_Wang.png)


Combining AdipoRon with Paclitaxel Unveils Synergistic Potential in Non-Small Cell Lung Cancer Cells via AMPK-ERK1/2 Signaling

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Drug Preparation
2.2. Chemical Reagents
2.3. Cell Proliferation Assay
2.4. PI Exclusion Assay
2.5. Cell Cycle Analysis
2.6. Colony Forming Assay
2.7. Cell Viability
2.8. Multi-Drug Synergy Analysis
2.9. Protein Extraction and Quantification
2.10. Immunoblotting
2.11. Statistical Analysis
3. Results
3.1. Combination of AdipoRon Plus Paclitaxel Enhances Cell Growth Inhibition and Impairs Clonogenic Potential in NSCLC Cells
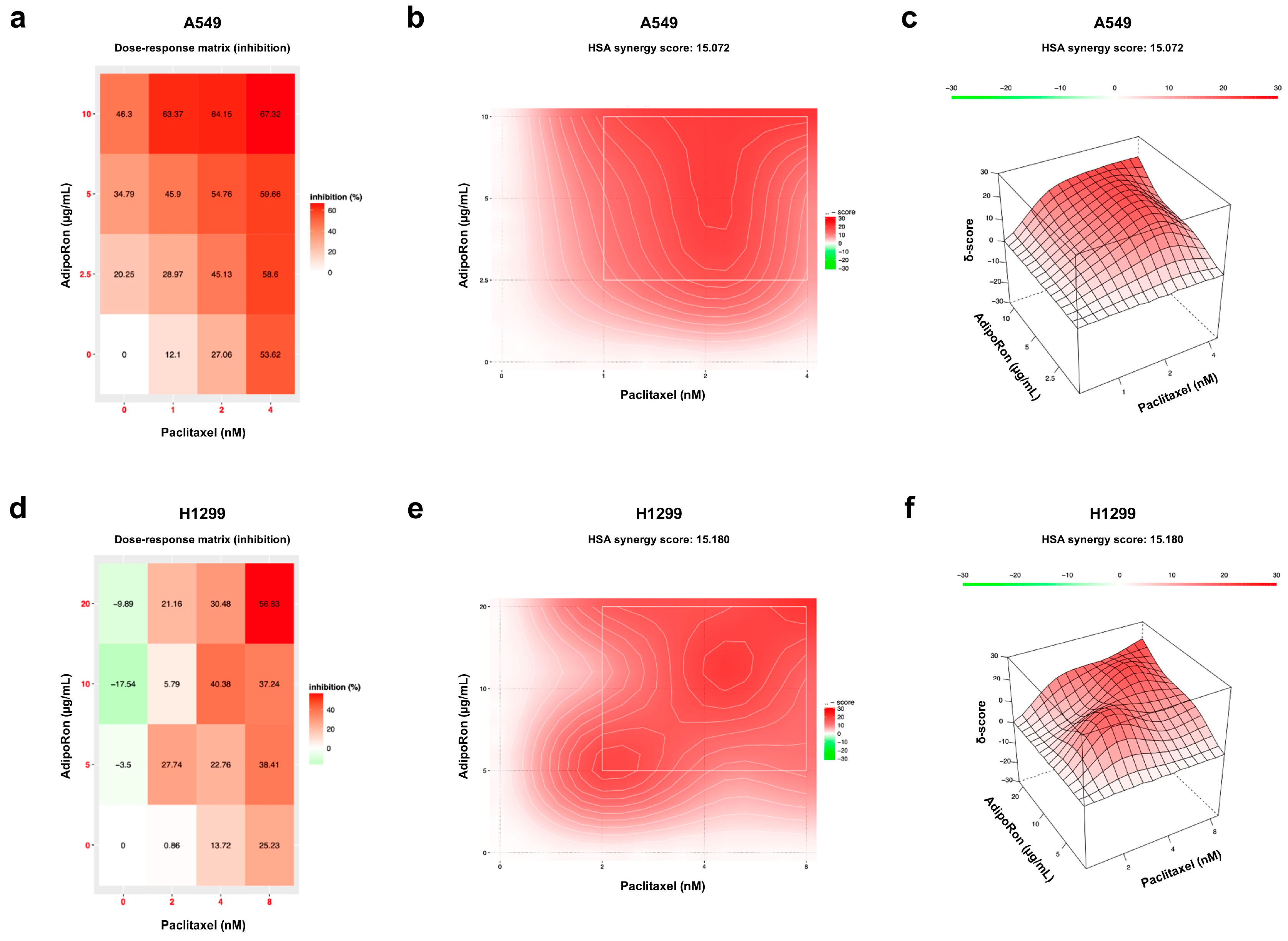
3.2. Combination of AdipoRon Plus Paclitaxel Displays Synergic Interplay in NSCLC Cells
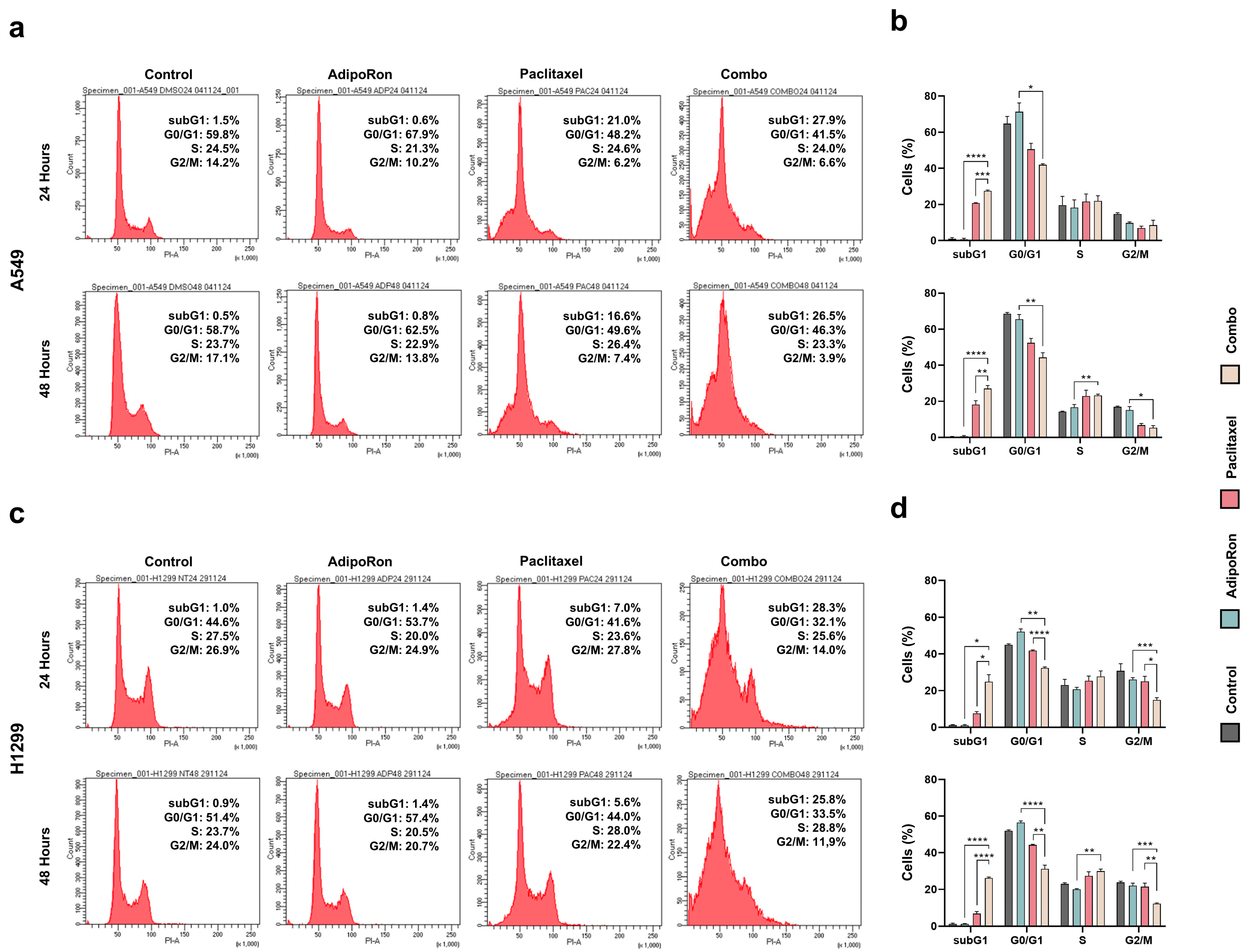
3.3. Combination of AdipoRon Plus Paclitaxel Affects Cell Cycle Progression in NSCLC Cells
3.4. Combination of AdipoRon Plus Paclitaxel Improves Chemotherapy-Induced Apoptosis in NSCLC Cells
3.5. Combination of AdipoRon Plus Paclitaxel Induces AMPK Activation in NSCLC Cells
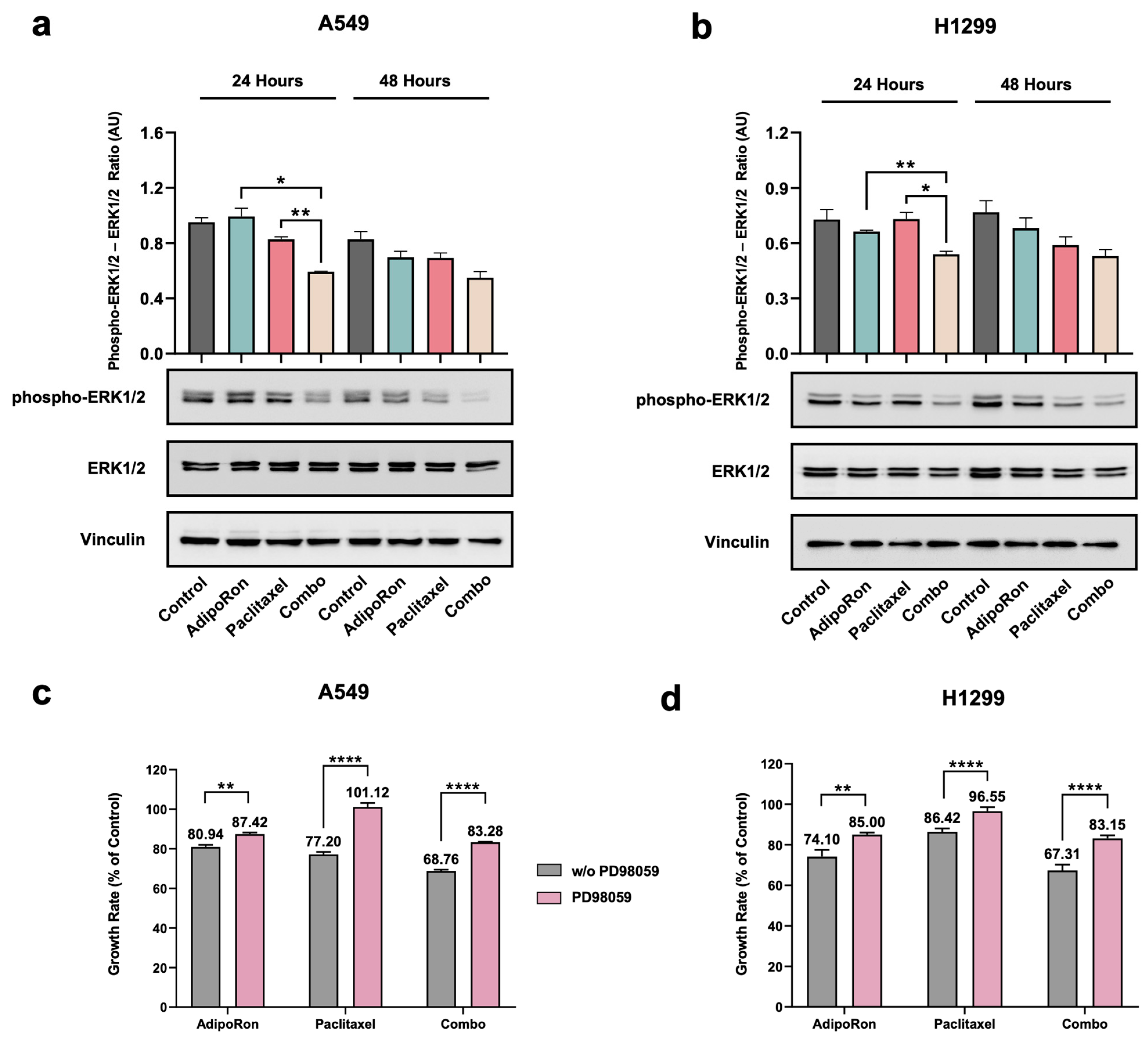
3.6. Combination of AdipoRon Plus Paclitaxel Downregulates ERK1/2 in NSCLC Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| NSCLC | Non-Small Cell Lung Cancer |
| AMPK | AMP-activated protein Kinase |
| ERK1/2 | Extracellular Signal-Regulated Kinase 1/2 |
| FDA | Food and Drug Administration |
| PDAC | Pancreatic Ductal Adenocarcinoma |
| MAPK | Mitogen-Activated Protein Kinases |
| mTOR | Mammalian Target Of Rapamycin |
| NF-κB | Nuclear Factor Kappa-light-chain-enhancer of activated B cells |
| RIPKs | Receptor-Interacting Protein Kinase Family Members |
| ULK1 | Unc-51-Like autophagy-activating Kinase |
| PI | Propidium iodide |
| CFA | Colony Formation Assay |
| HAS | Highest Single Agent |
| PARP | Poly ADP Ribose Polymerase |
| MEK1/2 | Mitogen-Activated Protein Kinase Kinase-MAPKK |
| PI3K | Phosphatidylinositol 3-Kinase |
| AKT | Protein Kinase B |
References
- Raskova Kafkova, L.; Mierzwicka, J.M.; Chakraborty, P.; Jakubec, P.; Fischer, O.; Skarda, J.; Maly, P.; Raska, M. NSCLC: From tumorigenesis, immune checkpoint misuse to current and future targeted therapy. Front. Immunol. 2024, 15, 1342086. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.L.; Fitzgerald, B.G.; Paz-Ares, L.; Cappuzzo, F.; Janne, P.A.; Peters, S.; Hirsch, F.R. New promises and challenges in the treatment of advanced non-small-cell lung cancer. Lancet 2024, 404, 803–822. [Google Scholar] [CrossRef]
