
Laquinimod Supports Remyelination in Non-Supportive Environments


 and
and 
Abstract
:1. Introduction
2. Material and Methods
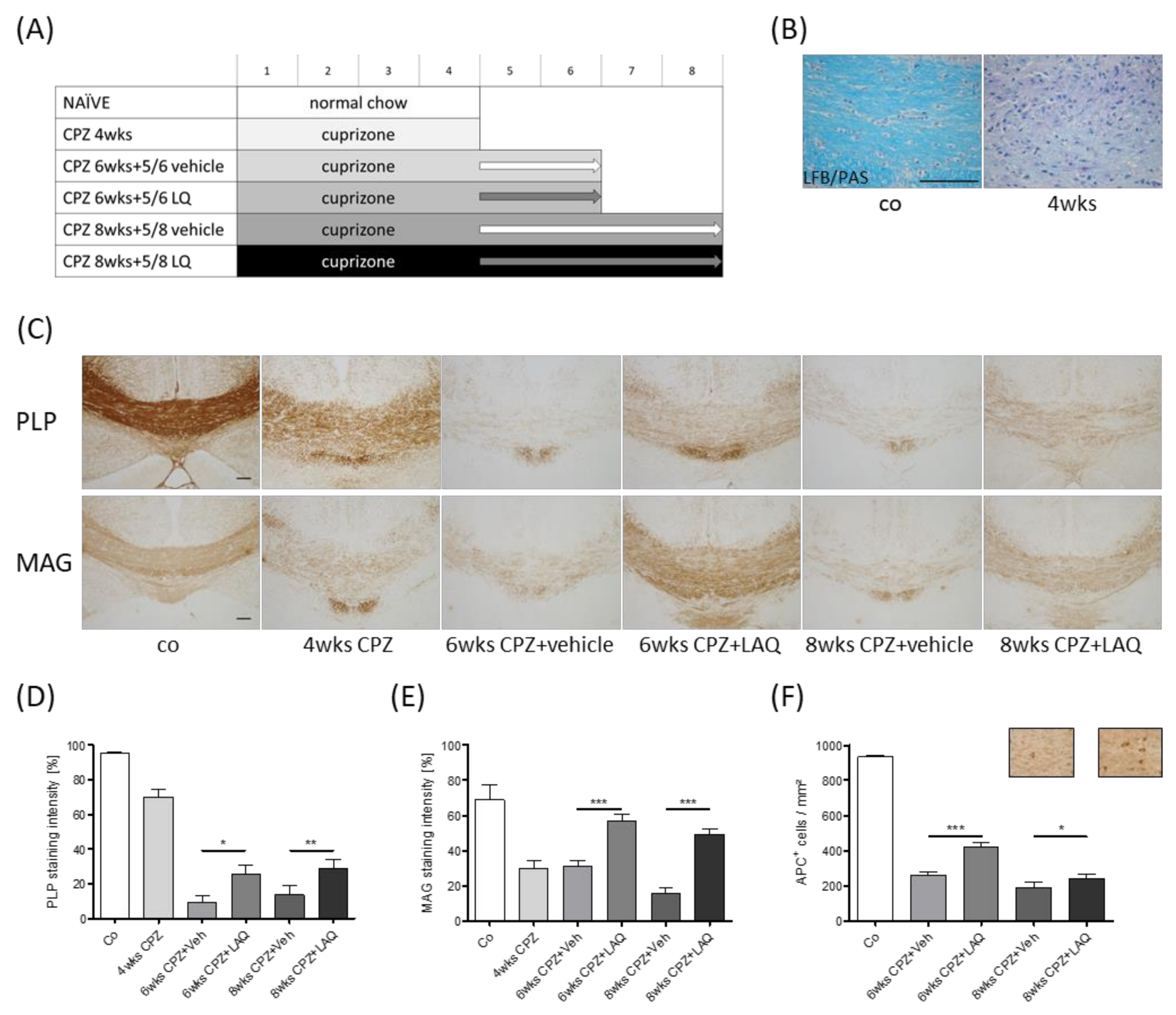
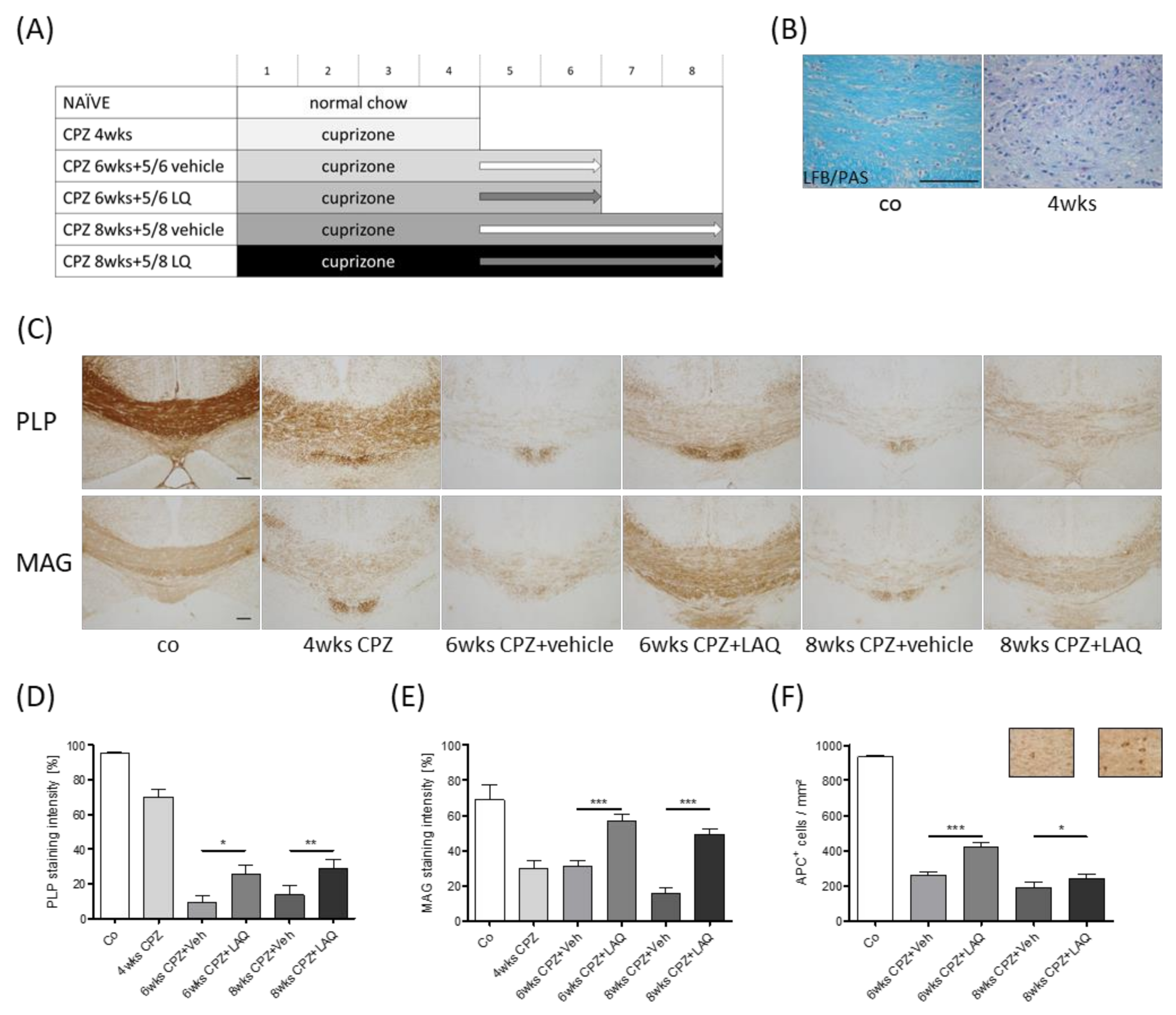
2.1. Animals and Experimental Setup
2.2. Tissue Preparation
2.3. Luxol Fast Blue (LFB) Periodic Acid–Schiff (PAS) Stain and Myelin Status Scoring
2.4. Immunohistochemistry and Densitometric Analyses
2.5. Immunofluorescence Double Labelling
2.6. Statistical Analyses
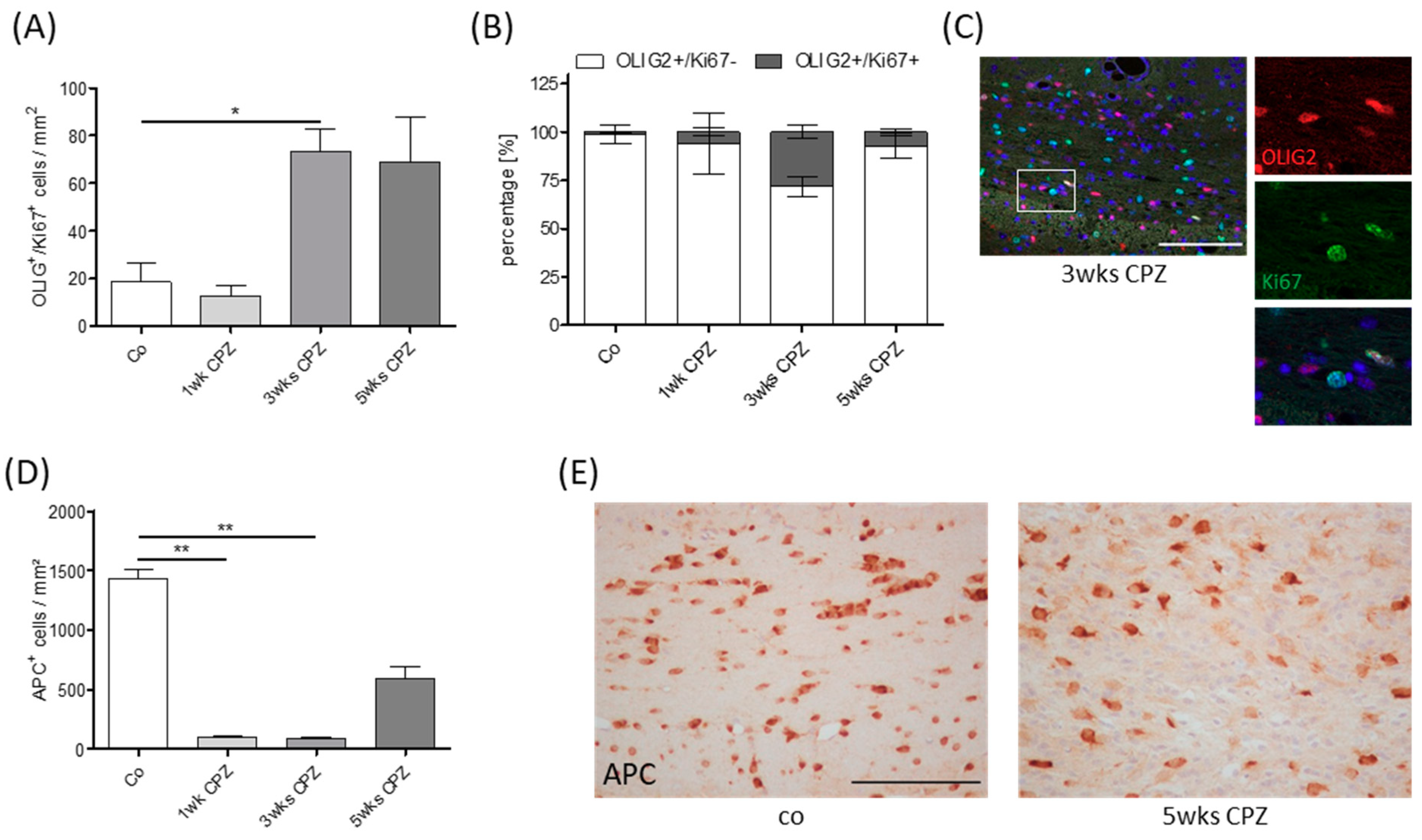
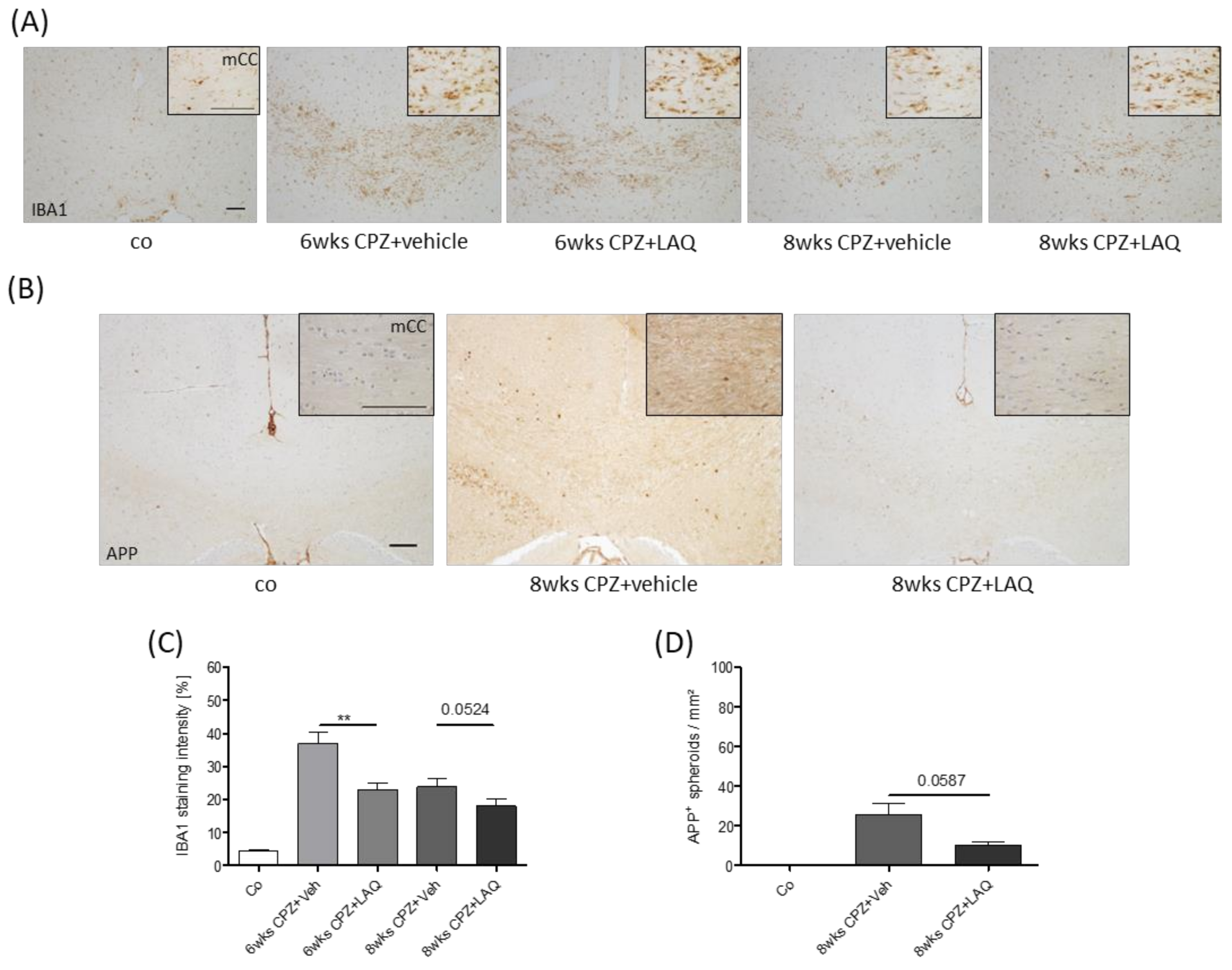
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Haider, L.; Simeonidou, C.; Steinberger, G.; Hametner, S.; Grigoriadis, N.; Deretzi, G.; Frischer, J.M. Multiple sclerosis deep grey matter: The relation between demyelination, neurodegeneration, inflammation and iron. J. Neurol Neurosurg Psychiatry 2014, 85, 1386–1395. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, B.; Matyszak, M.K.; Esiri, M.M.; Perry, V.H. Axonal damage in acute multiple sclerosis lesions. Brain 1997, 120, 393–399. [Google Scholar] [CrossRef]
- Kuhlmann, T.; Lingfeld, G.; Bitsch, A.; Schuchardt, J.; Bruck, W. Acute axonal damage in multiple sclerosis is most extensive in early disease stages and decreases over time. Brain 2002, 125, 2202–2212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, J.; Rauschka, H.; Lassmann, H. Inflammation in the nervous system: The human perspective. Glia 2001, 36, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Barnett, M.H.; Prineas, J.W. Relapsing and remitting multiple sclerosis: Pathology of the newly forming lesion. Ann. Neurol. 2004, 55, 458–468. [Google Scholar] [CrossRef]
- Lucchinetti, C.; Bruck, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. Heterogeneity of multiple sclerosis lesions: for the pathogenesis of demyelination. Ann. Neurol. 2000, 47, 707–717. [Google Scholar] [CrossRef]
- Naldi, P.; Collimedaglia, L.; Vecchio, D.; Rosso, M.G.; Perl, F.; Stecco, A.; Monaco, F.; Leone, M.A. Predictors of attack severity and duration in multiple sclerosis: prospective study. Open Neurol. J. 2011, 5, 75–82. [Google Scholar] [CrossRef]
- Vidal-Jordana, A.; Montalban, X. Multiple Sclerosis: Epidemiologic, Clinical, and Therapeutic Aspects. Neuroimaging Clin. N. Am. 2017, 27, 195–204. [Google Scholar] [CrossRef]
- Confavreux, C.; Vukusic, S. The clinical course of multiple sclerosis. Handb. Clin. Neurol. 2014, 122, 343–369. [Google Scholar]
- Tartaglia, M.C.; Narayanan, S.; Francis, S.J.; Santos, A.C.; De Stefano, N.; Lapierre, Y.; Arnold, D.L. The relationship between diffuse axonal damage and fatigue in multiple sclerosis. Arch. Neurol. 2004, 61, 201–207. [Google Scholar] [CrossRef]
- De Stefano, N.; Matthews, P.M.; Fu, L.; Narayanan, S.; Stanley, J.; Francis, G.S.; Antel, J.P.; Arnold, D.L. Axonal damage correlates with disability in patients with relapsing-remitting multiple sclerosis. Results of a longitudinal magnetic resonance spectroscopy study. Brain 1998, 121, 1469–1477. [Google Scholar] [CrossRef] [PubMed]
