

Proportions of Basement Membrane Proteins in Cerebrovascular Smooth Muscle Cells After Exposure to Hypercapnia and Amyloid Beta

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
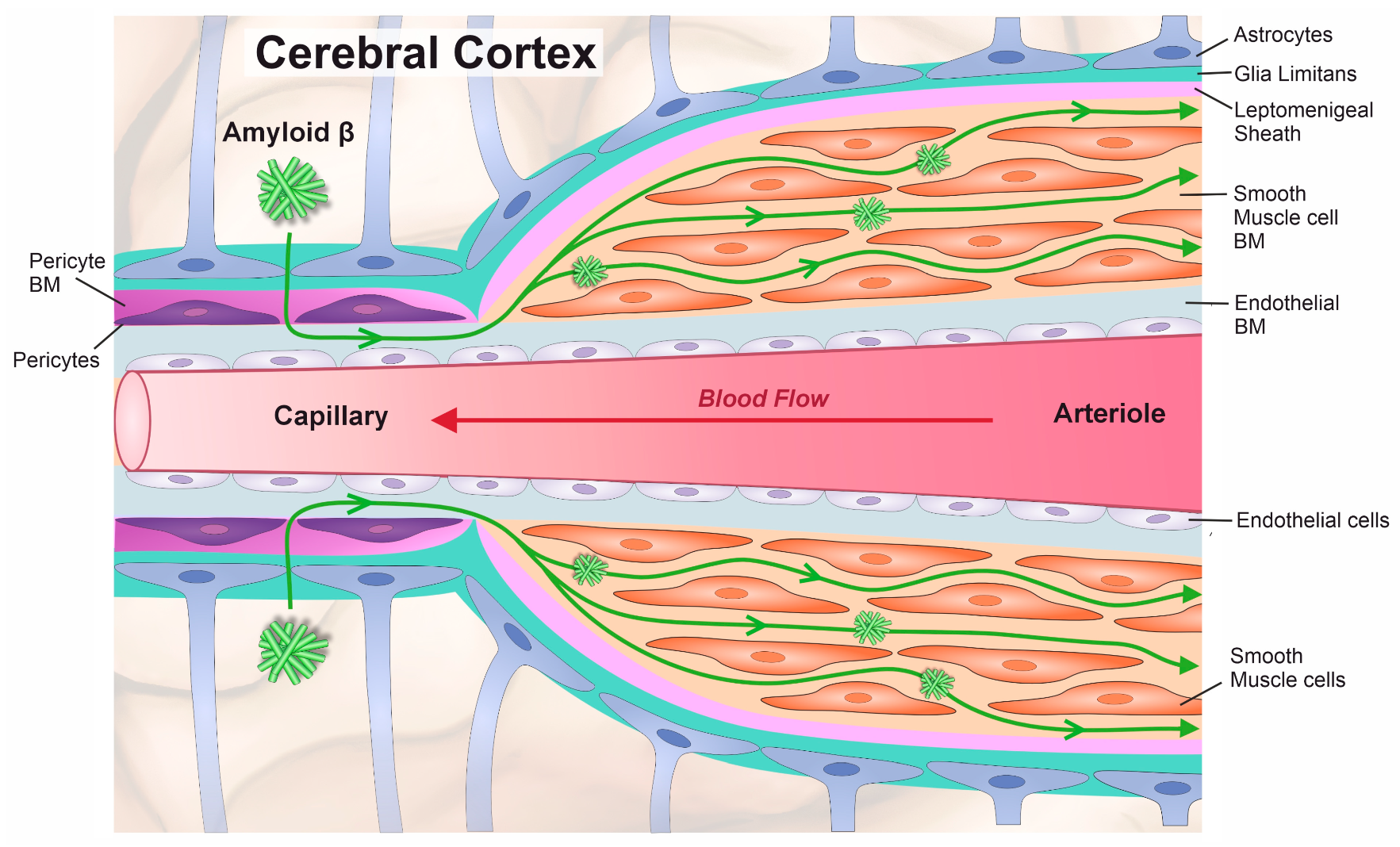
1. Introduction
2. Materials and Methods
2.1. Amyloid Beta Preparation
2.2. Cell Cultures
2.3. Immunocytochemistry for BM Components
2.4. Imaging and Analysis
2.5. MTS Assay
2.6. Data Analysis and Statistics
3. Results
3.1. Smooth Muscle Cells Produce the Highest Total Amount of Basement Membrane Proteins
3.2. Perlecan Levels Were Consistent Between Pericytes, Astrocytes, and Endothelial Cells
3.3. Pericytes and Astrocytes Express Similar Amounts of Laminin
3.4. Astrocytes and Endothelial Cells Express Similar Amounts of Fibronectin
3.5. Pericytes Expressed the Highest Amount of Collagen IV
3.6. Hypercapnia and Aβ Exposure Do Not Affect HBVSMC Viability
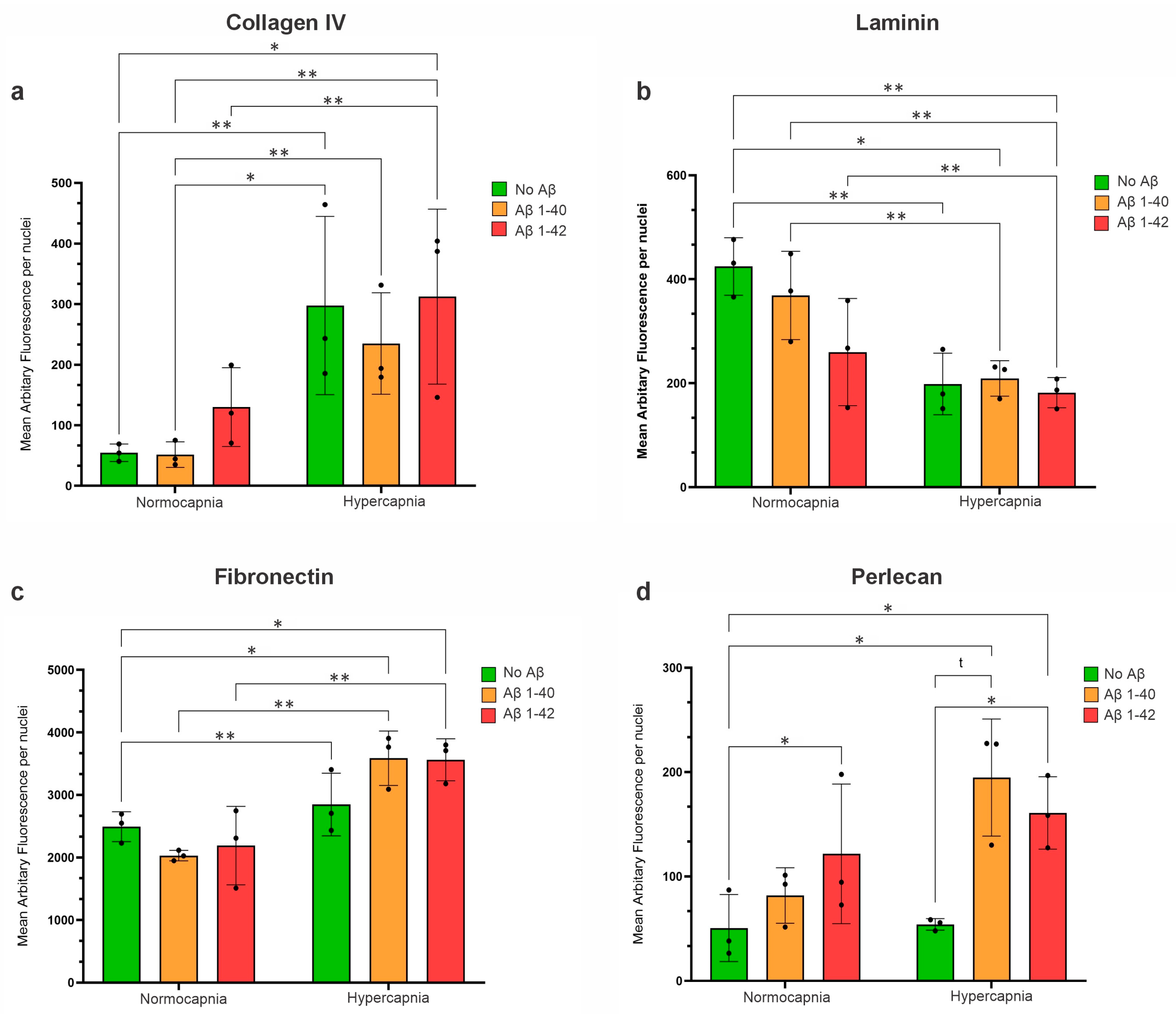
3.7. Hypercapnia Increased Collagen IV Expression in HBVSMCs
3.7.1. Hypercapnia Decreased Laminin Expression in HBVSMCs
3.7.2. Hypercapnia Increased Fibronectin Expression in HBVSMCs
3.7.3. Hypercapnic Conditions Increase the Expression of Perlecan in HBVSMCs, but Only in the Presence of Aβ
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BMs | Basement Membranes |
| SMCs | Smooth Muscle Cells |
| Aβ | Amyloid Beta |
| IPAD | Intramural Periarterial Drainage |
| AD | Alzheimer’s Disease |
| CAA | Cerebral Amyloid Angiopathy |
| BBB | Blood–Brain barrier |
| HSPGs | Heparin Sulphate Proteogylcans |
| HA | Human Astrocytes |
| HBVSMCs | Human brain Vascular Smooth Muscle Cells |
| HP | Human Pericytes |
| hCMEC/D3 | Human Cerebral Endothelial Cell Line |
| PBS | Phosphate-Buffered Saline |
| BSA | Bovine Serum Albumin |
| AF | Arbitary Fluorescence |
| CARASIL | Cerebral Autosomal Recessive Arteriopathy with Subcortical Infarcts and Leukoencephalopathy |
| MMP | Matrix Metalloproteinases |
| ECM | Extracellular Matrix |
| TIMPs | Tissue Inhibitor of Metalloproteinases |
References