- Mountzios, G.; Naidoo, J.; Wang, C.; Creelan, B.C.; Trotier, D.C.; Campbell, T.C.; Peters, S. Beyond Chemoimmunotherapy in Advanced Non–Small Cell Lung Cancer: New Frontiers, New Challenges. Am. Soc. Clin. Oncol. Educ. Book 2024, 44, e432526. [Google Scholar] [CrossRef] [PubMed]
- Nawara, H.M.; Afify, S.M.; Hassan, G.; Zahra, M.H.; Seno, A.; Seno, M. Paclitaxel-Based Chemotherapy Targeting Cancer Stem Cells from Mono- to Combination Therapy. Biomedicines 2021, 9, 500. [Google Scholar] [CrossRef] [PubMed]
- Alalawy, A.I. Key genes and molecular mechanisms related to Paclitaxel Resistance. Cancer Cell Int. 2024, 24, 244. [Google Scholar] [CrossRef]
- Sousa-Pimenta, M.; Estevinho, L.M.; Szopa, A.; Basit, M.; Khan, K.; Armaghan, M.; Ibrayeva, M.; Sonmez Gurer, E.; Calina, D.; Hano, C.; et al. Chemotherapeutic properties and side-effects associated with the clinical practice of terpene alkaloids: Paclitaxel, docetaxel, and cabazitaxel. Front. Pharmacol. 2023, 14, 1157306. [Google Scholar] [CrossRef]
- Khing, T.M.; Choi, W.S.; Kim, D.M.; Po, W.W.; Thein, W.; Shin, C.Y.; Sohn, U.D. The effect of paclitaxel on apoptosis, autophagy and mitotic catastrophe in AGS cells. Sci. Rep. 2021, 11, 23490. [Google Scholar] [CrossRef]
- Kogure, Y.; Iwasawa, S.; Saka, H.; Hamamoto, Y.; Kada, A.; Hashimoto, H.; Atagi, S.; Takiguchi, Y.; Ebi, N.; Inoue, A.; et al. Efficacy and safety of carboplatin with nab-paclitaxel versus docetaxel in older patients with squamous non-small-cell lung cancer (CAPITAL): A randomised, multicentre, open-label, phase 3 trial. Lancet Healthy Longev. 2021, 2, e791–e800. [Google Scholar] [CrossRef]
- Okada-Iwabu, M.; Yamauchi, T.; Iwabu, M.; Honma, T.; Hamagami, K.; Matsuda, K.; Yamaguchi, M.; Tanabe, H.; Kimura-Someya, T.; Shirouzu, M.; et al. A small-molecule AdipoR agonist for type 2 diabetes and short life in obesity. Nature 2013, 503, 493–499. [Google Scholar] [CrossRef]
- Barbalho, S.M.; Mendez-Sanchez, N.; Fornari Laurindo, L. AdipoRon and ADP355, adiponectin receptor agonists, in Metabolic-associated Fatty Liver Disease (MAFLD) and Nonalcoholic Steatohepatitis (NASH): A systematic review. Biochem. Pharmacol. 2023, 218, 115871. [Google Scholar] [CrossRef]
- Iwabu, M.; Okada-Iwabu, M.; Tanabe, H.; Ohuchi, N.; Miyata, K.; Kobori, T.; Odawara, S.; Kadowaki, Y.; Yokoyama, S.; Yamauchi, T.; et al. AdipoR agonist increases insulin sensitivity and exercise endurance in AdipoR-humanized mice. Commun. Biol. 2021, 4, 45. [Google Scholar] [CrossRef] [PubMed]
- Nigro, E.; Daniele, A.; Salzillo, A.; Ragone, A.; Naviglio, S.; Sapio, L. AdipoRon and Other Adiponectin Receptor Agonists as Potential Candidates in Cancer Treatments. Int. J. Mol. Sci. 2021, 22, 5569. [Google Scholar] [CrossRef] [PubMed]