- Kipp, M.; Amor, S. FTY720 on the way from the base camp to the summit of the mountain: Relevance for remyelination. Mult. Scler. 2012, 18, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Perier, O.; Gregoire, A. Electron microscopic features of multiple sclerosis lesions. Brain 1965, 88, 937–952. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Scheithauer, B. Ultrastructure of multiple sclerosis. Ultrastruct Pathol. 1994, 18, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Prineas, J.W.; Kwon, E.E.; Cho, E.S.; Sharer, L.R. Continual breakdown and regeneration of myelin in progressive multiple sclerosis plaques. Ann. N. Y. Acad. Sci. 1984, 436, 11–32. [Google Scholar] [CrossRef] [PubMed]
- Prineas, J.W.; Connell, F. Remyelination in multiple sclerosis. Ann. Neurol. 1979, 5, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Prineas, J.W.; Barnard, R.O.; Kwon, E.E.; Sharer, L.R.; Cho, E.S. Multiple sclerosis: Remyelination of nascent lesions. Ann. Neurol. 1993, 33, 137–151. [Google Scholar] [CrossRef]
- Trapp, B.D.; Stys, P.K. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurol. 2009, 8, 280–291. [Google Scholar] [CrossRef]
- Funfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Mobius, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.J. Sodium channels and multiple sclerosis: Roles in symptom production, damage and therapy. Brain Pathol. 2007, 17, 230–242. [Google Scholar] [CrossRef]
- Prineas, J.W.; Barnard, R.O.; Revesz, T.; Kwon, E.E.; Sharer, L.; Cho, E.S. Multiple sclerosis. Pathology of recurrent lesions. Brain 1993, 116, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Willoughby, E.W.; Grochowski, E.; Li, D.K.; Oger, J.; Kastrukoff, L.F.; Paty, D.W. Serial magnetic resonance scanning in multiple sclerosis: A second prospective study in relapsing patients. Ann. Neurol. 1989, 25, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.A.; Narayanan, S.; Arnold, D.L. Imaging of repeated episodes of demyelination and remyelination in multiple sclerosis. Neuroimage Clin. 2014, 6, 20–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, E.S.; Ludwin, S.K. The demonstration of recurrent demyelination and remyelination of axons in the central nervous system. Acta Neuropathol. 1981, 53, 93–98. [Google Scholar] [CrossRef]
- Mason, J.L.; Langaman, C.; Morell, P.; Suzuki, K.; Matsushima, G.K. Episodic demyelination and subsequent remyelination within the murine central nervous system: Changes in axonal calibre. Neuropathol. Appl. Neurobiol. 2001, 27, 50–58. [Google Scholar] [CrossRef]
- Penderis, J.; Shields, S.A.; Franklin, R.J. Impaired remyelination and depletion of oligodendrocyte progenitors does not occur following repeated episodes of focal demyelination in the rat central nervous system. Brain 2003, 126, 1382–1391. [Google Scholar] [CrossRef] [Green Version]