- Carare, R.O.; Bernardes-Silva, M.; Newman, T.A.; Page, A.M.; Nicoll, J.A.; Perry, V.H.; Weller, R.O. Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: Significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol. Appl. Neurobiol. 2008, 34, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.A.; Jayakody, N.; Johnston, D.A.; Bechmann, I.; Carare, R.O. Failure of perivascular drainage of beta-amyloid in cerebral amyloid angiopathy. Brain Pathol. 2014, 24, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.W.; Carare, R.O.; Schreiber, S.; Hawkes, C.A. The Cerebrovascular Basement Membrane: Role in the Clearance of beta-amyloid and Cerebral Amyloid Angiopathy. Front. Aging Neurosci. 2014, 6, 251. [Google Scholar] [CrossRef]
- Carare, R.O.; Aldea, R.; Bulters, D.; Alzetani, A.; Birch, A.A.; Richardson, G.; Weller, R.O. Vasomotion Drives Periarterial Drainage of Abeta from the Brain. Neuron 2020, 105, 400–401. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.A.; Gatherer, M.; Sharp, M.M.; Dorr, A.; Yuen, H.M.; Kalaria, R.; Weller, R.O.; Carare, R.O. Regional differences in the morphological and functional effects of aging on cerebral basement membranes and perivascular drainage of amyloid-beta from the mouse brain. Aging Cell 2013, 12, 224–236. [Google Scholar] [CrossRef]
- Kitaguchi, H.; Tomimoto, H.; Ihara, M.; Shibata, M.; Uemura, K.; Kalaria, R.N.; Kihara, T.; Asada-Utsugi, M.; Kinoshita, A.; Takahashi, R. Chronic cerebral hypoperfusion accelerates amyloid beta deposition in APPSwInd transgenic mice. Brain Res. 2009, 1294, 202–210. [Google Scholar] [CrossRef]
- Hosoki, S.; Hansra, G.K.; Jayasena, T.; Poljak, A.; Mather, K.A.; Catts, V.S.; Rust, R.; Sagare, A.; Kovacic, J.C.; Brodtmann, A.; et al. Molecular biomarkers for vascular cognitive impairment and dementia. Nat. Rev. Neurol. 2023, 19, 737–753. [Google Scholar] [CrossRef]
- Hawkes, C.A.; Hartig, W.; Kacza, J.; Schliebs, R.; Weller, R.O.; Nicoll, J.A.; Carare, R.O. Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathol. 2011, 121, 431–443. [Google Scholar] [CrossRef]
- Thomsen, M.S.; Routhe, L.J.; Moos, T. The vascular basement membrane in the healthy and pathological brain. J. Cereb. Blood Flow. Metab. 2017, 37, 3300–3317. [Google Scholar] [CrossRef]
- Keable, A.; Fenna, K.; Yuen, H.M.; Johnston, D.A.; Smyth, N.R.; Smith, C.; Al-Shahi Salman, R.; Samarasekera, N.; Nicoll, J.A.; Attems, J.; et al. Deposition of amyloid beta in the walls of human leptomeningeal arteries in relation to perivascular drainage pathways in cerebral amyloid angiopathy. Biochim. Biophys. Acta 2016, 1862, 1037–1046. [Google Scholar] [CrossRef]