- Akimoto, M.; Maruyama, R.; Kawabata, Y.; Tajima, Y.; Takenaga, K. Antidiabetic adiponectin receptor agonist AdipoRon suppresses tumour growth of pancreatic cancer by inducing RIPK1/ERK-dependent necroptosis. Cell Death Dis. 2018, 9, 804. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, E.; Kawahara, T.; Kinoshita, F.; Shiota, M.; Inokuchi, J.; Miyamoto, H.; Eto, M. The Role of Adipocytokines and their Receptors in Prostate Cancer: Adiponectin May Protect Against Progression. Anticancer Res. 2024, 44, 1369–1376. [Google Scholar] [CrossRef]
- Li, C.; Zhang, J.; Dionigi, G.; Liang, N.; Guan, H.; Sun, H. Uncovering the connection between obesity and thyroid cancer: The therapeutic potential of adiponectin receptor agonist in the AdipoR2-ULK axis. Cell Death Dis. 2024, 15, 708. [Google Scholar] [CrossRef]
- Messaggio, F.; Mendonsa, A.M.; Castellanos, J.; Nagathihalli, N.S.; Gorden, L.; Merchant, N.B.; VanSaun, M.N. Adiponectin receptor agonists inhibit leptin induced pSTAT3 and in vivo pancreatic tumor growth. Oncotarget 2017, 8, 85378–85391. [Google Scholar] [CrossRef]
- Sapio, L.; Nigro, E.; Ragone, A.; Salzillo, A.; Illiano, M.; Spina, A.; Polito, R.; Daniele, A.; Naviglio, S. AdipoRon Affects Cell Cycle Progression and Inhibits Proliferation in Human Osteosarcoma Cells. J. Oncol. 2020, 2020, 7262479. [Google Scholar] [CrossRef]
- Manley, S.J.; Olou, A.A.; Jack, J.L.; Ruckert, M.T.; Walsh, R.M.; Eades, A.E.; Bye, B.A.; Ambrose, J.; Messaggio, F.; Anant, S.; et al. Synthetic adiponectin-receptor agonist, AdipoRon, induces glycolytic dependence in pancreatic cancer cells. Cell Death Dis. 2022, 13, 114. [Google Scholar] [CrossRef]
- Sapio, L.; Ragone, A.; Spina, A.; Salzillo, A.; Naviglio, S. AdipoRon and Pancreatic Ductal Adenocarcinoma: A future perspective in overcoming chemotherapy-induced resistance? Cancer Drug Resist. 2022, 5, 625–636. [Google Scholar] [CrossRef]
- Ragone, A.; Salzillo, A.; Spina, A.; Naviglio, S.; Sapio, L. Integrating Gemcitabine-Based Therapy With AdipoRon Enhances Growth Inhibition in Human PDAC Cell Lines. Front. Pharmacol. 2022, 13, 837503. [Google Scholar] [CrossRef] [PubMed]
- Kafeel, S.; Ragone, A.; Salzillo, A.; Palmiero, G.; Naviglio, S.; Sapio, L. Adiponectin Receptor Agonist AdipoRon Inhibits Proliferation and Drives Glycolytic Dependence in Non-Small-Cell Lung Cancer Cells. Cancers 2024, 16, 2633. [Google Scholar] [CrossRef] [PubMed]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef]