- Ruther, B.J.; Scheld, M.; Dreymueller, D.; Clarner, T.; Kress, E.; Brandenburg, L.O.; Swartenbroekx, T.; Hoornaert, C.; Ponsaerts, P.; Fallier-Becker, P.; et al. Combination of cuprizone and experimental autoimmune encephalomyelitis to study inflammatory brain lesion formation and progression. Glia 2017, 65, 1900–1913. [Google Scholar] [CrossRef]
- Scheld, M.; Ruther, B.J.; Grosse-Veldmann, R.; Ohl, K.; Tenbrock, K.; Dreymuller, D.; Fallier-Becker, P.; Zendedel, A.; Beyer, C.; Clarner, T.; et al. Neurodegeneration Triggers Peripheral Immune Cell Recruitment into the Forebrain. J. Neurosci. 2016, 36, 1410–1415. [Google Scholar] [CrossRef]
- Baxi, E.G.; DeBruin, J.; Tosi, D.M.; Grishkan, I.V.; Smith, M.D.; Kirby, L.A.; Gocke, A.R. Transfer of myelin-reactive th17 cells impairs endogenous remyelination in the central nervous system of cuprizone-fed mice. J. Neurosci 2015, 35, 8626–8639. [Google Scholar] [CrossRef]
- Nyamoya, S.; Leopold, P.; Becker, B.; Beyer, C.; Hustadt, F.; Schmitz, C.; Michel, A.; Kipp, M. G-Protein-Coupled Receptor Gpr17 Expression in Two Multiple Sclerosis Remyelination Models. Mol. Neurobiol. 2019, 56, 1109–1123. [Google Scholar] [CrossRef]
- Kramann, N.; Menken, L.; Hayardeny, L.; Hanisch, U.K.; Bruck, W. Laquinimod prevents cuprizone-induced demyelination independent of Toll-like receptor signaling. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e233. [Google Scholar] [CrossRef] [PubMed]
- Brück, W.; Pförtner, R.; Pham, T.; Zhang, J.; Hayardeny, L.; Piryatinsky, V.; Hanisch, U.K.; Regen, T.; van Rossum, D.; Brakelmann, L.; et al. Reduced astrocytic NF-kappaB activation by laquinimod protects from cuprizone-induced demyelination. Acta Neuropathol. 2012, 124, 411–424. [Google Scholar]
- Clarner, T.; Janssen, K.; Nellessen, L.; Stangel, M.; Skripuletz, T.; Krauspe, B.; Hess, F.M.; Denecke, B.; Beutner, C.; Linnartz-Gerlach, B.; et al. CXCL10 triggers early microglial activation in the cuprizone model. J. Immunol. 2015, 194, 3400–3413. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, J.; Clarner, T.; Beyer, C.; Kipp, M. Anatomical Distribution of Cuprizone-Induced Lesions in C57BL6 Mice. J. Mol. Neurosci. 2015, 57, 166–175. [Google Scholar] [CrossRef]
- Hoflich, K.M.; Beyer, C.; Clarner, T.; Schmitz, C.; Nyamoya, S.; Kipp, M.; Hochstrasser, T. Acute axonal damage in three different murine models of multiple sclerosis: A comparative approach. Brain Res. 2016, 1650, 125–133. [Google Scholar] [CrossRef]
- Benardais, K.; Kotsiari, A.; Skuljec, J.; Koutsoudaki, P.N.; Gudi, V.; Singh, V.; Vulinovic, F.; Skripuletz, T.; Stangel, M. Cuprizone [bis(cyclohexylidenehydrazide)] is selectively toxic for mature oligodendrocytes. Neurotox Res. 2013, 24, 244–250. [Google Scholar] [CrossRef]
- Benetti, F.; Ventura, M.; Salmini, B.; Ceola, S.; Carbonera, D.; Mammi, S.; Zitolo, A.; D’Angelo, P.; Urso, E.; Maffia, M.; et al. Cuprizone neurotoxicity, copper deficiency and neurodegeneration. Neurotoxicology 2010, 31, 509–517. [Google Scholar] [CrossRef]