- Jakel, L.; Van Nostrand, W.E.; Nicoll, J.A.R.; Werring, D.J.; Verbeek, M.M. Animal models of cerebral amyloid angiopathy. Clin. Sci. 2017, 131, 2469–2488. [Google Scholar] [CrossRef] [PubMed]
- Yousif, L.F.; Di Russo, J.; Sorokin, L. Laminin isoforms in endothelial and perivascular basement membranes. Cell Adhes. Migr. 2013, 7, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Yurchenco, P.D. Basement membranes: Cell scaffoldings and signaling platforms. Cold Spring Harb. Perspect. Biol. 2011, 3, a004911. [Google Scholar] [CrossRef]
- Wang, J.; Yin, L.; Chen, Z. New insights into the altered fibronectin matrix and extrasynaptic transmission in the aging brain. J. Clin. Gerontol. Geriatr. 2011, 2, 35–41. [Google Scholar] [CrossRef]
- Kadler, K.E.; Hill, A.; Canty-Laird, E.G. Collagen fibrillogenesis: Fibronectin, integrins, and minor collagens as organizers and nucleators. Curr. Opin. Cell Biol. 2008, 20, 495–501. [Google Scholar] [CrossRef]
- Wright, S.A.; Lennon, R.; Greenhalgh, A.D. Basement membranes’ role in immune cell recruitment to the central nervous system. J. Inflamm. 2024, 21, 53. [Google Scholar] [CrossRef] [PubMed]
- Gould, D.B.; Phalan, F.C.; Breedveld, G.J.; van Mil, S.E.; Smith, R.S.; Schimenti, J.C.; Aguglia, U.; van der Knaap, M.S.; Heutink, P.; John, S.W.M. Mutations in Col4a1 Cause Perinatal Cerebral Hemorrhage and Porencephaly. Science 2005, 308, 1167–1171. [Google Scholar] [CrossRef]
- Hallmann, R.; Horn, N.; Selg, M.; Wendler, O.; Pausch, F.; Sorokin, L.M. Expression and Function of Laminins in the Embryonic and Mature Vasculature. Physiol. Rev. 2005, 85, 979–1000. [Google Scholar] [CrossRef]
- Menezes, M.J.; McClenahan, F.K.; Leiton, C.V.; Aranmolate, A.; Shan, X.; Colognato, H. The Extracellular Matrix Protein Laminin α2 Regulates the Maturation and Function of the Blood–Brain Barrier. J. Neurosci. 2014, 34, 15260–15280. [Google Scholar] [CrossRef]
- Göhring, W.; Sasaki, T.; Heldin, C.-H.; Timpl, R. Mapping of the binding of platelet-derived growth factor to distinct domains of the basement membrane proteins BM-40 and perlecan and distinction from the BM-40 collagen-binding epitope. Eur. J. Biochem. 1998, 255, 60–66. [Google Scholar] [CrossRef]