- Crowley, L.C.; Scott, A.P.; Marfell, B.J.; Boughaba, J.A.; Chojnowski, G.; Waterhouse, N.J. Measuring Cell Death by Propidium Iodide Uptake and Flow Cytometry. Cold Spring Harb. Protoc. 2016, 2016, pdb-prot087163. [Google Scholar] [CrossRef] [PubMed]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 3.0: An interactive analysis and consensus interpretation of multi-drug synergies across multiple samples. Nucleic Acids Res. 2022, 50, W739–W743. [Google Scholar] [CrossRef]
- Hertz, D.L.; Joerger, M.; Bang, Y.J.; Mathijssen, R.H.; Zhou, C.; Zhang, L.; Gandara, D.; Stahl, M.; Monk, B.J.; Jaehde, U.; et al. Paclitaxel therapeutic drug monitoring—International association of therapeutic drug monitoring and clinical toxicology recommendations. Eur. J. Cancer 2024, 202, 114024. [Google Scholar] [CrossRef]
- Ramzan, A.A.; Bitler, B.G.; Hicks, D.; Barner, K.; Qamar, L.; Behbakht, K.; Powell, T.; Jansson, T.; Wilson, H. Adiponectin receptor agonist AdipoRon induces apoptotic cell death and suppresses proliferation in human ovarian cancer cells. Mol. Cell. Biochem. 2019, 461, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Salzillo, A.; Ragone, A.; Spina, A.; Naviglio, S.; Sapio, L. Forskolin affects proliferation, migration and Paclitaxel-mediated cytotoxicity in non-small-cell lung cancer cell lines via adenylyl cyclase/cAMP axis. Eur. J. Cell Biol. 2023, 102, 151292. [Google Scholar] [CrossRef]
- Rajendran, V.; Jain, M.V. In Vitro Tumorigenic Assay: Colony Forming Assay for Cancer Stem Cells. Methods Mol. Biol. 2018, 1692, 89–95. [Google Scholar] [CrossRef]
- Biswal, S.; Mallick, B. Unlocking the potential of signature-based drug repurposing for anticancer drug discovery. Arch. Biochem. Biophys. 2024, 761, 110150. [Google Scholar] [CrossRef]
- Giri, A.K.; Ianevski, A. High-throughput screening for drug discovery targeting the cancer cell-microenvironment interactions in hematological cancers. Expert Opin. Drug Discov. 2021, 17, 181–190. [Google Scholar] [CrossRef]
- Duarte, D.; Vale, N. Evaluation of synergism in drug combinations and reference models for future orientations in oncology. Curr. Res. Pharmacol. Drug Discov. 2022, 3, 100110. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, M.; Ahmad, R.; Tantry, I.Q.; Ahmad, W.; Siddiqui, S.; Alam, M.; Abbas, K.; Moinuddin; Hassan, M.I.; Habib, S.; et al. Apoptosis: A Comprehensive Overview of Signaling Pathways, Morphological Changes, and Physiological Significance and Therapeutic Implications. Cells 2024, 13, 1838. [Google Scholar] [CrossRef]
- Zhao, S.; Tang, Y.; Wang, R.; Najafi, M. Mechanisms of cancer cell death induction by paclitaxel: An updated review. Apoptosis 2022, 27, 647–667. [Google Scholar] [CrossRef] [PubMed]
