M.; Abe, K.; et al. Mechanisms of action and resistance in histone methylation-targeted therapy. Nature 2024, 627, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, A.; Papazoglu, C.; Wu, Y.; Capurso, D.; Brodt, M.; Francis, D.; Bredel, M.; Vogel, H.; Mills, A.A. CHD5 is a tumor suppressor at human 1p36. Cell 2007, 128, 459–475. [Google Scholar] [CrossRef] [PubMed]
- Mashtalir, N.; D’Avino, A.R.; Michel, B.C.; Luo, J.; Pan, J.; Otto, J.E.; Zullow, H.J.; McKenzie, Z.M.; Kubiak, R.L.; St Pierre, R.; et al. Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell 2018, 175, 1272–1288.e20. [Google Scholar] [CrossRef]
- Wong, A.K.; Shanahan, F.; Chen, Y.; Lian, L.; Ha, P.; Hendricks, K.; Ghaffari, S.; Iliev, D.; Penn, B.; Woodland, A.M.; et al. BRG1, a component of the SWI-SNF complex, is mutated in multiple human tumor cell lines. Cancer Res. 2000, 60, 6171–6177. [Google Scholar] [PubMed]
- Li, J.; Wang, W.; Zhang, Y.; Cieslik, M.; Guo, J.; Tan, M.; Green, M.D.; Wang, W.; Lin, H.; Li, W.; et al. Epigenetic driver mutations in ARID1A shape cancer immune phenotype and immunotherapy. J. Clin. Investig. 2020, 130, 2712–2726. [Google Scholar] [CrossRef] [PubMed]
- Cucchi, D.G.J.; Denys, B.; Kaspers, G.J.L.; Janssen, J.; Ossenkoppele, G.J.; de Haas, V.; Zwaan, C.M.; van den Heuvel-Eibrink, M.M.; Philippe, J.; Csikos, T.; et al. RNA-based FLT3-ITD allelic ratio is associated with outcome and ex vivo response to FLT3 inhibitors in pediatric AML. Blood 2018, 131, 2485–2489. [Google Scholar] [CrossRef]
- Yu, B.D.; Hess, J.L.; Horning, S.E.; Brown, G.A.; Korsmeyer, S.J. Altered Hox expression and segmental identity in Mll-mutant mice. Nature 1995, 378, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Corral, J.; Lavenir, I.; Impey, H.; Warren, A.J.; Forster, A.; Larson, T.A.; Bell, S.; McKenzie, A.N.; King, G.; Rabbitts, T.H. An Mll-AF9 fusion gene made by homologous recombination causes acute leukemia in chimeric mice: A method to create fusion oncogenes. Cell 1996, 85, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Alawieh, D.; Cysique-Foinlan, L.; Willekens, C.; Renneville, A. RAS mutations in myeloid malignancies: Revisiting old questions with novel insights and therapeutic perspectives. Blood Cancer J. 2024, 14, 72. [Google Scholar] [CrossRef]
- Yang, L.; Rau, R.; Goodell, M.A. DNMT3A in haematological malignancies. Nat. Rev. Cancer 2015, 15, 152–165. [Google Scholar] [CrossRef]
- Heyes, E.; Wilhelmson, A.S.; Wenzel, A.; Manhart, G.; Eder, T.; Schuster, M.B.; Rzepa, E.; Pundhir, S.; D’Altri, T.; Frank, A.K.; et al. TET2 lesions enhance the aggressiveness of CEBPA-mutant acute myeloid leukemia by rebalancing GATA2 expression. Nat. Commun. 2023, 14, 6185. [Google Scholar] [CrossRef] [PubMed]
- Miyabayashi, T.; Teo, J.L.; Yamamoto, M.; McMillan, M.; Nguyen, C.; Kahn, M. Wnt/β-catenin/CBP signaling maintains long-term murine embryonic stem cell pluripotency. Proc. Natl. Acad. Sci. USA 2007, 104, 5668–5673. [Google Scholar] [CrossRef]
- ten Berge, D.; Kurek, D.; Blauwkamp, T.; Koole, W.; Maas, A.; Eroglu, E.; Siu, R.K.; Nusse, R. Embryonic stem cells require Wnt proteins to prevent differentiation to epiblast stem cells. Nat. Cell Biol. 2011, 13, 1070–1075. [Google Scholar] [CrossRef] [PubMed]
- Marson, A.; Foreman, R.; Chevalier, B.; Bilodeau, S.; Kahn, M.; Young, R.A.; Jaenisch, R. Wnt signaling promotes reprogramming of somatic cells to pluripotency. Cell Stem Cell 2008, 3, 132–135. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Lin, W.; Long, Y.; Yang, Y.; Zhang, H.; Wu, K.; Chu, Q. Notch signaling pathway: Architecture, disease, and therapeutics. Signal Transduct. Target. Ther. 2022, 7, 95. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Xue, C.; Zeng, Y.; Yuan, X.; Chu, Q.; Jiang, S.; Wang, J.; Zhang, Y.; Zhu, D.; Li, L. Notch signaling pathway in cancer: From mechanistic insights to targeted therapies. Signal Transduct. Target. Ther. 2024, 9, 128. [Google Scholar] [CrossRef]
- D’Assoro, A.B.; Leon-Ferre, R.; Braune, E.B.; Lendahl, U. Roles of Notch Signaling in the Tumor Microenvironment. Int. J. Mol. Sci. 2022, 23, 6241. [Google Scholar] [CrossRef]
- Baroja, I.; Kyriakidis, N.C.; Halder, G.; Moya, I.M. Expected and unexpected effects after systemic inhibition of Hippo transcriptional output in cancer. Nat. Commun. 2024, 15, 2700. [Google Scholar] [CrossRef]
- Ortega, A.; Vera, I.; Diaz, M.P.; Navarro, C.; Rojas, M.; Torres, W.; Parra, H.; Salazar, J.; De Sanctis, J.B.; Bermudez, V. The YAP/TAZ Signaling Pathway in the Tumor Microenvironment and Carcinogenesis: Current Knowledge and Therapeutic Promises. Int. J. Mol. Sci. 2021, 23, 430. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef]
- Laugesen, A.; Helin, K. Chromatin repressive complexes in stem cells, development, and cancer. Cell Stem Cell 2014, 14, 735–751. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Richart, L.; Aflaki, S.; Petitalot, A.; Burton, M.; Michaud, A.; Masliah-Planchon, J.; Kuhnowski, F.; Le Cam, S.; Balinas-Gavira, C.; et al. EZH2 mutations in follicular lymphoma distort H3K27me3 profiles and alter transcriptional responses to PRC2 inhibition. Nat. Commun. 2024, 15, 3452. [Google Scholar] [CrossRef] [PubMed]
- Laugesen, A.; Hojfeldt, J.W.; Helin, K. Role of the Polycomb Repressive Complex 2 (PRC2) in Transcriptional Regulation and Cancer. Cold Spring Harb. Perspect. Med. 2016, 6, a026575. [Google Scholar] [CrossRef] [PubMed]
- Beguelin, W.; Popovic, R.; Teater, M.; Jiang, Y.; Bunting, K.L.; Rosen, M.; Shen, H.; Yang, S.N.; Wang, L.; Ezponda, T.; et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 2013, 23, 677–692. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef]
- Kumar, V.E.; Nambiar, R.; De Souza, C.; Nguyen, A.; Chien, J.; Lam, K.S. Targeting Epigenetic Modifiers of Tumor Plasticity and Cancer Stem Cell Behavior. Cells 2022, 11, 1403. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Pei, D. The magic of four: Induction of pluripotent stem cells from somatic cells by Oct4, Sox2, Myc and Klf4. Cell Res. 2007, 17, 578–580. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Muller, M.; Hermann, P.C.; Liebau, S.; Weidgang, C.; Seufferlein, T.; Kleger, A.; Perkhofer, L. The role of pluripotency factors to drive stemness in gastrointestinal cancer. Stem Cell Res. 2016, 16, 349–357. [Google Scholar] [CrossRef]
- Dong, Y.; Tu, R.; Liu, H.; Qing, G. Regulation of cancer cell metabolism: Oncogenic MYC in the driver’s seat. Signal Transduct. Target. Ther. 2020, 5, 124. [Google Scholar] [CrossRef] [PubMed]
- Cazarin, J.; DeRollo, R.E.; Shahidan, S.; Burchett, J.B.; Mwangi, D.; Krishnaiah, S.; Hsieh, A.L.; Walton, Z.E.; Brooks, R.; Mello, S.S.; et al. MYC disrupts transcriptional and metabolic circadian oscillations in cancer and promotes enhanced biosynthesis. PLoS Genet. 2023, 19, e1010904. [Google Scholar] [CrossRef]
- Marques, C.; Unterkircher, T.; Kroon, P.; Oldrini, B.; Izzo, A.; Dramaretska, Y.; Ferrarese, R.; Kling, E.; Schnell, O.; Nelander, S.; et al. NF1 regulates mesenchymal glioblastoma plasticity and aggressiveness through the AP-1 transcription factor FOSL1. eLife 2021, 10, e64846. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; McKay, R.M.; Parada, L.F. Malignant glioma: Lessons from genomics, mouse models, and stem cells. Cell 2012, 149, 36–47. [Google Scholar] [CrossRef]
- Liu, F.; Hon, G.C.; Villa, G.R.; Turner, K.M.; Ikegami, S.; Yang, H.; Ye, Z.; Li, B.; Kuan, S.; Lee, A.Y.; et al. EGFR Mutation Promotes Glioblastoma through Epigenome and Transcription Factor Network Remodeling. Mol. Cell 2015, 60, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Furnari, F.B.; Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat. Rev. Cancer 2015, 15, 302–310. [Google Scholar] [CrossRef] [PubMed]
- White, R.M.; Zon, L.I. Melanocytes in development, regeneration, and cancer. Cell Stem Cell 2008, 3, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Uong, A.; Zon, L.I. Melanocytes in development and cancer. J. Cell Physiol. 2010, 222, 38–41. [Google Scholar] [CrossRef]
- Dave, N.; Guaita-Esteruelas, S.; Gutarra, S.; Frias, A.; Beltran, M.; Peiro, S.; de Herreros, A.G. Functional cooperation between Snail1 and twist in the regulation of ZEB1 expression during epithelial to mesenchymal transition. J. Biol. Chem. 2011, 286, 12024–12032. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef]
- Dow, L.E.; O’Rourke, K.P.; Simon, J.; Tschaharganeh, D.F.; van Es, J.H.; Clevers, H.; Lowe, S.W. Apc Restoration Promotes Cellular Differentiation and Reestablishes Crypt Homeostasis in Colorectal Cancer. Cell 2015, 161, 1539–1552. [Google Scholar] [CrossRef] [PubMed]