- Leoni, R.F.; Oliveira, I.A.; Pontes-Neto, O.M.; Santos, A.C.; Leite, J.P. Cerebral blood flow and vasoreactivity in aging: An arterial spin labeling study. Braz. J. Med. Biol. Res. 2017, 50, e5670. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Goodwin, J. Effect of aging on respiratory system physiology and immunology. Clin. Interv. Aging 2006, 1, 253–260. [Google Scholar] [CrossRef] [PubMed]
- MacGregor Sharp, M.; Saito, S.; Keable, A.; Gatherer, M.; Aldea, R.; Agarwal, N.; Simpson, J.E.; Wharton, S.B.; Weller, R.O.; Carare, R.O. Demonstrating a reduced capacity for removal of fluid from cerebral white matter and hypoxia in areas of white matter hyperintensity associated with age and dementia. Acta Neuropathol. Commun. 2020, 8, 131. [Google Scholar] [CrossRef]
- Vargas-George, S.; Dave, K.R. Models of cerebral amyloid angiopathy-related intracerebral hemorrhage. Brain Hemorrhages 2022, 3, 189–199. [Google Scholar] [CrossRef]
- Sun, X.; Chen, W.-D.; Wang, Y.-D. β-Amyloid: The Key Peptide in the Pathogenesis of Alzheimer’s Disease. Front. Pharmacol. 2015, 6, 221. [Google Scholar] [CrossRef]
- Neyazi, B.; Stein, K.-P.; Wilkens, L.; Maslehaty, H.; Dumitru, C.A.; Sandalcioglu, I.E. Age-dependent changes of collagen alpha-2(IV) expression in the extracellular matrix of brain arteriovenous malformations. Clin. Neurol. Neurosurg. 2020, 189, 105589. [Google Scholar] [CrossRef]
- Trout, A.L.; Rutkai, I.; Biose, I.J.; Bix, G.J. Review of Alterations in Perlecan-Associated Vascular Risk Factors in Dementia. Int. J. Mol. Sci. 2020, 21, 679. [Google Scholar] [CrossRef]
- Hill, J.; Rom, S.; Ramirez, S.H.; Persidsky, Y. Emerging roles of pericytes in the regulation of the neurovascular unit in health and disease. J. Neuroimmune Pharmacol. 2014, 9, 591–605. [Google Scholar] [CrossRef]
- Park, T.I.H.; Feisst, V.; Brooks, A.E.S.; Rustenhoven, J.; Monzo, H.J.; Feng, S.X.; Mee, E.W.; Bergin, P.S.; Oldfield, R.; Graham, E.S.; et al. Cultured pericytes from human brain show phenotypic and functional differences associated with differential CD90 expression. Sci. Rep. 2016, 6, 26587. [Google Scholar] [CrossRef]
- Lepelletier, F.X.; Mann, D.M.; Robinson, A.C.; Pinteaux, E.; Boutin, H. Early changes in extracellular matrix in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2017, 43, 167–182. [Google Scholar] [CrossRef]