- Sadria, M.; Seo, D.; Layton, A.T. The mixed blessing of AMPK signaling in Cancer treatments. BMC Cancer 2022, 22, 105. [Google Scholar] [CrossRef]
- Jiang, S.; Luo, Y.; Zhan, Z.; Tang, Z.; Zou, J.; Ying, Y.; Lin, H.; Huang, D.; Luo, L. AMP-activated protein kinase re-sensitizes A549 to paclitaxel via up-regulating solute carrier organic anion transporter family member 1B3 expression. Cell. Signal. 2022, 91, 110215. [Google Scholar] [CrossRef]
- Liu, G.; Yang, D.; Meng, J.; He, Q.; Wu, D. The combination of gemcitabine and albumin-bound paclitaxel effectively inhibits de novo lipogenesis in pancreatic cancer cells by targeting the AMPK/SREBP1 pathway. Biochem. Pharmacol. 2025, 232, 116721. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Zhang, Y.; Yu, Z.; Luo, Z. Metformin Suppresses Stemness of Non-Small-Cell Lung Cancer Induced by Paclitaxel through FOXO3a. Int. J. Mol. Sci. 2023, 24, 16611. [Google Scholar] [CrossRef]
- Song, Y.; Bi, Z.; Liu, Y.; Qin, F.; Wei, Y.; Wei, X. Targeting RAS-RAF-MEK-ERK signaling pathway in human cancer: Current status in clinical trials. Genes Dis. 2023, 10, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, R.; Satoh, R.; Takasaki, T. ERK: A Double-Edged Sword in Cancer. ERK-Dependent Apoptosis as a Potential Therapeutic Strategy for Cancer. Cells 2021, 10, 2509. [Google Scholar] [CrossRef]
- Huang, C.; Zhang, X.; Jiang, L.; Zhang, L.; Xiang, M.; Ren, H. FoxM1 Induced Paclitaxel Resistance via Activation of the FoxM1/PHB1/RAF-MEK-ERK Pathway and Enhancement of the ABCA2 Transporter. Mol. Ther. Oncolytics 2019, 14, 196–212. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, R.; Qin, X.; Mao, X.; Li, Q.; Fang, S. Cobimetinib Sensitizes Cervical Cancer to Paclitaxel via Suppressing Paclitaxel-Induced ERK Activation. Pharmacology 2022, 107, 398–405. [Google Scholar] [CrossRef]
- Yu-Wei, D.; Li, Z.S.; Xiong, S.M.; Huang, G.; Luo, Y.F.; Huo, T.Y.; Zhou, M.H.; Zheng, Y.W. Paclitaxel induces apoptosis through the TAK1–JNK activation pathway. FEBS Open Bio 2020, 10, 1655–1667. [Google Scholar] [CrossRef] [PubMed]
- Hwangbo, H.; Patterson, S.C.; Dai, A.; Plana, D.; Palmer, A.C. Additivity predicts the efficacy of most approved combination therapies for advanced cancer. Nature Cancer 2023, 4, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Avila, A.; Gollahon, L. Paclitaxel Induces Apoptosis in Breast Cancer Cells through Different Calcium—Regulating Mechanisms Depending on External Calcium Conditions. Int. J. Mol. Sci. 2014, 15, 2672–2694. [Google Scholar] [CrossRef]