- Slowik, A.; Schmidt, T.; Beyer, C.; Amor, S.; Clarner, T.; Kipp, M. The sphingosine 1-phosphate receptor agonist FTY720 is neuroprotective after cuprizone-induced CNS demyelination. Br. J. Pharm. 2015, 172, 80–92. [Google Scholar] [CrossRef]
- Cammer, W. The neurotoxicant, cuprizone, retards the differentiation of oligodendrocytes in vitro. J. Neurol. Sci. 1999, 168, 116–120. [Google Scholar] [CrossRef]
- Fischbach, F.; Nedelcu, J.; Leopold, P.; Zhan, J.; Clarner, T.; Nellessen, L.; Beissel, C.; van Heuvel, Y.; Goswami, A.; Weis, J.; et al. Cuprizone-induced graded oligodendrocyte vulnerability is regulated by the transcription factor DNA damage-inducible transcript 3. Glia 2019, 67, 263–276. [Google Scholar] [CrossRef]
- Faizi, M.; Salimi, A.; Seydi, E.; Naserzadeh, P.; Kouhnavard, M.; Rahimi, A.; Pourahmad, J. Toxicity of cuprizone a Cu(2+) chelating agent on isolated mouse brain mitochondria: A justification for demyelination and subsequent behavioral dysfunction. Toxicol. Mech. Methods 2016, 26, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Hoppel, C.L.; Tandler, B. Biochemical effects of cuprizone on mouse liver and heart mitochondria. Biochem. Pharm. 1973, 22, 2311–2318. [Google Scholar] [CrossRef]
- Baxi, E.G.; DeBruin, J.; Jin, J.; Strasburger, H.J.; Smith, M.D.; Orthmann-Murphy, J.L.; Schott, J.T.; Fairchild, A.N.; Bergles, D.E.; Calabresi, P.A. Lineage tracing reveals dynamic changes in oligodendrocyte precursor cells following cuprizone-induced demyelination. Glia 2017, 65, 2087–2098. [Google Scholar] [CrossRef] [PubMed]
- Gudi, V.; Moharregh-Khiabani, D.; Skripuletz, T.; Koutsoudaki, P.N.; Kotsiari, A.; Skuljec, J.; Trebst, C.; Stangel, M. Regional differences between grey and white matter in cuprizone induced demyelination. Brain Res. 2009, 1283, 127–138. [Google Scholar] [CrossRef]
- Xing, Y.L.; Roth, P.T.; Stratton, J.A.; Chuang, B.H.; Danne, J.; Ellis, S.L.; Ng, S.W.; Kilpatrick, T.J.; Merson, T.D. Adult neural precursor cells from the subventricular zone contribute significantly to oligodendrocyte regeneration and remyelination. J. Neurosci. 2014, 34, 14128–14146. [Google Scholar] [CrossRef]
- Moore, S.; Khalaj, A.J.; Yoon, J.; Patel, R.; Hannsun, G.; Yoo, T.; Sasidhar, M.; Martinez-Torres, L.; Hayardeny, L.; Tiwari-Woodruff, S.K. Therapeutic laquinimod treatment decreases inflammation, initiates axon remyelination, and improves motor deficit in a mouse model of multiple sclerosis. Brain Behav. 2013, 3, 664–682. [Google Scholar] [CrossRef]
- Wegner, C.; Stadelmann, C.; Pfortner, R.; Raymond, E.; Feigelson, S.; Alon, R.; Timan, B.; Hayardeny, L.; Bruck, W. Laquinimod interferes with migratory capacity of T cells and reduces IL-17 levels, inflammatory demyelination and acute axonal damage in mice with experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2010, 227, 133–143. [Google Scholar] [CrossRef]