- Merlini, M.; Meyer, E.P.; Ulmann-Schuler, A.; Nitsch, R.M. Vascular β-amyloid and early astrocyte alterations impair cerebrovascular function and cerebral metabolism in transgenic arcAβ mice. Acta Neuropathol. 2011, 122, 293–311. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.C.; Short, J.L.; Nicolazzo, J.A. Altered brain uptake of therapeutics in a triple transgenic mouse model of Alzheimer’s disease. Pharm. Res. 2013, 30, 2868–2879. [Google Scholar] [CrossRef] [PubMed]
- Okoye, M.I.; Watanabe, I. Ultrastructural features of cerebral amyloid angiopathy. Hum. Pathol. 1982, 13, 1127–1132. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Lin, C.; Sanan, D.A.; Mucke, L.; Masliah, E. Chronic Overproduction of Transforming Growth Factor-β1 by Astrocytes Promotes Alzheimer’s Disease-Like Microvascular Degeneration in Transgenic Mice. Am. J. Pathol. 2000, 156, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, S.; Fini, C.A.; Mabuchi, T.; Koziol, J.A.; Eggleston, L.L., Jr.; del Zoppo, G.J. Focal cerebral ischemia induces active proteases that degrade microvascular matrix. Stroke 2004, 35, 998–1004. [Google Scholar] [CrossRef]
- Katsu, M.; Niizuma, K.; Yoshioka, H.; Okami, N.; Sakata, H.; Chan, P.H. Hemoglobin-induced oxidative stress contributes to matrix metalloproteinase activation and blood-brain barrier dysfunction in vivo. J. Cereb. Blood Flow. Metab. 2010, 30, 1939–1950. [Google Scholar] [CrossRef]
- Hamann, G.F.; Okada, Y.; Fitridge, R.; del Zoppo, G.J. Microvascular basal lamina antigens disappear during cerebral ischemia and reperfusion. Stroke 1995, 26, 2120–2126. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Kanno, I.; Uemura, K.; Shishido, F.; Inugami, A.; Ogawa, T.; Murakami, M.; Suzuki, K. Reduction in regional cerebral metabolic rate of oxygen during human aging. Stroke 1986, 17, 1220–1228. [Google Scholar] [CrossRef]
- Takada, H.; Nagata, K.; Hirata, Y.; Satoh, Y.; Watahiki, Y.; Sugawara, J.; Yokoyama, E.; Kondoh, Y.; Shishido, F.; Inugami, A.; et al. Age-related decline of cerebral oxygen metabolism in normal population detected with positron emission tomography. Neurol. Res. 1992, 14 (Suppl. S2), 128–131. [Google Scholar] [CrossRef]
- Kato, T.; Manabe, R.I.; Igarashi, H.; Kametani, F.; Hirokawa, S.; Sekine, Y.; Fujita, N.; Saito, S.; Kawashima, Y.; Hatano, Y.; et al. Candesartan prevents arteriopathy progression in cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy model. J. Clin. Investig. 2021, 131, e140555. [Google Scholar] [CrossRef] [PubMed]