- Peng, Z.G.; Liu, D.C.; Yao, Y.B.; Feng, X.L.; Huang, X.; Tang, Y.L.; Yang, J.; Wang, X.X. Paclitaxel induces apoptosis in leukemia cells through a JNK activation-dependent pathway. Genet. Mol. Res. 2016, 15, 15013904. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tang, Y.; Tang, X.; Wang, Y.; Zhang, Z.; Yang, H. Paclitaxel Induces the Apoptosis of Prostate Cancer Cells via ROS-Mediated HIF-1alpha Expression. Molecules 2022, 27, 7183. [Google Scholar] [CrossRef]
- Huang, H.; Kung, F.-L.; Huang, Y.-W.; Hsu, C.-C.; Guh, J.-H.; Hsu, L.-C. Sensitization of cancer cells to paclitaxel-induced apoptosis by canagliflozin. Biochem. Pharmacol. 2024, 223, 116140. [Google Scholar] [CrossRef]
- Min, J.; Shen, H.; Xi, W.; Wang, Q.; Yin, L.; Zhang, Y.; Yu, Y.; Yang, Q.; Wang, Z.N. Synergistic Anticancer Activity of Combined Use of Caffeic Acid with Paclitaxel Enhances Apoptosis of Non-Small-Cell Lung Cancer H1299 Cells in Vivo and in Vitro. Cell. Physiol. Biochem. 2018, 48, 1433–1442. [Google Scholar] [CrossRef]
- Gielecińska, A.; Kciuk, M.; Yahya, E.B.; Ainane, T.; Mujwar, S.; Kontek, R. Apoptosis, necroptosis, and pyroptosis as alternative cell death pathways induced by chemotherapeutic agents? Biochim. Biophys. Acta (BBA) Rev. Cancer 2023, 1878, 189024. [Google Scholar] [CrossRef]
- Lim, P.T.; Goh, B.H.; Lee, W.-L. Taxol: Mechanisms of action against cancer, an update with current research. In Paclitaxel; Academic Press: Cambridge, MA, USA, 2022; pp. 47–71. [Google Scholar]
- Fisi, V.; Kátai, E.; Bogner, P.; Miseta, A.; Nagy, T. Timed, sequential administration of paclitaxel improves its cytotoxic effectiveness in a cell culture model. Cell Cycle 2016, 15, 1227–1233. [Google Scholar] [CrossRef]
- Obiorah, I.; Sengupta, S.; Fan, P.; Jordan, V.C. Delayed triggering of oestrogen induced apoptosis that contrasts with rapid paclitaxel-induced breast cancer cell death. Br. J. Cancer 2016, 115, e10. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Chen, D.; Zhang, W.; Zhao, J.; Zhi, L.; Huang, F.; Ji, H.; Zhang, J.; Liu, H.; Zou, L.; et al. Modulation of alternative splicing induced by paclitaxel in human lung cancer. Cell Death Dis. 2018, 9, 491. [Google Scholar] [CrossRef] [PubMed]
- Lv, C.; Qu, H.; Zhu, W.; Xu, K.; Xu, A.; Jia, B.; Qing, Y.; Li, H.; Wei, H.-J.; Zhao, H.-Y. Low-Dose Paclitaxel Inhibits Tumor Cell Growth by Regulating Glutaminolysis in Colorectal Carcinoma Cells. Front. Pharmacol. 2017, 8, 244. [Google Scholar] [CrossRef]
- Dziadyk, J.M.; Sui, M.H.; Zhu, X.M.; Fan, W.M. Paclitaxel-induced apoptosis may occur without a prior G/M-phase arrest. Anticancer Res. 2004, 24, 27–36. [Google Scholar] [PubMed]