- Comi, G.; Pulizzi, A.; Rovaris, M.; Abramsky, O.; Arbizu, T.; Boiko, A.; Gold, R.; Havrdova, E.; Komoly, S.; Selmaj, K.; et al. Effect of laquinimod on MRI-monitored disease activity in patients with relapsing-remitting multiple sclerosis: A multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet 2008, 371, 2085–2092. [Google Scholar] [CrossRef]
- Polman, C.; Barkhof, F.; Sandberg-Wollheim, M.; Linde, A.; Nordle, O.; Nederman, T. Treatment with laquinimod reduces development of active MRI lesions in relapsing MS. Neurology 2005, 64, 987–991. [Google Scholar] [CrossRef]
- Comi, G.; Jeffery, D.; Kappos, L.; Montalban, X.; Boyko, A.; Rocca, M.A.; Filippi, M.; Group, A.S. Placebo-controlled trial of oral laquinimod for multiple sclerosis. N. Engl. J. Med. 2012, 366, 1000–1009. [Google Scholar] [CrossRef]
- Schulze-Topphoff, U.; Shetty, A.; Varrin-Doyer, M.; Molnarfi, N.; Sagan, S.A.; Sobel, R.A. Laquinimod, a quinoline-3-carboxamide, induces type II myeloid cells that modulate central nervous system autoimmunity. PLoS ONE 2012, 7, e33797. [Google Scholar] [CrossRef] [PubMed]
- Jolivel, V.; Luessi, F.; Masri, J.; Kraus, S.H.; Hubo, M.; Poisa-Beiro, L.; Furlan, R. Modulation of dendritic cell properties by laquinimod as a mechanism for modulating multiple sclerosis. Brain 2013, 136, 1048–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruffini, F.; Rossi, S.; Bergamaschi, A.; Brambilla, E.; Finardi, A.; Motta, C.; Comi, G. Laquinimod prevents inflammation-induced synaptic alterations occurring in experimental autoimmune encephalomyelitis. Mult. Scler. 2013, 19, 1084–1094. [Google Scholar] [CrossRef] [PubMed]
- Ruffini, F.; Rossi, S.; Bergamaschi, A.; Brambilla, E.; Finardi, A.; Motta, C.; Comi, G. Laquinimod Treatment Improves Myelination Deficits at the Transcriptional and Ultrastructural Levels in the YAC128 Mouse Model. of Huntington Disease. Mol. Neurobiol. 2019, 56, 4464–4478. [Google Scholar]
- Ellrichmann, G.; Blusch, A.; Fatoba, O.; Brunner, J.; Reick, C.; Hayardeny, L.; Gold, R. Laquinimod treatment in the R6/2 mouse model. Sci Rep. 2017, 7, 4947. [Google Scholar] [CrossRef]
- Garcia-Miralles, M.; Hong, X.; Tan, L.J.; Caron, N.S.; Huang, Y.; To, X.V.; Lin, R.Y.; Franciosi, S.; Papapetropoulos, S.; Hayardeny, L.; et al. Laquinimod rescues striatal, cortical and white matter pathology and results in modest behavioural improvements in the YAC128 model of Huntington disease. Sci. Rep. 2016, 6, 31652. [Google Scholar] [CrossRef]
- Katsumoto, A.; Miranda, A.S.; Butovsky, O.; Teixeira, A.L.; Ransohoff, R.M.; Lamb, B.T. Laquinimod attenuates inflammation by modulating macrophage functions in traumatic brain injury mouse model. J. Neuroinflamm. 2018, 15, 26. [Google Scholar] [CrossRef]