- Howe, M.D.; Furr, J.W.; Munshi, Y.; Roy-O’Reilly, M.A.; Maniskas, M.E.; Koellhoffer, E.C.; d’Aigle, J.; Sansing, L.H.; McCullough, L.D.; Urayama, A. Transforming growth factor-beta promotes basement membrane fibrosis, alters perivascular cerebrospinal fluid distribution, and worsens neurological recovery in the aged brain after stroke. Geroscience 2019, 41, 543–559. [Google Scholar] [CrossRef]
- Howe, M.D.; Atadja, L.A.; Furr, J.W.; Maniskas, M.E.; Zhu, L.; McCullough, L.D.; Urayama, A. Fibronectin induces the perivascular deposition of cerebrospinal fluid–derived amyloid-β in aging and after stroke. Neurobiol. Aging 2018, 72, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C. Why Abeta42 Is Much More Toxic than Abeta40. ACS Chem. Neurosci. 2019, 10, 2843–2847. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.J.; Chen, Y.R. The coexistence of an equal amount of Alzheimer’s amyloid-β 40 and 42 forms structurally stable and toxic oligomers through a distinct pathway. FEBS J. 2014, 281, 2674–2687. [Google Scholar] [CrossRef]
- Krishtal, J.; Bragina, O.; Metsla, K.; Palumaa, P.; Tõugu, V. Toxicity of amyloid beta 1–40 and 1–42 on SH-SY5Y cell line. SpringerPlus 2015, 4, P19. [Google Scholar] [CrossRef]
- Hawkes, C.A.; Sullivan, P.M.; Hands, S.; Weller, R.O.; Nicoll, J.A.; Carare, R.O. Disruption of arterial perivascular drainage of amyloid-β from the brains of mice expressing the human APOE ε4 allele. PLoS ONE 2012, 7, e41636. [Google Scholar] [CrossRef]
- Haorah, J.; Ramirez, S.H.; Schall, K.; Smith, D.; Pandya, R.; Persidsky, Y. Oxidative stress activates protein tyrosine kinase and matrix metalloproteinases leading to blood-brain barrier dysfunction. J. Neurochem. 2007, 101, 566–576. [Google Scholar] [CrossRef]
- Rosenberg, G.A.; Sullivan, N.; Esiri, M.M. White matter damage is associated with matrix metalloproteinases in vascular dementia. Stroke 2001, 32, 1162–1168. [Google Scholar] [CrossRef]
- Duncombe, J.; Kitamura, A.; Hase, Y.; Ihara, M.; Kalaria, R.N.; Horsburgh, K. Chronic cerebral hypoperfusion: A key mechanism leading to vascular cognitive impairment and dementia. Closing the translational gap between rodent models and human vascular cognitive impairment and dementia. Clin. Sci. 2017, 131, 2451–2468. [Google Scholar] [CrossRef]