- Nong, H.; Song, X.; Li, Y.; Xu, Y.; Wang, F.; Wang, Y.; Zhang, J.; Chen, C.; Li, J. AdipoRon reduces cisplatin-induced ototoxicity in hair cells:possible relation to the regulation of mitochondrial biogenesis. Neurosci. Lett. 2024, 819, 137577. [Google Scholar] [CrossRef]
- Salmons, H.I.; Gow, C.; Limberg, A.K.; Bettencourt, J.W.; Carstens, M.F.; Payne, A.N.; Morrey, M.E.; Sanchez-Sotelo, J.; Berry, D.J.; Dudakovic, A.; et al. The Safety of Adiponectin Receptor Agonist AdipoRon in a Rabbit Model of Arthrofibrosis. Tissue Eng. Part C Methods 2023, 29, 154–159. [Google Scholar] [CrossRef]
- Sibaud, V.; Lebœuf, N.R.; Roche, H.; Belum, V.R.; Gladieff, L.; Deslandres, M.; Montastruc, M.; Eche, A.; Vigarios, E.; Dalenc, F.; et al. Dermatological adverse events with taxane chemotherapy. Eur. J. Dermatol. 2016, 26, 427–443. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, J.O.; Kim, N.; Lee, H.J.; Lee, Y.W.; Kim, H.I.; Kim, S.J.; Park, S.H.; Kim, H.S. Paclitaxel suppresses the viability of breast tumor MCF7 cells through the regulation of EF1α and FOXO3a by AMPK signaling. Int. J. Oncol. 2015, 47, 1874–1880. [Google Scholar] [CrossRef]
- McNamee, N.; Rajagopalan, P.; Tal-Mason, A.; Roytburd, S.; Sachdeva, U.M. AMPK Activation Serves as a Common Pro-Survival Pathway in Esophageal Adenocarcinoma Cells. Biomolecules 2024, 14, 1115. [Google Scholar] [CrossRef]
- Mekkawy, A.I.; Naguib, Y.W.; Alhaj-Suliman, S.O.; Wafa, E.I.; Ebeid, K.; Acri, T.; Salem, A.K. Paclitaxel anticancer activity is enhanced by the MEK 1/2 inhibitor PD98059 in vitro and by PD98059-loaded nanoparticles in BRAFV600E melanoma-bearing mice. Int. J. Pharm. 2021, 606, 120876. [Google Scholar] [CrossRef]
- Wiwatchaitawee, K.; Mekkawy, A.I.; Quarterman, J.C.; Naguib, Y.W.; Ebeid, K.; Geary, S.M.; Salem, A.K. The MEK 1/2 inhibitor PD98059 exhibits synergistic anti-endometrial cancer activity with paclitaxel in vitro and enhanced tissue distribution in vivo when formulated into PAMAM-coated PLGA-PEG nanoparticles. Drug Deliv. Transl. Res. 2021, 12, 1684–1696. [Google Scholar] [CrossRef] [PubMed]
- Okano, J.-i.; Nagahara, T.; Matsumoto, K.; Murawaki, Y. The growth inhibition of liver cancer cells by paclitaxel and the involvement of extracellular signal-regulated kinase and apoptosis. Oncol. Rep. 2007, 17, 1195–1200. [Google Scholar] [CrossRef] [PubMed]
- Brognard, J.; Dennis, P.A. Variable apoptotic response of NSCLC cells to inhibition of the MEK/ERK pathway by small molecules or dominant negative mutants. Cell Death Differ. 2002, 9, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Dong, X.; Yap, J.; Hu, J. The MAPK and AMPK signalings: Interplay and implication in targeted cancer therapy. J. Hematol. Oncol. 2020, 13, 113. [Google Scholar] [CrossRef]