- Schwitalla, S.; Fingerle, A.A.; Cammareri, P.; Nebelsiek, T.; Goktuna, S.I.; Ziegler, P.K.; Canli, O.; Heijmans, J.; Huels, D.J.; Moreaux, G.; et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 2013, 152, 25–38. [Google Scholar] [CrossRef]
- Verhagen, M.P.; Joosten, R.; Schmitt, M.; Valimaki, N.; Sacchetti, A.; Rajamaki, K.; Choi, J.; Procopio, P.; Silva, S.; van der Steen, B.; et al. Non-stem cell lineages as an alternative origin of intestinal tumorigenesis in the context of inflammation. Nat. Genet. 2024, 56, 1456–1467. [Google Scholar] [CrossRef] [PubMed]
- Storz, P. Acinar cell plasticity and development of pancreatic ductal adenocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 296–304. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Fontana, R.; Mestre-Farrera, A.; Yang, J. Update on Epithelial-Mesenchymal Plasticity in Cancer Progression. Annu. Rev. Pathol. Mech. Dis. 2024, 19, 133–156. [Google Scholar] [CrossRef]
- Feigin, M.E.; Muthuswamy, S.K. Polarity proteins regulate mammalian cell-cell junctions and cancer pathogenesis. Curr. Opin. Cell Biol. 2009, 21, 694–700. [Google Scholar] [CrossRef]
- Coradini, D.; Casarsa, C.; Oriana, S. Epithelial cell polarity and tumorigenesis: New perspectives for cancer detection and treatment. Acta Pharmacol. Sin. 2011, 32, 552–564. [Google Scholar] [CrossRef]
- Kaufhold, S.; Bonavida, B. Central role of Snail1 in the regulation of EMT and resistance in cancer: A target for therapeutic intervention. J. Exp. Clin. Cancer Res. 2014, 33, 62. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Guo, X.; Hong, W.; Liu, Q.; Wei, T.; Lu, C.; Gao, L.; Ye, D.; Zhou, Y.; Chen, J.; et al. Critical regulation of miR-200/ZEB2 pathway in Oct4/Sox2-induced mesenchymal-to-epithelial transition and induced pluripotent stem cell generation. Proc. Natl. Acad. Sci. USA 2013, 110, 2858–2863. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; Stephens, M.A.; Pathak, H.; Rangarajan, A. Transcription factors that mediate epithelial-mesenchymal transition lead to multidrug resistance by upregulating ABC transporters. Cell Death Dis. 2011, 2, e179. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Yang, L.; Chen, C.; Ashrafizadeh, M.; Tian, Y.; Sun, R. Wnt/β-catenin-driven EMT regulation in human cancers. Cell Mol. Life Sci. 2024, 81, 79. [Google Scholar] [CrossRef] [PubMed]
- Taipale, J.; Beachy, P.A. The Hedgehog and Wnt signalling pathways in cancer. Nature 2001, 411, 349–354. [Google Scholar] [CrossRef]
- Begicevic, R.R.; Falasca, M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362. [Google Scholar] [CrossRef] [PubMed]
- Jinesh, G.G.; Brohl, A.S. Classical epithelial-mesenchymal transition (EMT) and alternative cell death process-driven blebbishield metastatic-witch (BMW) pathways to cancer metastasis. Signal Transduct. Target. Ther. 2022, 7, 296. [Google Scholar] [CrossRef]
- Wang, G.; Xu, D.; Zhang, Z.; Li, X.; Shi, J.; Sun, J.; Liu, H.Z.; Li, X.; Zhou, M.; Zheng, T. The pan-cancer landscape of crosstalk between epithelial-mesenchymal transition and immune evasion relevant to prognosis and immunotherapy response. NPJ Precis. Oncol. 2021, 5, 56. [Google Scholar] [CrossRef]
- Zhou, H.M.; Zhang, J.G.; Zhang, X.; Li, Q. Targeting cancer stem cells for reversing therapy resistance: Mechanism, signaling, and prospective agents. Signal Transduct. Target. Ther. 2021, 6, 62. [Google Scholar] [CrossRef]
- Wu, H.T.; Zhong, H.T.; Li, G.W.; Shen, J.X.; Ye, Q.Q.; Zhang, M.L.; Liu, J. Oncogenic functions of the EMT-related transcription factor ZEB1 in breast cancer. J. Transl. Med. 2020, 18, 51. [Google Scholar] [CrossRef]
- Zhao, H.; Ming, T.; Tang, S.; Ren, S.; Yang, H.; Liu, M.; Tao, Q.; Xu, H. Wnt signaling in colorectal cancer: Pathogenic role and therapeutic target. Mol. Cancer 2022, 21, 144. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Yang, J.; Deng, S.; Xu, H.; Wu, D.; Zeng, Q.; Wang, S.; Hu, T.; Wu, F.; Zhou, H. TGF-beta signaling in the tumor metabolic microenvironment and targeted therapies. J. Hematol. Oncol. 2022, 15, 135. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Han, F.; Du, Y.; Shi, H.; Zhou, W. Hypoxic microenvironment in cancer: Molecular mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2023, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.N.; Ahn, D.H.; Kang, N.; Yeo, C.D.; Kim, Y.K.; Lee, K.Y.; Kim, T.J.; Lee, S.H.; Park, M.S.; Yim, H.W.; et al. TGF-beta induced EMT and stemness characteristics are associated with epigenetic regulation in lung cancer. Sci. Rep. 2020, 10, 10597. [Google Scholar] [CrossRef]
- Dardare, J.; Witz, A.; Merlin, J.L.; Bochnakian, A.; Toussaint, P.; Gilson, P.; Harle, A. Epithelial to Mesenchymal Transition in Patients with Pancreatic Ductal Adenocarcinoma: State-of-the-Art and Therapeutic Opportunities. Pharmaceuticals 2021, 14, 740. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zheng, Y.; Yang, F.; Zhu, L.; Zhu, X.Q.; Wang, Z.F.; Wu, X.L.; Zhou, C.H.; Yan, J.Y.; Hu, B.Y.; et al. The molecular biology of pancreatic adenocarcinoma: Translational challenges and clinical perspectives. Signal Transduct. Target. Ther. 2021, 6, 249. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Su, Y.; Zhong, S.; Cong, L.; Liu, B.; Yang, J.; Tao, Y.; He, Z.; Chen, C.; Jiang, Y. Exosomes: Key players in cancer and potential therapeutic strategy. Signal Transduct. Target. Ther. 2020, 5, 145. [Google Scholar] [CrossRef] [PubMed]
- Scioli, M.G.; Terriaca, S.; Fiorelli, E.; Storti, G.; Fabbri, G.; Cervelli, V.; Orlandi, A. Extracellular Vesicles and Cancer Stem Cells in Tumor Progression: New Therapeutic Perspectives. Int. J. Mol. Sci. 2021, 22, 10572. [Google Scholar] [CrossRef]
- Su, C.; Zhang, J.; Yarden, Y.; Fu, L. The key roles of cancer stem cell-derived extracellular vesicles. Signal Transduct. Target. Ther. 2021, 6, 109. [Google Scholar] [CrossRef]
- Zeijlemaker, W.; Grob, T.; Meijer, R.; Hanekamp, D.; Kelder, A.; Carbaat-Ham, J.C.; Oussoren-Brockhoff, Y.J.M.; Snel, A.N.; Veldhuizen, D.; Scholten, W.J.; et al. CD34+CD38− leukemic stem cell frequency to predict outcome in acute myeloid leukemia. Leukemia 2019, 33, 1102–1112. [Google Scholar] [CrossRef]
- Gerber, J.M.; Smith, B.D.; Ngwang, B.; Zhang, H.; Vala, M.S.; Morsberger, L.; Galkin, S.; Collector, M.I.; Perkins, B.; Levis, M.J.; et al. A clinically relevant population of leukemic CD34+CD38− cells in acute myeloid leukemia. Blood 2012, 119, 3571–3577. [Google Scholar] [CrossRef]
- Glumac, P.M.; LeBeau, A.M. The role of CD133 in cancer: A concise review. Clin. Transl. Med. 2018, 7, 18. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.M.; Nesterova, A.; Ryan, M.C.; Duniho, S.; Jonas, M.; Anderson, M.; Zabinski, R.F.; Sutherland, M.K.; Gerber, H.P.; Van Orden, K.L.; et al. CD133/prominin-1 is a potential therapeutic target for antibody-drug conjugates in hepatocellular and gastric cancers. Br. J. Cancer 2008, 99, 100–109. [Google Scholar] [CrossRef]
- Tang, X.; Zuo, C.; Fang, P.; Liu, G.; Qiu, Y.; Huang, Y.; Tang, R. Targeting Glioblastoma Stem Cells: A Review on Biomarkers, Signal Pathways and Targeted Therapy. Front. Oncol. 2021, 11, 701291. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Chai, S.; Wang, P.; Zhang, C.; Yang, Y.; Yang, Y.; Wang, K. Aldehyde dehydrogenases and cancer stem cells. Cancer Lett. 2015, 369, 50–57. [Google Scholar] [CrossRef]
- Wei, Y.; Li, Y.; Chen, Y.; Liu, P.; Huang, S.; Zhang, Y.; Sun, Y.; Wu, Z.; Hu, M.; Wu, Q.; et al. ALDH1: A potential therapeutic target for cancer stem cells in solid tumors. Front. Oncol. 2022, 12, 1026278. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Budhu, A.; Forgues, M.; Wang, X.W. Activation of hepatic stem cell marker EpCAM by Wnt-β-catenin signaling in hepatocellular carcinoma. Cancer Res. 2007, 67, 10831–10839. [Google Scholar] [CrossRef] [PubMed]
- Gires, O.; Klein, C.A.; Baeuerle, P.A. On the abundance of EpCAM on cancer stem cells. Nat. Rev. Cancer 2009, 9, 143. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Gires, O. EpCAM (CD326) finding its role in cancer. Br. J. Cancer 2007, 96, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Jin, X.; Jung, J.E.; Beck, S.; Kim, H. Cell surface Nestin is a biomarker for glioma stem cells. Biochem. Biophys. Res. Commun. 2013, 433, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.; Zhang, Y.; Sun, L.; Ma, Q.; Yang, J.; Ai, L.; Xue, J.; Chen, G.; Zhang, H.; Ji, C.; et al. Association of human breast cancer CD44(-)/CD24(-) cells with delayed distant metastasis. eLife 2021, 10, e65418. [Google Scholar] [CrossRef]