- Hussain, R.Z.; Miller-Little, W.A.; Lambracht-Washington, D.; Jaramillo, T.C.; Takahashi, M.; Zhang, S.; Fu, M.; Cutter, G.R.; Hayardeny, L.; Powell, C.M.; et al. Laquinimod has no effects on brain volume or cellular CNS composition in the F1 3xTg-AD/C3H mouse model of Alzheimer’s disease. J. Neuroimmunol. 2017, 309, 100–110. [Google Scholar] [CrossRef]
- Vollmer, T.L.; Sorensen, P.S.; Selmaj, K.; Zipp, F.; Havrdova, E.; Cohen, J.A.; Sasson, N.; Gilgun-Sherki, Y.; Arnold, D.L.; Group, B.S. A randomized placebo-controlled phase III trial of oral laquinimod for multiple sclerosis. J. Neurol. 2014, 261, 773–783. [Google Scholar] [CrossRef]
- Rodrigues, F.B.; Wild, E.J. Clinical Trials Corner: September 2017. J. Huntingt. Dis. 2017, 6, 255–263. [Google Scholar] [CrossRef] [Green Version]
- Wilmes, A.T.; Reinehr, S.; Kuhn, S.; Pedreiturria, X.; Petrikowski, L.; Faissner, S.; Ayzenberg, I.; Stute, G.; Gold, R.; Dick, H.B.; et al. Laquinimod protects the optic nerve and retina in an experimental autoimmune encephalomyelitis model. J. Neuroinflamm. 2018, 15, 183. [Google Scholar] [CrossRef] [PubMed]
- Luhder, F.; Kebir, H.; Odoardi, F.; Litke, T.; Sonneck, M.; Alvarez, J.I.; Winchenbach, J.; Eckert, N.; Hayardeny, L.; Sorani, E.; et al. Laquinimod enhances central nervous system barrier functions. Neurobiol. Dis. 2017, 102, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Gentile, A.; Musella, A.; De Vito, F.; Fresegna, D.; Bullitta, S.; Rizzo, F.R.; Centonze, D.; Mandolesi, G. Laquinimod ameliorates excitotoxic damage by regulating glutamate re-uptake. J. Neuroinflamm. 2018, 15, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaye, J.; Piryatinsky, V.; Birnberg, T.; Hingaly, T.; Raymond, E.; Kashi, R.; Amit-Romach, E.; Caballero, I.S.; Towfic, F.; Ator, M.A.; et al. Laquinimod arrests experimental autoimmune encephalomyelitis by activating the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA 2016, 113, E6145–E6152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawker, K.; O’Connor, P.; Freedman, M.S.; Calabresi, P.A.; Antel, J.; Simon, J.; Hauser, S.; Waubant, E.; Vollmer, T.; Panitch, H.; et al. Rituximab in patients with primary progressive multiple sclerosis: Results of a randomized double-blind placebo-controlled multicenter trial. Ann. Neurol. 2009, 66, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.; Miller, D.H.; Freedman, M.S.; Cree, B.A.C.; Wolinsky, J.S.; Weiner, H.; Lubetzki, C.; Hartung, H.P.; Montalban, X.; Uitdehaag, B.M.J.; et al. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): A phase 3, randomised, double-blind, placebo-controlled trial. Lancet 2016, 387, 1075–1084. [Google Scholar] [CrossRef]
- Rumbach, L.; Racadot, E.; Armspach, J.P.; Namer, I.J.; Bonneville, J.F.; Wijdenes, J.; Marescaux, C.; Herve, P.; Chambron, J. Biological assessment and MRI monitoring of the therapeutic efficacy of a monoclonal anti-T CD4 antibody in multiple sclerosis patients. Mult. Scler. 1996, 1, 207–212. [Google Scholar] [CrossRef]
- Revesz, T.; Kidd, D.; Thompson, A.J.; Barnard, R.O.; McDonald, W.I. A comparison of the pathology of primary and secondary progressive multiple sclerosis. Brain 1994, 117, 759–765. [Google Scholar] [CrossRef]