- Kawashima, I.; Mitsumori, T.; Nozaki, Y.; Yamamoto, T.; Shobu-Sueki, Y.; Nakajima, K.; Kirito, K. Negative regulation of the LKB1/AMPK pathway by ERK in human acute myeloid leukemia cells. Exp. Hematol. 2015, 43, 524–533.e1. [Google Scholar] [CrossRef]
- Tong, S.; Zhou, T.; Meng, Y.; Xu, D.; Chen, J. AMPK decreases ERK1/2 activity and cancer cell sensitivity to nutrition deprivation by mediating a positive feedback loop involving eEF2K. Oncol. Lett. 2020, 20, 61–66. [Google Scholar] [CrossRef]
- Stulpinas, A.; Sereika, M.; Vitkeviciene, A.; Imbrasaite, A.; Krestnikova, N.; Kalvelyte, A.V. Crosstalk between protein kinases AKT and ERK1/2 in human lung tumor-derived cell models. Front. Oncol. 2022, 12, 1045521. [Google Scholar] [CrossRef]
- Zhao, Y.; Hu, X.; Liu, Y.; Dong, S.; Wen, Z.; He, W.; Zhang, S.; Huang, Q.; Shi, M. ROS signaling under metabolic stress: Cross-talk between AMPK and AKT pathway. Mol. Cancer 2017, 16, 79. [Google Scholar] [CrossRef]
- Luce, A.; Lombardi, A.; Ferri, C.; Zappavigna, S.; Tathode, M.S.; Miles, A.K.; Boocock, D.J.; Vadakekolathu, J.; Bocchetti, M.; Alfano, R.; et al. A Proteomic Approach Reveals That miR-423-5p Modulates Glucidic and Amino Acid Metabolism in Prostate Cancer Cells. Int. J. Mol. Sci. 2022, 24, 617. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kafeel, S.; Palmiero, G.; Salzillo, A.; Ragone, A.; Naviglio, S.; Sapio, L. Combining AdipoRon with Paclitaxel Unveils Synergistic Potential in Non-Small Cell Lung Cancer Cells via AMPK-ERK1/2 Signaling. Cells 2025, 14, 602. https://doi.org/10.3390/cells14080602
Kafeel S, Palmiero G, Salzillo A, Ragone A, Naviglio S, Sapio L. Combining AdipoRon with Paclitaxel Unveils Synergistic Potential in Non-Small Cell Lung Cancer Cells via AMPK-ERK1/2 Signaling. Cells. 2025; 14(8):602. https://doi.org/10.3390/cells14080602
Chicago/Turabian StyleKafeel, Sanober, Giuseppina Palmiero, Alessia Salzillo, Angela Ragone, Silvio Naviglio, and Luigi Sapio. 2025. "Combining AdipoRon with Paclitaxel Unveils Synergistic Potential in Non-Small Cell Lung Cancer Cells via AMPK-ERK1/2 Signaling" Cells 14, no. 8: 602. https://doi.org/10.3390/cells14080602
APA StyleKafeel, S., Palmiero, G., Salzillo, A., Ragone, A., Naviglio, S., & Sapio, L. (2025). Combining AdipoRon with Paclitaxel Unveils Synergistic Potential in Non-Small Cell Lung Cancer Cells via AMPK-ERK1/2 Signaling. Cells, 14(8), 602. https://doi.org/10.3390/cells14080602