- Bahmad, H.F.; Cheaito, K.; Chalhoub, R.M.; Hadadeh, O.; Monzer, A.; Ballout, F.; El-Hajj, A.; Mukherji, D.; Liu, Y.N.; Daoud, G.; et al. Sphere-Formation Assay: Three-Dimensional in vitro Culturing of Prostate Cancer Stem/Progenitor Sphere-Forming Cells. Front. Oncol. 2018, 8, 347. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, C.Y.; Liu, T.; Wu, B.; Zhou, F.; Xiong, J.X.; Wu, H.S.; Tao, J.; Zhao, G.; Yang, M.; et al. Persistence of side population cells with high drug efflux capacity in pancreatic cancer. World J. Gastroenterol. 2008, 14, 925–930. [Google Scholar] [CrossRef]
- Del Vecchio, V.; La Noce, M.; Tirino, V. ALDH Activity Assay: A Method for Cancer Stem Cell (CSC) Identification and Isolation. Methods Mol. Biol. 2024, 2777, 83–89. [Google Scholar] [CrossRef]
- Kirstetter, P.; Anderson, K.; Porse, B.T.; Jacobsen, S.E.; Nerlov, C. Activation of the canonical Wnt pathway leads to loss of hematopoietic stem cell repopulation and multilineage differentiation block. Nat. Immunol. 2006, 7, 1048–1056. [Google Scholar] [CrossRef] [PubMed]
- Klaus, A.; Birchmeier, W. Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer 2008, 8, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: Clinical update. Nat. Rev. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef]
- Kumar, V.; Vashishta, M.; Kong, L.; Wu, X.; Lu, J.J.; Guha, C.; Dwarakanath, B.S. The Role of Notch, Hedgehog, and Wnt Signaling Pathways in the Resistance of Tumors to Anticancer Therapies. Front. Cell Dev. Biol. 2021, 9, 650772. [Google Scholar] [CrossRef]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef]
- Holland, J.D.; Klaus, A.; Garratt, A.N.; Birchmeier, W. Wnt signaling in stem and cancer stem cells. Curr. Opin. Cell Biol. 2013, 25, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Jun, S.; Jung, Y.S.; Suh, H.N.; Wang, W.; Kim, M.J.; Oh, Y.S.; Lien, E.M.; Shen, X.; Matsumoto, Y.; McCrea, P.D.; et al. LIG4 mediates Wnt signalling-induced radioresistance. Nat. Commun. 2016, 7, 10994. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.; Tang, D.; Morita, Y.; Sperka, T.; Omrani, O.; Lechel, A.; Sakk, V.; Kraus, J.; Kestler, H.A.; Kuhl, M.; et al. Wnt activity and basal niche position sensitize intestinal stem and progenitor cells to DNA damage. EMBO J. 2017, 36, 2920–2921. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Guttridge, D.C.; You, Z.; Zhang, Z.; Fribley, A.; Mayo, M.W.; Kitajewski, J.; Wang, C.Y. Wnt-1 signaling inhibits apoptosis by activating β-catenin/T cell factor-mediated transcription. J. Cell Biol. 2001, 152, 87–96. [Google Scholar] [CrossRef]
- Correa, S.; Binato, R.; Du Rocher, B.; Castelo-Branco, M.T.; Pizzatti, L.; Abdelhay, E. Wnt/β-catenin pathway regulates ABCB1 transcription in chronic myeloid leukemia. BMC Cancer 2012, 12, 303. [Google Scholar] [CrossRef] [PubMed]
- Stein, U.; Fleuter, C.; Siegel, F.; Smith, J.; Kopacek, A.; Scudiero, D.A.; Hite, K.M.; Schlag, P.M.; Shoemaker, R.H.; Walther, W. Impact of mutant beta-catenin on ABCB1 expression and therapy response in colon cancer cells. Br. J. Cancer 2012, 106, 1395–1405. [Google Scholar] [CrossRef]
- Zhao, F.; Xiao, C.; Evans, K.S.; Theivanthiran, T.; DeVito, N.; Holtzhausen, A.; Liu, J.; Liu, X.; Boczkowski, D.; Nair, S.; et al. Paracrine Wnt5a-beta-Catenin Signaling Triggers a Metabolic Program that Drives Dendritic Cell Tolerization. Immunity 2018, 48, 147.e7–160.e7. [Google Scholar] [CrossRef]
- Suryawanshi, A.; Hussein, M.S.; Prasad, P.D.; Manicassamy, S. Wnt Signaling Cascade in Dendritic Cells and Regulation of Anti-tumor Immunity. Front. Immunol. 2020, 11, 122. [Google Scholar] [CrossRef]
- Li, X.; Xiang, Y.; Li, F.; Yin, C.; Li, B.; Ke, X. WNT/β-Catenin Signaling Pathway Regulating T Cell-Inflammation in the Tumor Microenvironment. Front. Immunol. 2019, 10, 2293. [Google Scholar] [CrossRef]
- Yu, F.; Yu, C.; Li, F.; Zuo, Y.; Wang, Y.; Yao, L.; Wu, C.; Wang, C.; Ye, L. Wnt/β-catenin signaling in cancers and targeted therapies. Signal Transduct. Target. Ther. 2021, 6, 307. [Google Scholar] [CrossRef]
- Jing, J.; Wu, Z.; Wang, J.; Luo, G.; Lin, H.; Fan, Y.; Zhou, C. Hedgehog signaling in tissue homeostasis, cancers, and targeted therapies. Signal Transduct. Target. Ther. 2023, 8, 315. [Google Scholar] [CrossRef]
- Wang, C.Y.; Chang, Y.C.; Kuo, Y.L.; Lee, K.T.; Chen, P.S.; Cheung, C.H.A.; Chang, C.P.; Phan, N.N.; Shen, M.R.; Hsu, H.P. Mutation of the PTCH1 gene predicts recurrence of breast cancer. Sci. Rep. 2019, 9, 16359. [Google Scholar] [CrossRef]
- Faiao-Flores, F.; Alves-Fernandes, D.K.; Pennacchi, P.C.; Sandri, S.; Vicente, A.L.; Scapulatempo-Neto, C.; Vazquez, V.L.; Reis, R.M.; Chauhan, J.; Goding, C.R.; et al. Targeting the hedgehog transcription factors GLI1 and GLI2 restores sensitivity to vemurafenib-resistant human melanoma cells. Oncogene 2017, 36, 1849–1861. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.Y.; Sugumar, V.; Alshanon, A.F.; Wong, W.F.; Fung, S.Y.; Looi, C.Y. Defining the Role of GLI/Hedgehog Signaling in Chemoresistance: Implications in Therapeutic Approaches. Cancers 2021, 13, 4746. [Google Scholar] [CrossRef] [PubMed]
- Po, A.; Citarella, A.; Catanzaro, G.; Besharat, Z.M.; Trocchianesi, S.; Gianno, F.; Sabato, C.; Moretti, M.; De Smaele, E.; Vacca, A.; et al. Hedgehog-GLI signalling promotes chemoresistance through the regulation of ABC transporters in colorectal cancer cells. Sci. Rep. 2020, 10, 13988. [Google Scholar] [CrossRef]
- Meng, E.; Hanna, A.; Samant, R.S.; Shevde, L.A. The Impact of Hedgehog Signaling Pathway on DNA Repair Mechanisms in Human Cancer. Cancers 2015, 7, 1333–1348. [Google Scholar] [CrossRef] [PubMed]
- Kanda, S.; Mitsuyasu, T.; Nakao, Y.; Kawano, S.; Goto, Y.; Matsubara, R.; Nakamura, S. Anti-apoptotic role of the sonic hedgehog signaling pathway in the proliferation of ameloblastoma. Int. J. Oncol. 2013, 43, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Ma, L.; Zhang, Z.; Liu, X.; Gao, H.; Zhuang, Y.; Yang, P.; Kornmann, M.; Tian, X.; Yang, Y. Hedgehog Signaling Regulates Epithelial-Mesenchymal Transition in Pancreatic Cancer Stem-Like Cells. J. Cancer 2016, 7, 408–417. [Google Scholar] [CrossRef]
- Ohta, H.; Aoyagi, K.; Fukaya, M.; Danjoh, I.; Ohta, A.; Isohata, N.; Saeki, N.; Taniguchi, H.; Sakamoto, H.; Shimoda, T.; et al. Cross talk between hedgehog and epithelial-mesenchymal transition pathways in gastric pit cells and in diffuse-type gastric cancers. Br. J. Cancer 2009, 100, 389–398. [Google Scholar] [CrossRef]
- Wang, J.; Cui, B.; Li, X.; Zhao, X.; Huang, T.; Ding, X. The emerging roles of Hedgehog signaling in tumor immune microenvironment. Front. Oncol. 2023, 13, 1171418. [Google Scholar] [CrossRef]
- Giammona, A.; Crivaro, E.; Stecca, B. Emerging Roles of Hedgehog Signaling in Cancer Immunity. Int. J. Mol. Sci. 2023, 24, 1321. [Google Scholar] [CrossRef]
- Wang, M.; Yu, F.; Zhang, Y.; Li, P. Novel insights into Notch signaling in tumor immunity: Potential targets for cancer immunotherapy. Front. Immunol. 2024, 15, 1352484. [Google Scholar] [CrossRef]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol. Cancer 2023, 22, 138. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Wang, Y.; Zhou, C.; Mei, W.; Zeng, C. PI3K/Akt/mTOR Pathway and Its Role in Cancer Therapeutics: Are We Making Headway? Front. Oncol. 2022, 12, 819128. [Google Scholar] [CrossRef]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef] [PubMed]
- Son, B.; Lee, W.; Kim, H.; Shin, H.; Park, H.H. Targeted therapy of cancer stem cells: Inhibition of mTOR in pre-clinical and clinical research. Cell Death Dis. 2024, 15, 696. [Google Scholar] [CrossRef] [PubMed]
- Karami Fath, M.; Ebrahimi, M.; Nourbakhsh, E.; Zia Hazara, A.; Mirzaei, A.; Shafieyari, S.; Salehi, A.; Hoseinzadeh, M.; Payandeh, Z.; Barati, G. PI3K/Akt/mTOR signaling pathway in cancer stem cells. Pathol. Res. Pract. 2022, 237, 154010. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Xu, X.Y. PI3K/Akt/mTOR signaling pathway in cancer stem cells: From basic research to clinical application. Am. J. Cancer Res. 2015, 5, 1602–1609. [Google Scholar]
- Heddleston, J.M.; Li, Z.; Lathia, J.D.; Bao, S.; Hjelmeland, A.B.; Rich, J.N. Hypoxia inducible factors in cancer stem cells. Br. J. Cancer 2010, 102, 789–795. [Google Scholar] [CrossRef]
- Keith, B.; Simon, M.C. Hypoxia-inducible factors, stem cells, and cancer. Cell 2007, 129, 465–472. [Google Scholar] [CrossRef]