- Stevenson, V.L.; Miller, D.H.; Rovaris, M.; Barkhof, F.; Brochet, B.; Dousset, V.; Dousset, V.; Filippi, M.; Montalban, X.; Polman, C.H.; et al. Primary and transitional progressive MS: A clinical and MRI cross-sectional study. Neurology 1999, 52, 839–845. [Google Scholar] [CrossRef]
- Correale, J.; Gaitan, M.I.; Ysrraelit, M.C.; Fiol, M.P. Progressive multiple sclerosis: From pathogenic mechanisms to treatment. Brain 2017, 140, 527–546. [Google Scholar] [CrossRef]
- Draheim, T.; Liessem, A.; Scheld, M.; Wilms, F.; Weissflog, M.; Denecke, B.; Kensler, T.W.; Zendedel, A.; Beyer, C.; Kipp, M.; et al. Activation of the astrocytic Nrf2/ARE system ameliorates the formation of demyelinating lesions in a multiple sclerosis animal model. Glia 2016, 64, 2219–2230. [Google Scholar] [CrossRef] [PubMed]
- McMahon, E.J.; Suzuki, K.; Matsushima, G.K. Peripheral macrophage recruitment in cuprizone-induced CNS demyelination despite an intact blood-brain barrier. J. Neuroimmunol. 2002, 130, 32–45. [Google Scholar] [CrossRef]
- Verden, D.; Macklin, W.B. Neuroprotection by central nervous system remyelination: Molecular, cellular, and functional considerations. J. Neurosci. Res. 2016, 94, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Mayer, R.F. Conduction velocity in the central nervous system of the cat during experimental demyelination and remyelination. Int. J. Neurosci. 1971, 1, 287–308. [Google Scholar] [CrossRef] [PubMed]
- Irvine, K.A.; Blakemore, W.F. Remyelination protects axons from demyelination-associated axon degeneration. Brain 2008, 131, 1464–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manrique-Hoyos, N.; Jurgens, T.; Gronborg, M.; Kreutzfeldt, M.; Schedensack, M.; Kuhlmann, T.; Schrick, C.; Bruck, W.; Urlaub, H.; Simons, M.; et al. Late motor decline after accomplished remyelination: Impact for progressive multiple sclerosis. Ann. Neurol. 2012, 71, 227–244. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, A.F.; Miron, V.E. The pro-remyelination properties of microglia in the central nervous system. Nat. Rev. Neurol. 2019, 15, 447–458. [Google Scholar] [CrossRef]
- Olah, M.; Amor, S.; Brouwer, N.; Vinet, J.; Eggen, B.; Biber, K.; Boddeke, H.W. Identification of a microglia phenotype supportive of remyelination. Glia 2012, 60, 306–321. [Google Scholar] [CrossRef]
- Kotter, M.R.; Li, W.W.; Zhao, C.; Franklin, R.J. Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. J. Neurosci. 2006, 26, 328–332. [Google Scholar] [CrossRef]
- Lampron, A.; Larochelle, A.; Laflamme, N.; Prefontaine, P.; Plante, M.M.; Sanchez, M.G.; Yong, V.W.; Stys, P.K.; Tremblay, M.E.; Rivest, S. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J. Exp. Med. 2015, 212, 481–495. [Google Scholar] [CrossRef] [Green Version]