- Horstmann, S.; Kalb, P.; Koziol, J.; Gardner, H.; Wagner, S. Profiles of Matrix Metalloproteinases, Their Inhibitors, and Laminin in Stroke Patients. Stroke 2003, 34, 2165–2170. [Google Scholar] [CrossRef]
- Dewing, J.M.; Carare, R.O.; Lotery, A.J.; Ratnayaka, J.A. The Diverse Roles of TIMP-3: Insights into Degenerative Diseases of the Senescent Retina and Brain. Cells 2019, 9, 39. [Google Scholar] [CrossRef]
- Manousopoulou, A.; Gatherer, M.; Smith, C.; Nicoll, J.A.R.; Woelk, C.H.; Johnson, M.; Kalaria, R.; Attems, J.; Garbis, S.D.; Carare, R.O. Systems proteomic analysis reveals that clusterin and tissue inhibitor of metalloproteinases 3 increase in leptomeningeal arteries affected by cerebral amyloid angiopathy. Neuropathol. Appl. Neurobiol. 2017, 43, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Leitner, D.; Kavanagh, T.; Kanshin, E.; Balcomb, K.; Pires, G.; Thierry, M.; Suazo, J.I.; Schneider, J.; Ueberheide, B.; Drummond, E.; et al. Differences in the cerebral amyloid angiopathy proteome in Alzheimer’s disease and mild cognitive impairment. Acta Neuropathol. 2024, 148, 9. [Google Scholar] [CrossRef] [PubMed]
- Snow, A.D.; Cummings, J.A.; Lake, T. The Unifying Hypothesis of Alzheimer’s Disease: Heparan Sulfate Proteoglycans/Glycosaminoglycans Are Key as First Hypothesized Over 30 Years Ago. Front. Aging Neurosci. 2021, 13, 710683. [Google Scholar] [CrossRef]
- Boche, D.; Zotova, E.; Weller, R.O.; Love, S.; Neal, J.W.; Pickering, R.M.; Wilkinson, D.; Holmes, C.; Nicoll, J.A. Consequence of Abeta immunization on the vasculature of human Alzheimer’s disease brain. Brain 2008, 131, 3299–3310. [Google Scholar] [PubMed]
- Sakai, K.; Boche, D.; Carare, R.; Johnston, D.; Holmes, C.; Love, S.; Nicoll, J.A. Abeta immunotherapy for Alzheimer’s disease: Effects on apoE and cerebral vasculopathy. Acta Neuropathol. 2014, 128, 777–789. [Google Scholar] [CrossRef]
- Frieser, M.; Nockel, H.; Pausch, F.; Roder, C.; Hahn, A.; Deutzmann, R.; Sorokin, L.M. Cloning of the mouse laminin alpha 4 cDNA. Expression in a subset of endothelium. Eur. J. Biochem. 1997, 246, 727–735. [Google Scholar] [CrossRef]