- Tong, W.W.; Tong, G.H.; Liu, Y. Cancer stem cells and hypoxia-inducible factors (Review). Int. J. Oncol. 2018, 53, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Thews, O.; Gassner, B.; Kelleher, D.K.; Gekle, M. Activity of drug efflux transporters in tumor cells under hypoxic conditions. Adv. Exp. Med. Biol. 2008, 614, 157–164. [Google Scholar] [CrossRef]
- Tafech, A.; Stephanou, A. On the Importance of Acidity in Cancer Cells and Therapy. Biology 2024, 13, 225. [Google Scholar] [CrossRef] [PubMed]
- Vander Linden, C.; Corbet, C. Therapeutic Targeting of Cancer Stem Cells: Integrating and Exploiting the Acidic Niche. Front. Oncol. 2019, 9, 159. [Google Scholar] [CrossRef] [PubMed]
- Rolver, M.G.; Camacho-Roda, J.; Dai, Y.; Flinck, M.; Ialchina, R.; Hindkaer, J.; Dyhr, R.T.; Bodilsen, A.N.; Prasad, N.S.; Baldan, J.; et al. Tumor microenvironment acidosis favors pancreatic cancer stem cell properties and in vivo metastasis. iScience 2025, 28, 111956. [Google Scholar] [CrossRef]
- Cardone, R.A.; Casavola, V.; Reshkin, S.J. The role of disturbed pH dynamics and the Na+/H+ exchanger in metastasis. Nat. Rev. Cancer 2005, 5, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Harguindey, S.; Orive, G.; Luis Pedraz, J.; Paradiso, A.; Reshkin, S.J. The role of pH dynamics and the Na+/H+ antiporter in the etiopathogenesis and treatment of cancer. Two faces of the same coin—One single nature. Biochim. Biophys. Acta 2005, 1756, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Peppicelli, S.; Andreucci, E.; Ruzzolini, J.; Laurenzana, A.; Margheri, F.; Fibbi, G.; Del Rosso, M.; Bianchini, F.; Calorini, L. The acidic microenvironment as a possible niche of dormant tumor cells. Cell Mol. Life Sci. 2017, 74, 2761–2771. [Google Scholar] [CrossRef]
- Cheng, G.M.; To, K.K. Adverse Cell Culture Conditions Mimicking the Tumor Microenvironment Upregulate ABCG2 to Mediate Multidrug Resistance and a More Malignant Phenotype. ISRN Oncol. 2012, 2012, 746025. [Google Scholar] [CrossRef]
- Visioli, F.; Wang, Y.; Alam, G.N.; Ning, Y.; Rados, P.V.; Nor, J.E.; Polverini, P.J. Glucose-regulated protein 78 (Grp78) confers chemoresistance to tumor endothelial cells under acidic stress. PLoS ONE 2014, 9, e101053. [Google Scholar] [CrossRef]
- Thews, O.; Nowak, M.; Sauvant, C.; Gekle, M. Hypoxia-induced extracellular acidosis increases p-glycoprotein activity and chemoresistance in tumors in vivo via p38 signaling pathway. Adv. Exp. Med. Biol. 2011, 701, 115–122. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, K.M.; Rennert, P.D.; Freeman, G.J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 2015, 14, 561–584. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Galassi, C.; Musella, M.; Manduca, N.; Maccafeo, E.; Sistigu, A. The Immune Privilege of Cancer Stem Cells: A Key to Understanding Tumor Immune Escape and Therapy Failure. Cells 2021, 10, 2361. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.R.; Fang, Y.; Lyu, Z.; Zhu, Y.; Yang, L. Exploring the dynamic interplay between cancer stem cells and the tumor microenvironment: Implications for novel therapeutic strategies. J. Transl. Med. 2023, 21, 686. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Hsu, W.H.; Han, J.; Xia, Y.; DePinho, R.A. Cancer Stemness Meets Immunity: From Mechanism to Therapy. Cell Rep. 2021, 34, 108597. [Google Scholar] [CrossRef] [PubMed]
- Vito, A.; El-Sayes, N.; Mossman, K. Hypoxia-Driven Immune Escape in the Tumor Microenvironment. Cells 2020, 9, 992. [Google Scholar] [CrossRef]
- Guo, T.; Xu, J. Cancer-associated fibroblasts: A versatile mediator in tumor progression, metastasis, and targeted therapy. Cancer Metastasis Rev. 2024, 43, 1095–1116. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Yang, J.; Liu, J.; Wang, Y.; Mu, J.; Zeng, Q.; Deng, S.; Zhou, H. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct. Target. Ther. 2021, 6, 218. [Google Scholar] [CrossRef]
- Prager, B.C.; Xie, Q.; Bao, S.; Rich, J.N. Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell 2019, 24, 41–53. [Google Scholar] [CrossRef]
- Lathia, J.D.; Heddleston, J.M.; Venere, M.; Rich, J.N. Deadly teamwork: Neural cancer stem cells and the tumor microenvironment. Cell Stem Cell 2011, 8, 482–485. [Google Scholar] [CrossRef]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Cabarcas, S.M.; Mathews, L.A.; Farrar, W.L. The cancer stem cell niche--there goes the neighborhood? Int. J. Cancer 2011, 129, 2315–2327. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ye, Y.; Zhu, L.; Xiao, X.; Zhou, B.; Gu, Y.; Si, H.; Liang, H.; Liu, M.; Li, J.; et al. Niche stiffness sustains cancer stemness via TAZ and NANOG phase separation. Nat. Commun. 2023, 14, 238. [Google Scholar] [CrossRef]
- Dzobo, K.; Dandara, C. The Extracellular Matrix: Its Composition, Function, Remodeling, and Role in Tumorigenesis. Biomimetics 2023, 8, 146. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.R.; Zhao, J.T.; Xie, Z.Z. Integrin-mediated cancer progression as a specific target in clinical therapy. Biomed. Pharmacother. 2022, 155, 113745. [Google Scholar] [CrossRef]
- Cooper, J.; Giancotti, F.G. Integrin Signaling in Cancer: Mechanotransduction, Stemness, Epithelial Plasticity, and Therapeutic Resistance. Cancer Cell 2019, 35, 347–367. [Google Scholar] [CrossRef]
- Nonnast, E.; Mira, E.; Manes, S. The role of laminins in cancer pathobiology: A comprehensive review. J. Transl. Med. 2025, 23, 83. [Google Scholar] [CrossRef] [PubMed]
- Givant-Horwitz, V.; Davidson, B.; Reich, R. Laminin-induced signaling in tumor cells. Cancer Lett. 2005, 223, 1–10. [Google Scholar] [CrossRef]
- Lokeshwar, V.B.; Mirza, S.; Jordan, A. Targeting hyaluronic acid family for cancer chemoprevention and therapy. Adv. Cancer Res. 2014, 123, 35–65. [Google Scholar] [CrossRef]
- Bhattacharyya, M.; Jariyal, H.; Srivastava, A. Hyaluronic acid: More than a carrier, having an overpowering extracellular and intracellular impact on cancer. Carbohydr. Polym. 2023, 317, 121081. [Google Scholar] [CrossRef] [PubMed]
- Michalczyk, M.; Humeniuk, E.; Adamczuk, G.; Korga-Plewko, A. Hyaluronic Acid as a Modern Approach in Anticancer Therapy-Review. Int. J. Mol. Sci. 2022, 24, 103. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.W.; Wong, C.K.; Ozturk, S.; Papageorgis, P.; Raghunathan, R.; Alekseyev, Y.; Gower, A.C.; Reinhard, B.M.; Abdolmaleky, H.M.; Thiagalingam, S. Tumor Cell-Derived Periostin Regulates Cytokines That Maintain Breast Cancer Stem Cells. Mol. Cancer Res. 2016, 14, 103–113. [Google Scholar] [CrossRef]
- Dorafshan, S.; Razmi, M.; Safaei, S.; Gentilin, E.; Madjd, Z.; Ghods, R. Periostin: Biology and function in cancer. Cancer Cell Int. 2022, 22, 315. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gonzalez, L.; Alonso, J. Periostin: A Matricellular Protein With Multiple Functions in Cancer Development and Progression. Front. Oncol. 2018, 8, 225. [Google Scholar] [CrossRef]
- Seno, M. A Landscape of Cancer Initiation and Cancer Stem Cells. Cancers 2025, 17, 203. [Google Scholar] [CrossRef] [PubMed]
- Leck, L.Y.W.; Abd El-Aziz, Y.S.; McKelvey, K.J.; Park, K.C.; Sahni, S.; Lane, D.J.R.; Skoda, J.; Jansson, P.J. Cancer stem cells: Masters of all traits. Biochim. Biophys. Acta Mol. Basis Dis. 2024, 1871, 167549. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Kreso, A.; Jamieson, C.H. Cancer stem cells and self-renewal. Clin. Cancer Res. 2010, 16, 3113–3120. [Google Scholar] [CrossRef]
- Rich, J.N. Cancer stem cells: Understanding tumor hierarchy and heterogeneity. Medicine 2016, 95, S2–S7. [Google Scholar] [CrossRef]
- Lionetti, M.C.; Fumagalli, M.R.; La Porta, C.A.M. Cancer stem cells, plasticity, and drug resistance. Cancer Drug Resist. 2020, 3, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Nandy, S.B.; Lakshmanaswamy, R. Cancer Stem Cells and Metastasis. Prog. Mol. Biol. Transl. Sci. 2017, 151, 137–176. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, F.; Huang, B.; Wang, N. Forces in stem cells and cancer stem cells. Cells Dev. 2022, 170, 203776. [Google Scholar] [CrossRef]
- Liu, N.; Li, S.; Wu, N.; Cho, K.S. Acetylation and deacetylation in cancer stem-like cells. Oncotarget 2017, 8, 89315–89325. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, E.N.; Scaffidi, P. Epigenetics and Cancer Stem Cells: Unleashing, Hijacking, and Restricting Cellular Plasticity. Trends Cancer 2017, 3, 372–386. [Google Scholar] [CrossRef] [PubMed]
- Verona, F.; Pantina, V.D.; Modica, C.; Lo Iacono, M.; D’Accardo, C.; Porcelli, G.; Cricchio, D.; Turdo, A.; Gaggianesi, M.; Di Franco, S. Targeting epigenetic alterations in cancer stem cells. Front. Mol. Med. 2022, 2, 1011882. [Google Scholar] [CrossRef]