- Hagemeyer, N.; Hanft, K.M.; Akriditou, M.A.; Unger, N.; Park, E.S.; Stanley, E.R.; Staszewski, O.; Dimou, L.; Prinz, M. Microglia contribute to normal myelinogenesis and to oligodendrocyte progenitor maintenance during adulthood. Acta Neuropathol. 2017, 134, 441–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wlodarczyk, A.; Holtman, I.R.; Krueger, M.; Yogev, N.; Bruttger, J.; Khorooshi, R.; Benmamar-Badel, A.; de Boer-Bergsma, J.J.; Martin, N.A.; Karram, K.; et al. A novel microglial subset plays a key role in myelinogenesis in developing brain. Embo J. 2017, 36, 3292–3308. [Google Scholar] [CrossRef] [PubMed]
- Skripuletz, T.; Hackstette, D.; Bauer, K.; Gudi, V.; Pul, R.; Voss, E.; Berger, K.; Kipp, M.; Baumgartner, W.; Stangel, M. Astrocytes regulate myelin clearance through recruitment of microglia during cuprizone-induced demyelination. Brain 2013, 136, 147–167. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the brain sections are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
| Name | Host/Clone | Order Number | RRID | Supplier |
|---|---|---|---|---|
| MAG | Mouse monoclonal | ab89780 | AB_2042411 | Abcam |
| IBA1 | Rabbit polyclonal | 019-19741 | AB_839504 | WAKO |
| APP (A4) | Mouse monoclonal | MAB348 | AB_94882 | Millipore |
| APC (Ab-7) | Mouse monoclonal | OP80 | AB_2057371 | Millipore |
| PLP | Mouse monoclonal | MCA839G | AB_2237198 | Biorad |
| OLIG2 | Mouse monoclonal | MABN50 | AB_10807410 | Millipore |
| Ki67 | Rabbit monoclonal | ab16667 | AB_302459 | Abcam |
| Name | Host/Clone | Order Number | RRID | Supplier |
|---|---|---|---|---|
| Biotinylated Goat Anti-Rabbit IgG | Goat Polyclonal | BA-1000 | AB_2313606 | Vector Laboratories |
| Biotinylated Horse Anti-Mouse IgG | Horse Polyclonal | BA-2000 | AB_2313581 | Vector Laboratories |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Donkey Polyclonal | A21206 | AB_2535792 | Thermo Fisher Scientific |
| Goat anti-Mouse IgG2a Cross-Adsorbed Secondary Antibody, Alexa Fluor 546 | Goat Polyclonal | A21133 | AB_2535772 | Thermo Fisher Scientific |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nyamoya, S.; Steinle, J.; Chrzanowski, U.; Kaye, J.; Schmitz, C.; Beyer, C.; Kipp, M. Laquinimod Supports Remyelination in Non-Supportive Environments. Cells 2019, 8, 1363. https://doi.org/10.3390/cells8111363
Nyamoya S, Steinle J, Chrzanowski U, Kaye J, Schmitz C, Beyer C, Kipp M. Laquinimod Supports Remyelination in Non-Supportive Environments. Cells. 2019; 8(11):1363. https://doi.org/10.3390/cells8111363
Chicago/Turabian StyleNyamoya, Stella, Julia Steinle, Uta Chrzanowski, Joel Kaye, Christoph Schmitz, Cordian Beyer, and Markus Kipp. 2019. "Laquinimod Supports Remyelination in Non-Supportive Environments" Cells 8, no. 11: 1363. https://doi.org/10.3390/cells8111363
APA StyleNyamoya, S., Steinle, J., Chrzanowski, U., Kaye, J., Schmitz, C., Beyer, C., & Kipp, M. (2019). Laquinimod Supports Remyelination in Non-Supportive Environments. Cells, 8(11), 1363. https://doi.org/10.3390/cells8111363