- Sorokin, L.M.; Pausch, F.; Frieser, M.; Kroger, S.; Ohage, E.; Deutzmann, R. Developmental regulation of the laminin alpha5 chain suggests a role in epithelial and endothelial cell maturation. Dev. Biol. 1997, 189, 285–300. [Google Scholar] [CrossRef]
- Wu, C.; Ivars, F.; Anderson, P.; Hallmann, R.; Vestweber, D.; Nilsson, P.; Robenek, H.; Tryggvason, K.; Song, J.; Korpos, E.; et al. Endothelial basement membrane laminin alpha5 selectively inhibits T lymphocyte extravasation into the brain. Nat. Med. 2009, 15, 519–527. [Google Scholar] [CrossRef]
- Hannocks, M.J.; Pizzo, M.E.; Huppert, J.; Despande, T.; Abbott, N.J.; Thorne, R.G.; Sorokin, L. Molecular characterization of perivascular drainage pathways in the murine brain. J. Cereb. Blood Flow. Metab. 2017, 38, 669–686. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.J.; Nelson, L.; Slade, J.Y.; Oakley, A.E.; Khundakar, A.A.; Kalaria, R.N. Morphometry of the hippocampal microvasculature in post-stroke and age-related dementias. Neuropathol. Appl. Neurobiol. 2014, 40, 284–295. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dewing, J.M.; Keable, A.; Laslo, A.; Chinezu, L.; Ivanescu, A.; Ratnayaka, J.A.; Kalaria, R.; Slevin, M.; Verma, A.; Carare, R.O. Proportions of Basement Membrane Proteins in Cerebrovascular Smooth Muscle Cells After Exposure to Hypercapnia and Amyloid Beta. Cells 2025, 14, 614. https://doi.org/10.3390/cells14080614
Dewing JM, Keable A, Laslo A, Chinezu L, Ivanescu A, Ratnayaka JA, Kalaria R, Slevin M, Verma A, Carare RO. Proportions of Basement Membrane Proteins in Cerebrovascular Smooth Muscle Cells After Exposure to Hypercapnia and Amyloid Beta. Cells. 2025; 14(8):614. https://doi.org/10.3390/cells14080614
Chicago/Turabian StyleDewing, Jennifer M., Abby Keable, Alexandru Laslo, Laura Chinezu, Adrian Ivanescu, J. Arjuna Ratnayaka, Raj Kalaria, Mark Slevin, Ajay Verma, and Roxana O. Carare. 2025. "Proportions of Basement Membrane Proteins in Cerebrovascular Smooth Muscle Cells After Exposure to Hypercapnia and Amyloid Beta" Cells 14, no. 8: 614. https://doi.org/10.3390/cells14080614
APA StyleDewing, J. M., Keable, A., Laslo, A., Chinezu, L., Ivanescu, A., Ratnayaka, J. A., Kalaria, R., Slevin, M., Verma, A., & Carare, R. O. (2025). Proportions of Basement Membrane Proteins in Cerebrovascular Smooth Muscle Cells After Exposure to Hypercapnia and Amyloid Beta. Cells, 14(8), 614. https://doi.org/10.3390/cells14080614