- Esteller, M.; Dawson, M.A.; Kadoch, C.; Rassool, F.V.; Jones, P.A.; Baylin, S.B. The Epigenetic Hallmarks of Cancer. Cancer Discov. 2024, 14, 1783–1809. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.G. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012, 22, 457–472. [Google Scholar] [CrossRef]
- Eun, K.; Ham, S.W.; Kim, H. Cancer stem cell heterogeneity: Origin and new perspectives on CSC targeting. BMB Rep. 2017, 50, 117–125. [Google Scholar] [CrossRef]
- Pietras, A. Cancer stem cells in tumor heterogeneity. Adv. Cancer Res. 2011, 112, 255–281. [Google Scholar] [CrossRef] [PubMed]
- Bautch, V.L. Cancer: Tumour stem cells switch sides. Nature 2010, 468, 770–771. [Google Scholar] [CrossRef]
- Malanchi, I.; Santamaria-Martinez, A.; Susanto, E.; Peng, H.; Lehr, H.A.; Delaloye, J.F.; Huelsken, J. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 2011, 481, 85–89. [Google Scholar] [CrossRef]
- Bjerkvig, R.; Johansson, M.; Miletic, H.; Niclou, S.P. Cancer stem cells and angiogenesis. Semin. Cancer Biol. 2009, 19, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Flores, D.G.; Ledur, P.F.; Abujamra, A.L.; Brunetto, A.L.; Schwartsmann, G.; Lenz, G.; Roesler, R. Cancer stem cells and the biology of brain tumors. Curr. Stem Cell Res. Ther. 2009, 4, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Piccirillo, S.G.; Binda, E.; Fiocco, R.; Vescovi, A.L.; Shah, K. Brain cancer stem cells. J. Mol. Med. 2009, 87, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Denysenko, T.; Gennero, L.; Roos, M.A.; Melcarne, A.; Juenemann, C.; Faccani, G.; Morra, I.; Cavallo, G.; Reguzzi, S.; Pescarmona, G.; et al. Glioblastoma cancer stem cells: Heterogeneity, microenvironment and related therapeutic strategies. Cell Biochem. Funct. 2010, 28, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Ohlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523. [Google Scholar] [CrossRef]
- Qin, X.; Guo, H.; Wang, X.; Zhu, X.; Yan, M.; Wang, X.; Xu, Q.; Shi, J.; Lu, E.; Chen, W.; et al. Exosomal miR-196a derived from cancer-associated fibroblasts confers cisplatin resistance in head and neck cancer through targeting CDKN1B and ING5. Genome Biol. 2019, 20, 12. [Google Scholar] [CrossRef]
- Qin, X.; Yan, M.; Zhang, J.; Wang, X.; Shen, Z.; Lv, Z.; Li, Z.; Wei, W.; Chen, W. TGFβ3-mediated induction of Periostin facilitates head and neck cancer growth and is associated with metastasis. Sci. Rep. 2016, 6, 20587. [Google Scholar] [CrossRef]
- Alvarez-Teijeiro, S.; Garcia-Inclan, C.; Villaronga, M.A.; Casado, P.; Hermida-Prado, F.; Granda-Diaz, R.; Rodrigo, J.P.; Calvo, F.; Del-Rio-Ibisate, N.; Gandarillas, A.; et al. Factors Secreted by Cancer-Associated Fibroblasts that Sustain Cancer Stem Properties in Head and Neck Squamous Carcinoma Cells as Potential Therapeutic Targets. Cancers 2018, 10, 334. [Google Scholar] [CrossRef]
- Jiang, J.; Ye, F.; Yang, X.; Zong, C.; Gao, L.; Yang, Y.; Zhao, Q.; Han, Z.; Wei, L. Peri-tumor associated fibroblasts promote intrahepatic metastasis of hepatocellular carcinoma by recruiting cancer stem cells. Cancer Lett. 2017, 404, 19–28. [Google Scholar] [CrossRef]
- Wang, S.S.; Gao, X.L.; Liu, X.; Gao, S.Y.; Fan, Y.L.; Jiang, Y.P.; Ma, X.R.; Jiang, J.; Feng, H.; Chen, Q.M.; et al. CD133+ cancer stem-like cells promote migration and invasion of salivary adenoid cystic carcinoma by inducing vasculogenic mimicry formation. Oncotarget 2016, 7, 29051–29062. [Google Scholar] [CrossRef] [PubMed]
- Murai, T.; Matsuda, S. Targeting the PI3K-Akt-mTOR signaling pathway involved in vasculogenic mimicry promoted by cancer stem cells. Am. J. Cancer Res. 2023, 13, 5039–5046. [Google Scholar] [PubMed]
- Lizarraga-Verdugo, E.; Avendano-Felix, M.; Bermudez, M.; Ramos-Payan, R.; Perez-Plasencia, C.; Aguilar-Medina, M. Cancer Stem Cells and Its Role in Angiogenesis and Vasculogenic Mimicry in Gastrointestinal Cancers. Front. Oncol. 2020, 10, 413. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Shen, L.; Luo, M.; Zhang, K.; Li, J.; Yang, Q.; Zhu, F.; Zhou, D.; Zheng, S.; Chen, Y.; et al. Circulating tumor cells: Biology and clinical significance. Signal Transduct. Target. Ther. 2021, 6, 404. [Google Scholar] [CrossRef]
- Gu, X.; Wei, S.; Lv, X. Circulating tumor cells: From new biological insights to clinical practice. Signal Transduct. Target. Ther. 2024, 9, 226. [Google Scholar] [CrossRef]
- Aramini, B.; Masciale, V.; Arienti, C.; Dominici, M.; Stella, F.; Martinelli, G.; Fabbri, F. Cancer Stem Cells (CSCs), Circulating Tumor Cells (CTCs) and Their Interplay with Cancer Associated Fibroblasts (CAFs): A New World of Targets and Treatments. Cancers 2022, 14, 2408. [Google Scholar] [CrossRef]
- Li, W.; Ma, H.; Zhang, J.; Zhu, L.; Wang, C.; Yang, Y. Unraveling the roles of CD44/CD24 and ALDH1 as cancer stem cell markers in tumorigenesis and metastasis. Sci. Rep. 2017, 7, 13856. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Niu, M.; Yuan, X.; Wu, K.; Liu, A. CD44 as a tumor biomarker and therapeutic target. Exp. Hematol. Oncol. 2020, 9, 36. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Guo, X.; Yang, K.; Yang, Y.; Zhou, W.; Huang, Y.; Liang, X.; Su, J.; Jiang, L.; Li, J.; et al. EpCAM-targeting CAR-T cell immunotherapy is safe and efficacious for epithelial tumors. Sci. Adv. 2023, 9, eadg9721. [Google Scholar] [CrossRef]
- Zeng, S.; Jin, N.; Yu, B.; Ren, Q.; Yan, Z.; Fu, S. Chimeric antigen receptor-T cells targeting epithelial cell adhesion molecule antigens are effective in the treatment of colorectal cancer. BMC Gastroenterol. 2024, 24, 249. [Google Scholar] [CrossRef]
- Ooki, A.; VandenBussche, C.J.; Kates, M.; Hahn, N.M.; Matoso, A.; McConkey, D.J.; Bivalacqua, T.J.; Hoque, M.O. CD24 regulates cancer stem cell (CSC)-like traits and a panel of CSC-related molecules serves as a non-invasive urinary biomarker for the detection of bladder cancer. Br. J. Cancer 2018, 119, 961–970. [Google Scholar] [CrossRef]
- Jang, Y.; Kang, S.; Han, H.H.; Kim, B.G.; Cho, N.H. CD24 induced cellular quiescence-like state and chemoresistance in ovarian cancer cells via miR-130a/301a-dependent CDK19 downregulation. Cell Death Discov. 2024, 10, 81. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhu, G.; Yang, L.; Yang, Y. Targeting CD24 as a novel immunotherapy for solid cancers. Cell Commun. Signal 2023, 21, 312. [Google Scholar] [CrossRef] [PubMed]
- Ran, Y.; Hossain, F.; Pannuti, A.; Lessard, C.B.; Ladd, G.Z.; Jung, J.I.; Minter, L.M.; Osborne, B.A.; Miele, L.; Golde, T.E. gamma-Secretase inhibitors in cancer clinical trials are pharmacologically and functionally distinct. EMBO Mol. Med. 2017, 9, 950–966. [Google Scholar] [CrossRef]
- Feng, M.; Santhanam, R.K.; Xing, H.; Zhou, M.; Jia, H. Inhibition of γ-secretase/Notch pathway as a potential therapy for reversing cancer drug resistance. Biochem. Pharmacol. 2024, 220, 115991. [Google Scholar] [CrossRef]
- Slack, R.J.; Macdonald, S.J.F.; Roper, J.A.; Jenkins, R.G.; Hatley, R.J.D. Emerging therapeutic opportunities for integrin inhibitors. Nat. Rev. Drug Discov. 2022, 21, 60–78. [Google Scholar] [CrossRef]
- Chen, H.C.; Mueller, N.; Stott, K.; Kapeni, C.; Rivers, E.; Sauer, C.M.; Beke, F.; Walsh, S.J.; Ashman, N.; O’Brien, L.; et al. Novel immunotherapeutics against LGR5 to target multiple cancer types. EMBO Mol. Med. 2024, 16, 2233–2261. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Liu, C.; Li, B.; Li, J.; Zhang, P.; Huang, Y.; Li, L. Cell surface patching via CXCR4-targeted nanothreads for cancer metastasis inhibition. Nat. Commun. 2024, 15, 2763. [Google Scholar] [CrossRef] [PubMed]
- Leo, M.; Sabatino, L. Targeting CXCR4 and CD47 Receptors: An Overview of New and Old Molecules for a Biological Personalized Anticancer Therapy. Int. J. Mol. Sci. 2022, 23, 12499. [Google Scholar] [CrossRef] [PubMed]
- Takahashi-Yanaga, F.; Kahn, M. Targeting Wnt signaling: Can we safely eradicate cancer stem cells? Clin. Cancer Res. 2010, 16, 3153–3162. [Google Scholar] [CrossRef]
- Wend, P.; Holland, J.D.; Ziebold, U.; Birchmeier, W. Wnt signaling in stem and cancer stem cells. Semin. Cell Dev. Biol. 2010, 21, 855–863. [Google Scholar] [CrossRef]
- Liu, Y.; Qi, X.; Donnelly, L.; Elghobashi-Meinhardt, N.; Long, T.; Zhou, R.W.; Sun, Y.; Wang, B.; Li, X. Mechanisms and inhibition of Porcupine-mediated Wnt acylation. Nature 2022, 607, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.; Panchal, S.; Patel, B. Porcupine inhibitors: Novel and emerging anti-cancer therapeutics targeting the Wnt signaling pathway. Pharmacol. Res. 2021, 167, 105532. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Pan, S.; Hsieh, M.H.; Ng, N.; Sun, F.; Wang, T.; Kasibhatla, S.; Schuller, A.G.; Li, A.G.; Cheng, D.; et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc. Natl. Acad. Sci. USA 2013, 110, 20224–20229. [Google Scholar] [CrossRef]
- Huang, S.M.; Mishina, Y.M.; Liu, S.; Cheung, A.; Stegmeier, F.; Michaud, G.A.; Charlat, O.; Wiellette, E.; Zhang, Y.; Wiessner, S.; et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009, 461, 614–620. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G. Wnt/β-catenin signalling: Function, biological mechanisms, and therapeutic opportunities. Signal Transduct. Target. Ther. 2022, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, T.; Nasu, K.; Miyabe, S.; Kouji, H.; Katoh, A.; Uemura, N.; Narahara, H. β-catenin signaling inhibitors ICG-001 and C-82 improve fibrosis in preclinical models of endometriosis. Sci. Rep. 2019, 9, 20056. [Google Scholar] [CrossRef]
- Lin, H.H.; Feng, W.C.; Lu, L.C.; Shao, Y.Y.; Hsu, C.H.; Cheng, A.L. Inhibition of the Wnt/β-catenin signaling pathway improves the anti-tumor effects of sorafenib against hepatocellular carcinoma. Cancer Lett. 2016, 381, 58–66. [Google Scholar] [CrossRef]
- DeVito, N.C.; Sturdivant, M.; Thievanthiran, B.; Xiao, C.; Plebanek, M.P.; Salama, A.K.S.; Beasley, G.M.; Holtzhausen, A.; Novotny-Diermayr, V.; Strickler, J.H.; et al. Pharmacological Wnt ligand inhibition overcomes key tumor-mediated resistance pathways to anti-PD-1 immunotherapy. Cell Rep. 2021, 35, 109071. [Google Scholar] [CrossRef] [PubMed]
- Diamond, J.R.; Becerra, C.; Richards, D.; Mita, A.; Osborne, C.; O’Shaughnessy, J.; Zhang, C.; Henner, R.; Kapoun, A.M.; Xu, L.; et al. Phase Ib clinical trial of the anti-frizzled antibody vantictumab (OMP-18R5) plus paclitaxel in patients with locally advanced or metastatic HER2-negative breast cancer. Breast Cancer Res. Treat. 2020, 184, 53–62. [Google Scholar] [CrossRef]
- Liao, H.; Li, X.; Zhao, L.; Wang, Y.; Wang, X.; Wu, Y.; Zhou, X.; Fu, W.; Liu, L.; Hu, H.G.; et al. A PROTAC peptide induces durable β-catenin degradation and suppresses Wnt-dependent intestinal cancer. Cell Discov. 2020, 6, 35. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Mir, R.; Galande, S. Epigenetic Regulation of the Wnt/beta-Catenin Signaling Pathway in Cancer. Front. Genet. 2021, 12, 681053. [Google Scholar] [CrossRef]
- Nguyen, N.M.; Cho, J. Hedgehog Pathway Inhibitors as Targeted Cancer Therapy and Strategies to Overcome Drug Resistance. Int. J. Mol. Sci. 2022, 23, 1733. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Song, Q.; Day, B.W. Phase I and phase II sonidegib and vismodegib clinical trials for the treatment of paediatric and adult MB patients: A systemic review and meta-analysis. Acta Neuropathol. Commun. 2019, 7, 123. [Google Scholar] [CrossRef]
- Danial, C.; Sarin, K.Y.; Oro, A.E.; Chang, A.L. An Investigator-Initiated Open-Label Trial of Sonidegib in Advanced Basal Cell Carcinoma Patients Resistant to Vismodegib. Clin. Cancer Res. 2016, 22, 1325–1329. [Google Scholar] [CrossRef]
- Bruzzese, A.; Martino, E.A.; Labanca, C.; Mendicino, F.; Lucia, E.; Olivito, V.; Fimognari, F.; Neri, A.; Morabito, F.; Vigna, E.; et al. Glasdegib for the treatment of acute myeloid leukemia. Expert. Opin. Pharmacother. 2023, 24, 1537–1543. [Google Scholar] [CrossRef]
- Harada, K.; Ohashi, R.; Naito, K.; Kanki, K. Hedgehog Signal Inhibitor GANT61 Inhibits the Malignant Behavior of Undifferentiated Hepatocellular Carcinoma Cells by Targeting Non-Canonical GLI Signaling. Int. J. Mol. Sci. 2020, 21, 3126. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, J.J.; Kim, J.; Gardner, D.; Beachy, P.A. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc. Natl. Acad. Sci. USA 2010, 107, 13432–13437. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Liu, M.; Wang, Q.; Shen, Y.; Mei, H.; Li, D.; Liu, W. Itraconazole exerts its anti-melanoma effect by suppressing Hedgehog, Wnt, and PI3K/mTOR signaling pathways. Oncotarget 2017, 8, 28510–28525. [Google Scholar] [CrossRef]
- Becher, O.J. HDAC inhibitors to the rescue in sonic hedgehog medulloblastoma. Neuro-Oncol. 2019, 21, 1091–1092. [Google Scholar] [CrossRef]
- Pannuti, A.; Foreman, K.; Rizzo, P.; Osipo, C.; Golde, T.; Osborne, B.; Miele, L. Targeting Notch to target cancer stem cells. Clin. Cancer Res. 2010, 16, 3141–3152. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sullenger, B.A.; Rich, J.N. Notch signaling in cancer stem cells. Adv. Exp. Med. Biol. 2012, 727, 174–185. [Google Scholar] [CrossRef]
- Shih Ie, M.; Wang, T.L. Notch signaling, γ-secretase inhibitors, and cancer therapy. Cancer Res. 2007, 67, 1879–1882. [Google Scholar] [CrossRef] [PubMed]
- Massard, C.; Azaro, A.; Soria, J.C.; Lassen, U.; Le Tourneau, C.; Sarker, D.; Smith, C.; Ohnmacht, U.; Oakley, G.; Patel, B.K.R.; et al. First-in-human study of LY3039478, an oral Notch signaling inhibitor in advanced or metastatic cancer. Ann. Oncol. 2018, 29, 1911–1917. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.C.; Eisenberg, P.D.; Manikhas, G.; Chugh, R.; Gubens, M.A.; Stagg, R.J.; Kapoun, A.M.; Xu, L.; Dupont, J.; Sikic, B. A phase I dose escalation and expansion study of the anticancer stem cell agent demcizumab (anti-DLL4) in patients with previously treated solid tumors. Clin. Cancer Res. 2014, 20, 6295–6303. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, A.; Moore, K.N.; Gordon, M.; Chugh, R.; Diamond, J.R.; Aljumaily, R.; Mendelson, D.; Kapoun, A.M.; Xu, L.; Stagg, R.; et al. A first-in-human phase 1a study of the bispecific anti-DLL4/anti-VEGF antibody navicixizumab (OMP-305B83) in patients with previously treated solid tumors. Investig. New Drugs 2019, 37, 461–472. [Google Scholar] [CrossRef]
- Smith, D.C.; Chugh, R.; Patnaik, A.; Papadopoulos, K.P.; Wang, M.; Kapoun, A.M.; Xu, L.; Dupont, J.; Stagg, R.J.; Tolcher, A. A phase 1 dose escalation and expansion study of Tarextumab (OMP-59R5) in patients with solid tumors. Investig. New Drugs 2019, 37, 722–730. [Google Scholar] [CrossRef]
- Vigolo, M.; Urech, C.; Lamy, S.; Monticone, G.; Zabaleta, J.; Hossain, F.; Wyczechowska, D.; Del Valle, L.; O’Regan, R.M.; Miele, L.; et al. The Efficacy of CB-103, a First-in-Class Transcriptional Notch Inhibitor, in Preclinical Models of Breast Cancer. Cancers 2023, 15, 3957. [Google Scholar] [CrossRef]
- Hanna, G.J.; Stathis, A.; Lopez-Miranda, E.; Racca, F.; Quon, D.; Leyvraz, S.; Hess, D.; Keam, B.; Rodon, J.; Ahn, M.J.; et al. A Phase I Study of the Pan-Notch Inhibitor CB-103 for Patients with Advanced Adenoid Cystic Carcinoma and Other Tumors. Cancer Res. Commun. 2023, 3, 1853–1861. [Google Scholar] [CrossRef]
- Kim, K.J.; Kim, J.W.; Sung, J.H.; Suh, K.J.; Lee, J.Y.; Kim, S.H.; Lee, J.O.; Kim, J.W.; Kim, Y.J.; Kim, J.H.; et al. PI3K-targeting strategy using alpelisib to enhance the antitumor effect of paclitaxel in human gastric cancer. Sci. Rep. 2020, 10, 12308. [Google Scholar] [CrossRef]
- Garrido-Castro, A.C.; Saura, C.; Barroso-Sousa, R.; Guo, H.; Ciruelos, E.; Bermejo, B.; Gavila, J.; Serra, V.; Prat, A.; Pare, L.; et al. Phase 2 study of buparlisib (BKM120), a pan-class I PI3K inhibitor, in patients with metastatic triple-negative breast cancer. Breast Cancer Res. 2020, 22, 120. [Google Scholar] [CrossRef]
- Yam, C.; Xu, X.; Davies, M.A.; Gimotty, P.A.; Morrissette, J.J.D.; Tetzlaff, M.T.; Wani, K.M.; Liu, S.; Deng, W.; Buckley, M.; et al. A Multicenter Phase I Study Evaluating Dual PI3K and BRAF Inhibition with PX-866 and Vemurafenib in Patients with Advanced BRAF V600-Mutant Solid Tumors. Clin. Cancer Res. 2018, 24, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Laranjeira, A.B.A.; Hollingshead, M.G.; Nguyen, D.; Kinders, R.J.; Doroshow, J.H.; Yang, S.X. DNA damage, demethylation and anticancer activity of DNA methyltransferase (DNMT) inhibitors. Sci. Rep. 2023, 13, 5964. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Liu, X.; Zeng, Y.; Liu, J.; Wu, F. DNA methyltransferase inhibitors combination therapy for the treatment of solid tumor: Mechanism and clinical application. Clin. Epigenetics 2021, 13, 166. [Google Scholar] [CrossRef] [PubMed]
- Billam, M.; Sobolewski, M.D.; Davidson, N.E. Effects of a novel DNA methyltransferase inhibitor zebularine on human breast cancer cells. Breast Cancer Res. Treat. 2010, 120, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zhang, M.; Zhou, Y.; Guo, W.; Yi, M.; Zhang, Z.; Ding, Y.; Wang, Y. The application of histone deacetylases inhibitors in glioblastoma. J. Exp. Clin. Cancer Res. 2020, 39, 138. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, D.; Rampias, T. HDAC Inhibitors: Dissecting Mechanisms of Action to Counter Tumor Heterogeneity. Cancers 2021, 13, 3575. [Google Scholar] [CrossRef]
- Qi, J.; Shi, Y. Selective Targeting of Different Bromodomains by Small Molecules. Cancer Cell 2020, 37, 764–766. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Zhang, Z.C.; Wu, Y.Y.; Pi, Y.N.; Lou, S.H.; Liu, T.B.; Lou, G.; Yang, C. Bromodomain and extraterminal (BET) proteins: Biological functions, diseases, and targeted therapy. Signal Transduct. Target. Ther. 2023, 8, 420. [Google Scholar] [CrossRef]
- Wu, D.; Khan, F.A.; Zhang, K.; Pandupuspitasari, N.S.; Negara, W.; Guan, K.; Sun, F.; Huang, C. Retinoic acid signaling in development and differentiation commitment and its regulatory topology. Chem. Biol. Interact. 2024, 387, 110773. [Google Scholar] [CrossRef]
- Brown, G. Retinoic acid receptor regulation of decision-making for cell differentiation. Front. Cell Dev. Biol. 2023, 11, 1182204. [Google Scholar] [CrossRef] [PubMed]
- Marcinkowska, E.; Wallace, G.R.; Brown, G. The Use of 1alpha,25-Dihydroxyvitamin D(3) as an Anticancer Agent. Int. J. Mol. Sci. 2016, 17, 729. [Google Scholar] [CrossRef]
- Duffy, M.J.; Murray, A.; Synnott, N.C.; O’Donovan, N.; Crown, J. Vitamin D analogues: Potential use in cancer treatment. Crit. Rev. Oncol. Hematol. 2017, 112, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Schiera, G.; Di Liegro, C.M.; Di Liegro, I. Involvement of Thyroid Hormones in Brain Development and Cancer. Cancers 2021, 13, 2693. [Google Scholar] [CrossRef]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Investig. 2010, 120, 142–156. [Google Scholar] [CrossRef]
- De Los Santos-Jimenez, J.; Rosales, T.; Ko, B.; Campos-Sandoval, J.A.; Alonso, F.J.; Marquez, J.; DeBerardinis, R.J.; Mates, J.M. Metabolic Adjustments following Glutaminase Inhibition by CB-839 in Glioblastoma Cell Lines. Cancers 2023, 15, 531. [Google Scholar] [CrossRef]
- Restall, I.J.; Cseh, O.; Richards, L.M.; Pugh, T.J.; Luchman, H.A.; Weiss, S. Brain Tumor Stem Cell Dependence on Glutaminase Reveals a Metabolic Vulnerability through the Amino Acid Deprivation Response Pathway. Cancer Res. 2020, 80, 5478–5490. [Google Scholar] [CrossRef]
- Cioce, M.; Pulito, C.; Strano, S.; Blandino, G.; Fazio, V.M. Metformin: Metabolic Rewiring Faces Tumor Heterogeneity. Cells 2020, 9, 2439. [Google Scholar] [CrossRef] [PubMed]
- Moufarrij, S.; Srivastava, A.; Gomez, S.; Hadley, M.; Palmer, E.; Austin, P.T.; Chisholm, S.; Diab, N.; Roche, K.; Yu, A.; et al. Combining DNMT and HDAC6 inhibitors increases anti-tumor immune signaling and decreases tumor burden in ovarian cancer. Sci. Rep. 2020, 10, 3470. [Google Scholar] [CrossRef]
- Huang, J.L.; Chen, S.Y.; Lin, C.S. Targeting Cancer Stem Cells through Epigenetic Modulation of Interferon Response. J. Pers. Med. 2022, 12, 556. [Google Scholar] [CrossRef] [PubMed]
- Beziaud, L.; Young, C.M.; Alonso, A.M.; Norkin, M.; Minafra, A.R.; Huelsken, J. IFNgamma-induced stem-like state of cancer cells as a driver of metastatic progression following immunotherapy. Cell Stem Cell 2023, 30, 818–831.e6. [Google Scholar] [CrossRef]
- Lee, K.W.; Yam, J.W.P.; Mao, X. Dendritic Cell Vaccines: A Shift from Conventional Approach to New Generations. Cells 2023, 12, 2147. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Tang, D.G.; Rycaj, K. Cancer stem cells: Regulation programs, immunological properties and immunotherapy. Semin. Cancer Biol. 2018, 52, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Cui, P.; Li, R.; Huang, Z.; Wu, Z.; Tao, H.; Zhang, S.; Hu, Y. Comparative effectiveness of pembrolizumab vs. nivolumab in patients with recurrent or advanced NSCLC. Sci. Rep. 2020, 10, 13160. [Google Scholar] [CrossRef] [PubMed]
- Fessas, P.; Lee, H.; Ikemizu, S.; Janowitz, T. A molecular and preclinical comparison of the PD-1-targeted T-cell checkpoint inhibitors nivolumab and pembrolizumab. Semin. Oncol. 2017, 44, 136–140. [Google Scholar] [CrossRef]
- Chao, M.P.; Takimoto, C.H.; Feng, D.D.; McKenna, K.; Gip, P.; Liu, J.; Volkmer, J.P.; Weissman, I.L.; Majeti, R. Therapeutic Targeting of the Macrophage Immune Checkpoint CD47 in Myeloid Malignancies. Front. Oncol. 2019, 9, 1380. [Google Scholar] [CrossRef]
- Masoumi, J.; Jafarzadeh, A.; Abdolalizadeh, J.; Khan, H.; Philippe, J.; Mirzaei, H.; Mirzaei, H.R. Cancer stem cell-targeted chimeric antigen receptor (CAR)-T cell therapy: Challenges and prospects. Acta Pharm. Sin. B 2021, 11, 1721–1739. [Google Scholar] [CrossRef]
- Alhabbab, R.Y. Targeting Cancer Stem Cells by Genetically Engineered Chimeric Antigen Receptor T Cells. Front. Genet. 2020, 11, 312. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, Y.; He, Z.; Li, L.; Liu, S.; Jiang, M.; Zhao, B.; Deng, M.; Wang, W.; Mi, X.; et al. Breakthrough of solid tumor treatment: CAR-NK immunotherapy. Cell Death Discov. 2024, 10, 40. [Google Scholar] [CrossRef]
- Xiao, L.; Cen, D.; Gan, H.; Sun, Y.; Huang, N.; Xiong, H.; Jin, Q.; Su, L.; Liu, X.; Wang, K.; et al. Adoptive Transfer of NKG2D CAR mRNA-Engineered Natural Killer Cells in Colorectal Cancer Patients. Mol. Ther. 2019, 27, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Benner, B.; Good, L.; Quiroga, D.; Schultz, T.E.; Kassem, M.; Carson, W.E.; Cherian, M.A.; Sardesai, S.; Wesolowski, R. Pexidartinib, a Novel Small Molecule CSF-1R Inhibitor in Use for Tenosynovial Giant Cell Tumor: A Systematic Review of Pre-Clinical and Clinical Development. Drug Des. Dev. Ther. 2020, 14, 1693–1704. [Google Scholar] [CrossRef]
- Rolfo, C.; Giovannetti, E.; Martinez, P.; McCue, S.; Naing, A. Applications and clinical trial landscape using Toll-like receptor agonists to reduce the toll of cancer. NPJ Precis. Oncol. 2023, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Kaczanowska, S.; Joseph, A.M.; Davila, E. TLR agonists: Our best frenemy in cancer immunotherapy. J. Leukoc. Biol. 2013, 93, 847–863. [Google Scholar] [CrossRef]
- Ciardiello, D.; Elez, E.; Tabernero, J.; Seoane, J. Clinical development of therapies targeting TGFβ: Current knowledge and future perspectives. Ann. Oncol. 2020, 31, 1336–1349. [Google Scholar] [CrossRef]
- Huang, C.Y.; Chung, C.L.; Hu, T.H.; Chen, J.J.; Liu, P.F.; Chen, C.L. Recent progress in TGF-βinhibitors for cancer therapy. Biomed. Pharmacother. 2021, 134, 111046. [Google Scholar] [CrossRef] [PubMed]
- Fox, E.; Bates, S.E. Tariquidar (XR9576): A P-glycoprotein drug efflux pump inhibitor. Expert. Rev. Anticancer Ther. 2007, 7, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Zechner, M.; Castro Jaramillo, C.A.; Zubler, N.S.; Taddio, M.F.; Mu, L.; Altmann, K.H.; Kramer, S.D. In Vitro and In Vivo Evaluation of ABCG2 (BCRP) Inhibitors Derived from Ko143. J. Med. Chem. 2023, 66, 6782–6797. [Google Scholar] [CrossRef] [PubMed]
- Packeiser, E.M.; Engels, L.; Nolte, I.; Goericke-Pesch, S.; Murua Escobar, H. MDR1 Inhibition Reverses Doxorubicin-Resistance in Six Doxorubicin-Resistant Canine Prostate and Bladder Cancer Cell Lines. Int. J. Mol. Sci. 2023, 24, 8136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Patel, S.B.; King, M.R. Micelle-in-Liposomes for Sustained Delivery of Anticancer Agents That Promote Potent TRAIL-Induced Cancer Cell Apoptosis. Molecules 2020, 26, 157. [Google Scholar] [CrossRef]
- Zhang, Z.; Xu, X.; Du, J.; Chen, X.; Xue, Y.; Zhang, J.; Yang, X.; Chen, X.; Xie, J.; Ju, S. Redox-responsive polymer micelles co-encapsulating immune checkpoint inhibitors and chemotherapeutic agents for glioblastoma therapy. Nat. Commun. 2024, 15, 1118. [Google Scholar] [CrossRef]
- Blanpain, C.; Mohrin, M.; Sotiropoulou, P.A.; Passegue, E. DNA-damage response in tissue-specific and cancer stem cells. Cell Stem Cell 2011, 8, 16–29. [Google Scholar] [CrossRef]
- Sperka, T.; Wang, J.; Rudolph, K.L. DNA damage checkpoints in stem cells, ageing and cancer. Nat. Rev. Mol. Cell Biol. 2012, 13, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Shkundina, I.S.; Gall, A.A.; Dick, A.; Cocklin, S.; Mazin, A.V. New RAD51 Inhibitors to Target Homologous Recombination in Human Cells. Genes 2021, 12, 920. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Sinha, S.; Kundu, C.N. Targeting cancer stem cells in the tumor microenvironment: An emerging role of PARP inhibitors. Pharmacol. Res. 2022, 184, 106425. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.E. DNA damage responses in cancer stem cells: Implications for cancer therapeutic strategies. World J. Biol. Chem. 2015, 6, 57–64. [Google Scholar] [CrossRef]
- Abad, E.; Graifer, D.; Lyakhovich, A. DNA damage response and resistance of cancer stem cells. Cancer Lett. 2020, 474, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L. Targeting replication stress to tackle cancer stem cells. Cell Death Dis. 2021, 12, 315. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Sullivan, M.J.; Wynn, R.R.; Demagall, A.; Hendrix, A.S.; Sindhwani, P.; Petros, F.G.; Nadiminty, N. PARP inhibitors chemopotentiate and synergize with cisplatin to inhibit bladder cancer cell survival and tumor growth. BMC Cancer 2022, 22, 312. [Google Scholar] [CrossRef]
- Huang, T.T.; Lampert, E.J.; Coots, C.; Lee, J.M. Targeting the PI3K pathway and DNA damage response as a therapeutic strategy in ovarian cancer. Cancer Treat. Rev. 2020, 86, 102021. [Google Scholar] [CrossRef]
- Cheng, T.; Rodrigues, N.; Shen, H.; Yang, Y.; Dombkowski, D.; Sykes, M.; Scadden, D.T. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science 2000, 287, 1804–1808. [Google Scholar] [CrossRef] [PubMed]
- Rampioni Vinciguerra, G.L.; Sonego, M.; Segatto, I.; Dall’Acqua, A.; Vecchione, A.; Baldassarre, G.; Belletti, B. CDK4/6 Inhibitors in Combination Therapies: Better in Company Than Alone: A Mini Review. Front. Oncol. 2022, 12, 891580. [Google Scholar] [CrossRef]
- Kurppa, K.J.; Liu, Y.; To, C.; Zhang, T.; Fan, M.; Vajdi, A.; Knelson, E.H.; Xie, Y.; Lim, K.; Cejas, P.; et al. Treatment-Induced Tumor Dormancy through YAP-Mediated Transcriptional Reprogramming of the Apoptotic Pathway. Cancer Cell 2020, 37, 104–122.e2. [Google Scholar] [CrossRef] [PubMed]
- Mai, Y.; Su, J.; Yang, C.; Xia, C.; Fu, L. The strategies to cure cancer patients by eradicating cancer stem-like cells. Mol. Cancer 2023, 22, 171. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; Chan, D.K.; Shank, J.J.; Griffith, K.A.; Fan, H.; Szulawski, R.; Yang, K.; Reynolds, R.K.; Johnston, C.; McLean, K.; et al. Phase II clinical trial of metformin as a cancer stem cell-targeting agent in ovarian cancer. JCI Insight 2020, 5, e133247. [Google Scholar] [CrossRef]
- Yap, T.A.; Daver, N.; Mahendra, M.; Zhang, J.; Kamiya-Matsuoka, C.; Meric-Bernstam, F.; Kantarjian, H.M.; Ravandi, F.; Collins, M.E.; Francesco, M.E.D.; et al. Complex I inhibitor of oxidative phosphorylation in advanced solid tumors and acute myeloid leukemia: Phase I trials. Nat. Med. 2023, 29, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.H.; Choi, D.S.; Ensor, J.E.; Kaipparettu, B.A.; Bass, B.L.; Chang, J.C. The autophagy inhibitor chloroquine targets cancer stem cells in triple negative breast cancer by inducing mitochondrial damage and impairing DNA break repair. Cancer Lett. 2016, 376, 249–258. [Google Scholar] [CrossRef]
- Marsh, T.; Tolani, B.; Debnath, J. The pleiotropic functions of autophagy in metastasis. J. Cell Sci. 2021, 134, jcs247056. [Google Scholar] [CrossRef]
- Abbott, A. Stem cells head to the clinic: Treatments for cancer, diabetes and Parkinson’s disease could soon be here. Nature 2025, 637, 18–20. [Google Scholar] [CrossRef]
- Zhang, C.L.; Huang, T.; Wu, B.L.; He, W.X.; Liu, D. Stem cells in cancer therapy: Opportunities and challenges. Oncotarget 2017, 8, 75756–75766. [Google Scholar] [CrossRef]
- Lee, J.Y.; Hong, S.H. Hematopoietic Stem Cells and Their Roles in Tissue Regeneration. Int. J. Stem Cells 2020, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Arwert, E.N.; Hoste, E.; Watt, F.M. Epithelial stem cells, wound healing and cancer. Nat. Rev. Cancer 2012, 12, 170–180. [Google Scholar] [CrossRef]
- Horsley, V. Skin in the Game: Stem Cells in Repair, Cancer, and Homeostasis. Cell 2020, 181, 492–494. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Deng, P.; Li, L.; He, Y.; Wang, J.; Hao, D.; Yang, H. Advances in genetically modified neural stem cell therapy for central nervous system injury and neurological diseases. Stem Cell Res. Ther. 2024, 15, 482. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Y.; Gou, W.; Lu, Q.; Peng, J.; Lu, S. Role of mesenchymal stem cells in bone regeneration and fracture repair: A review. Int. Orthop. 2013, 37, 2491–2498. [Google Scholar] [CrossRef] [PubMed]
- Li, T.T.; Wang, Z.R.; Yao, W.Q.; Linghu, E.Q.; Wang, F.S.; Shi, L. Stem Cell Therapies for Chronic Liver Diseases: Progress and Challenges. Stem Cells Transl. Med. 2022, 11, 900–911. [Google Scholar] [CrossRef] [PubMed]
- King, A.; Balaji, S.; Keswani, S.G.; Crombleholme, T.M. The Role of Stem Cells in Wound Angiogenesis. Adv. Wound Care 2014, 3, 614–625. [Google Scholar] [CrossRef]
- Chu, D.T.; Nguyen, T.T.; Tien, N.L.B.; Tran, D.K.; Jeong, J.H.; Anh, P.G.; Thanh, V.V.; Truong, D.T.; Dinh, T.C. Recent Progress of Stem Cell Therapy in Cancer Treatment: Molecular Mechanisms and Potential Applications. Cells 2020, 9, 563. [Google Scholar] [CrossRef]
- Stuckey, D.W.; Shah, K. Stem cell-based therapies for cancer treatment: Separating hope from hype. Nat. Rev. Cancer 2014, 14, 683–691. [Google Scholar] [CrossRef]
- Enow, J.A.; Sheikh, H.I.; Rahman, M.M. Tumor Tropism of DNA Viruses for Oncolytic Virotherapy. Viruses 2023, 15, 2262. [Google Scholar] [CrossRef]
- Labusca, L.; Herea, D.D.; Mashayekhi, K. Stem cells as delivery vehicles for regenerative medicine-challenges and perspectives. World J. Stem Cells 2018, 10, 43–56. [Google Scholar] [CrossRef]
- Joshi, S.; Allabun, S.; Ojo, S.; Alqahtani, M.S.; Shukla, P.K.; Abbas, M.; Wechtaisong, C.; Almohiy, H.M. Enhanced Drug Delivery System Using Mesenchymal Stem Cells and Membrane-Coated Nanoparticles. Molecules 2023, 28, 2130. [Google Scholar] [CrossRef] [PubMed]
- Minev, T.; Balbuena, S.; Gill, J.M.; Marincola, F.M.; Kesari, S.; Lin, F. Mesenchymal stem cells—The secret agents of cancer immunotherapy: Promises, challenges, and surprising twists. Oncotarget 2024, 15, 793–805. [Google Scholar] [CrossRef] [PubMed]

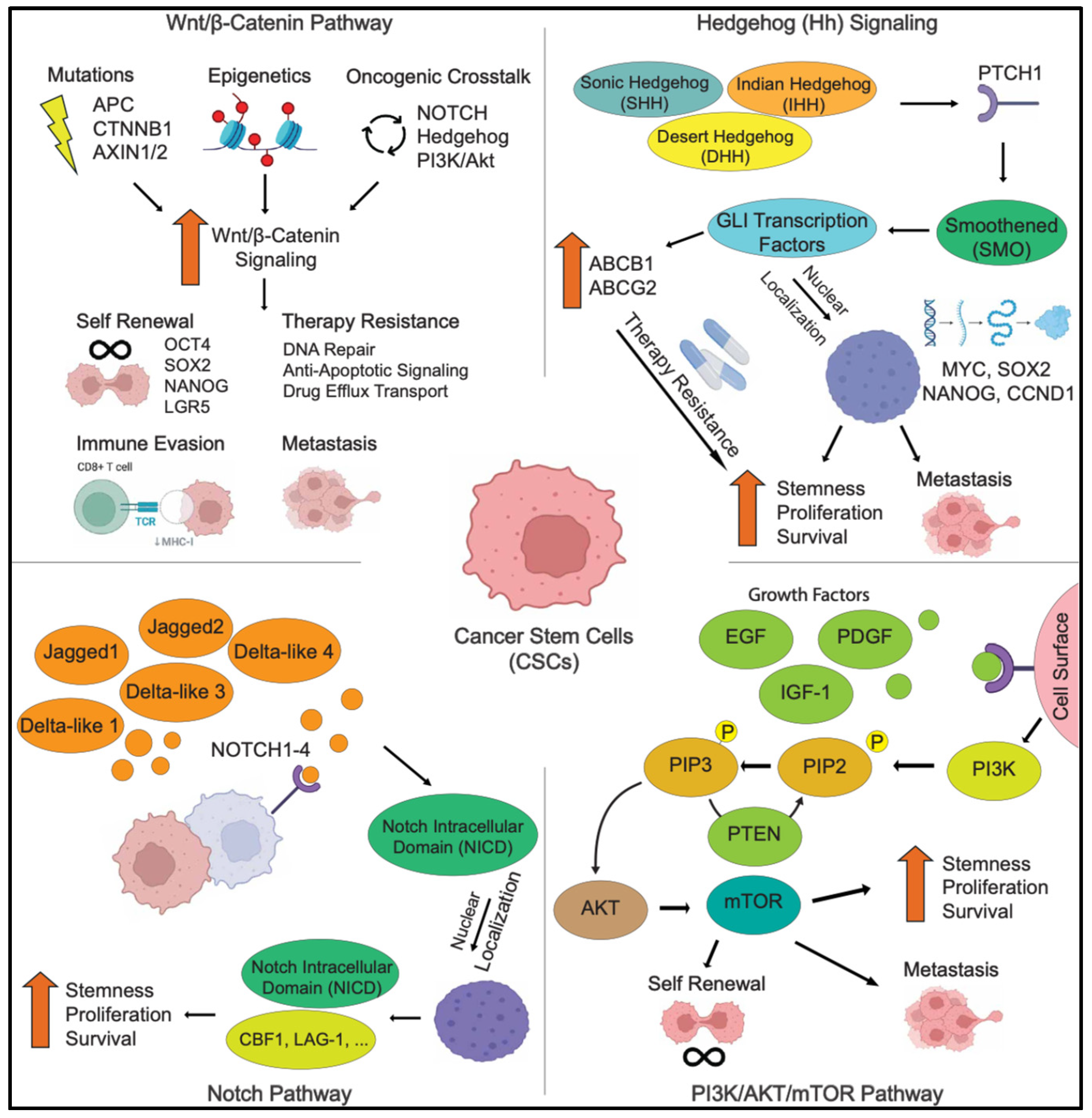
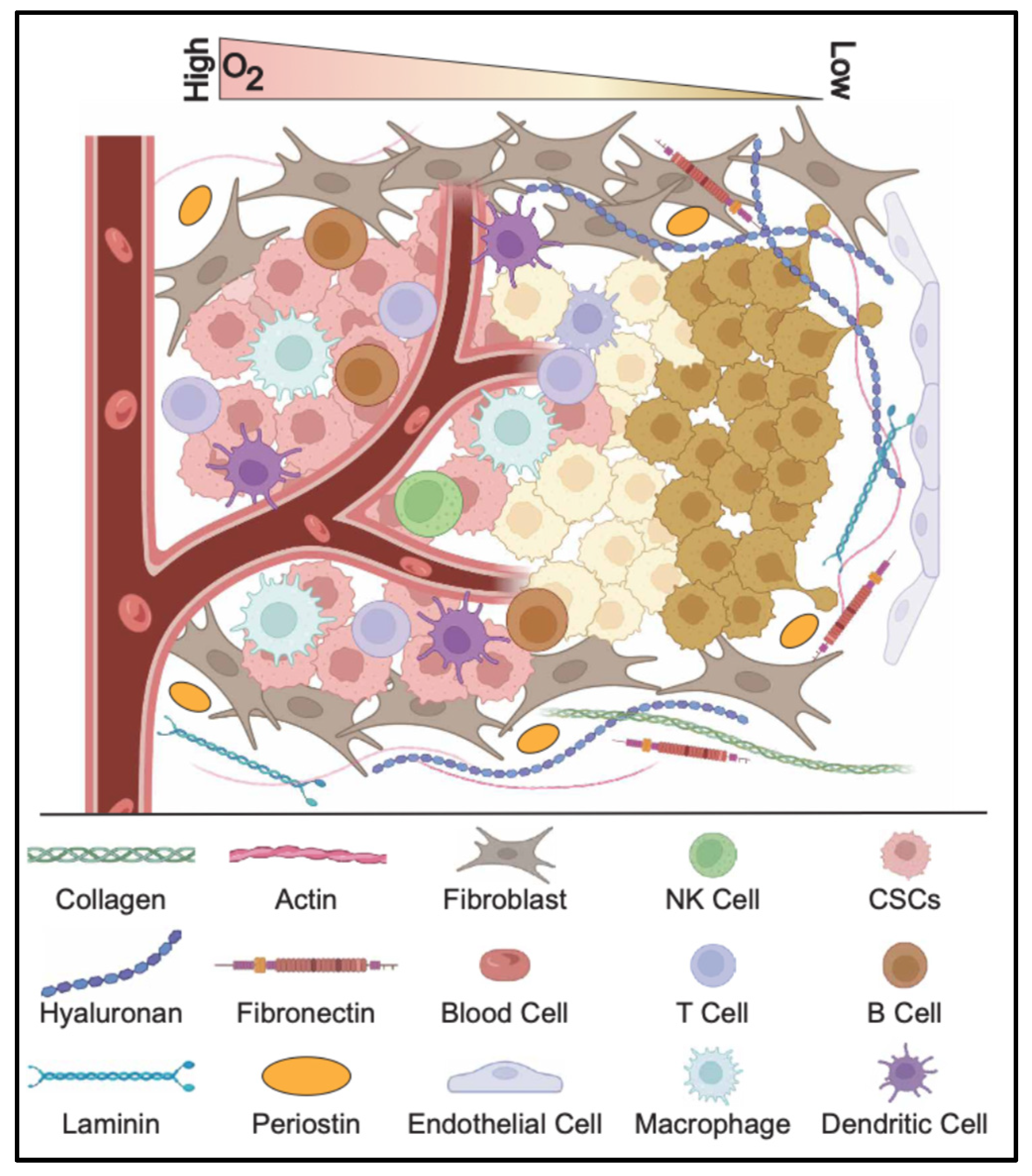


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Key Components | Functions in CSCs | Therapeutic Strategies |
|---|---|---|---|
| WNT/β-CATENIN | Wnt ligands Frizzled (FZD) receptors LRP5/6 β-catenin TCF/LEF | Maintains CSC self-renewal Promotes therapy resistance Enhances EMT and metastasis | Porcupine inhibitors (LGK974, ETC-159) β-catenin inhibitors (PRI-724, ICG-001) Frizzled receptor blockers (OMP-18R5, vantictumab) |
| HEDGEHOG (HH) | SHH/IHH/DHH ligands Patched (PTCH) Smoothened (SMO) GLI1/2 | Regulates CSC survival Enhances drug resistance and EMT Drives tumor progression | SMO inhibitors (Vismodegib, Sonidegib, Glasdegib) GLI inhibitors (GANT61, Arsenic Trioxide) Hh ligand inhibitors (5E1, Robotnikinin) |
| NOTCH | Notch1-4 receptors Jagged/Delta ligands NICD (Notch Intracellular Domain) | Maintains CSC populations Increases chemoresistance Promotes tumor angiogenesis and immune evasion | γ-secretase inhibitors (DAPT, MK-0752, RO4929097) Notch ligand inhibitors (Demcizumab, Tarextumab) Notch transcription inhibitors (CB-103, SAHM1) |
| PI3K/AKT/MTOR | PI3K AKT mTORC1/mTORC2 PTEN | Regulates CSC metabolism and survival Enhances drug resistance via ABC transporters Promotes tumor invasion and metastasis | PI3K inhibitors (Alpelisib, Buparlisib, PX-866) AKT inhibitors (Capivasertib, MK-2206, Ipatasertib) mTOR inhibitors (Rapamycin, Everolimus, AZD8055) |
| TGF-β | TGF-β ligands TGF-β receptors SMAD2/3/4 | Drives EMT and metastasis Induces therapy resistance and immune suppression | TGF-β inhibitors (Galunisertib, Fresolimumab) SMAD inhibitors |
| JAK/STAT | IL-6 JAK1/2 STAT3/5 | Promotes inflammation-induced CSC expansion Enhances immune evasion | JAK inhibitors (Ruxolitinib, Tofacitinib) STAT3 inhibitors (WP1066, Stattic) |
| HIPPO/YAP | MST1/2 LATS1/2 YAP/TAZ | Enhances CSC self-renewal Regulates stemness and drug resistance | YAP inhibitors (Verteporfin, CA3) |
| NF-KB | IKK IκB p65/RelA | Enhances CSC survival and immune evasion | IKK inhibitors (Bortezomib, Bay 11-7082) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haddadin, L.; Sun, X. Stem Cells in Cancer: From Mechanisms to Therapeutic Strategies. Cells 2025, 14, 538. https://doi.org/10.3390/cells14070538
Haddadin L, Sun X. Stem Cells in Cancer: From Mechanisms to Therapeutic Strategies. Cells. 2025; 14(7):538. https://doi.org/10.3390/cells14070538
Chicago/Turabian StyleHaddadin, Laurence, and Xueqin Sun. 2025. "Stem Cells in Cancer: From Mechanisms to Therapeutic Strategies" Cells 14, no. 7: 538. https://doi.org/10.3390/cells14070538
APA StyleHaddadin, L., & Sun, X. (2025). Stem Cells in Cancer: From Mechanisms to Therapeutic Strategies. Cells, 14(7), 538. https://doi.org/10.3390/cells14070538