Mitophagy in Parkinson’s Disease: From Pathogenesis to Treatment

Abstract
:1. Introduction
2. Mitophagy Pathways
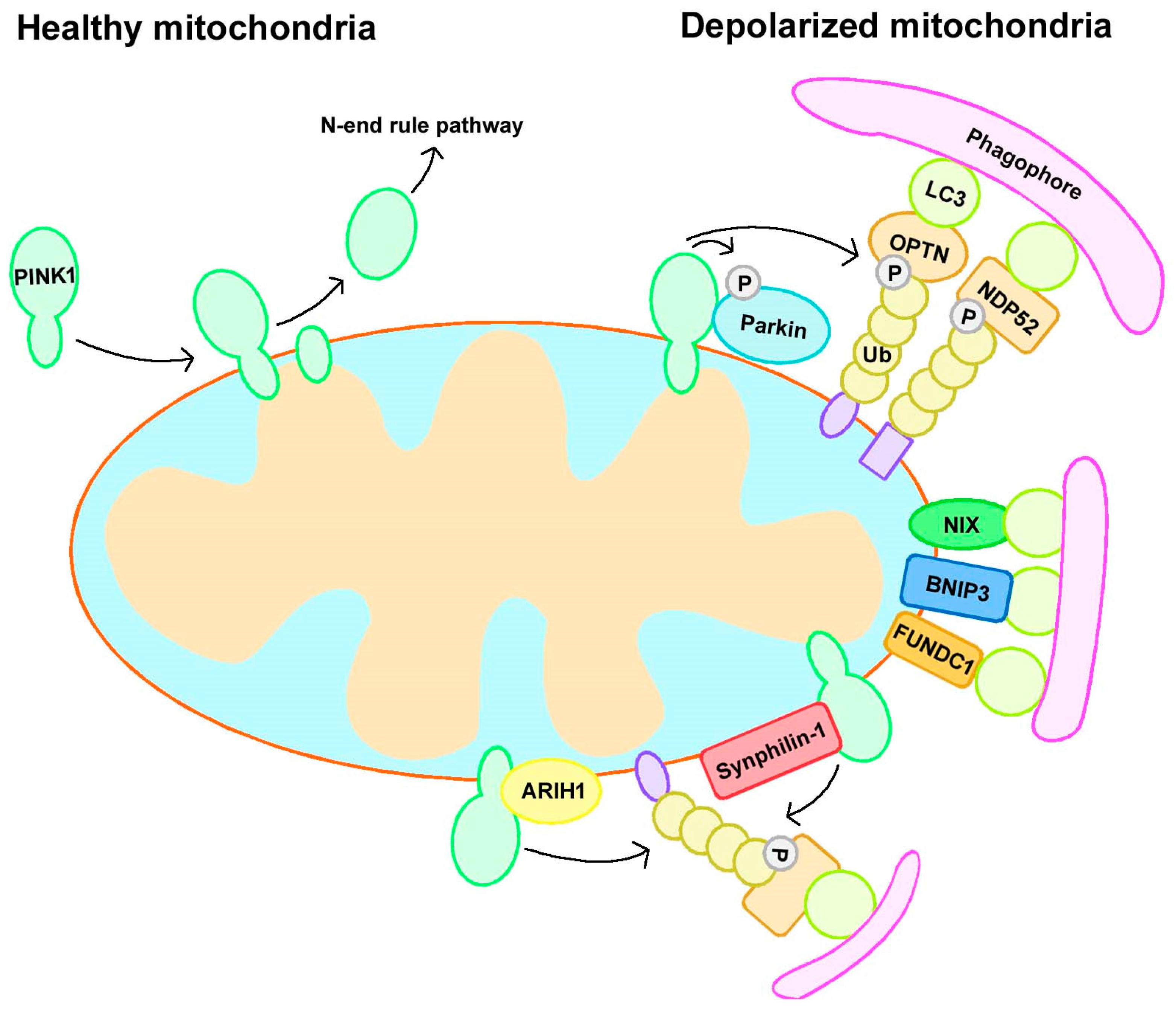
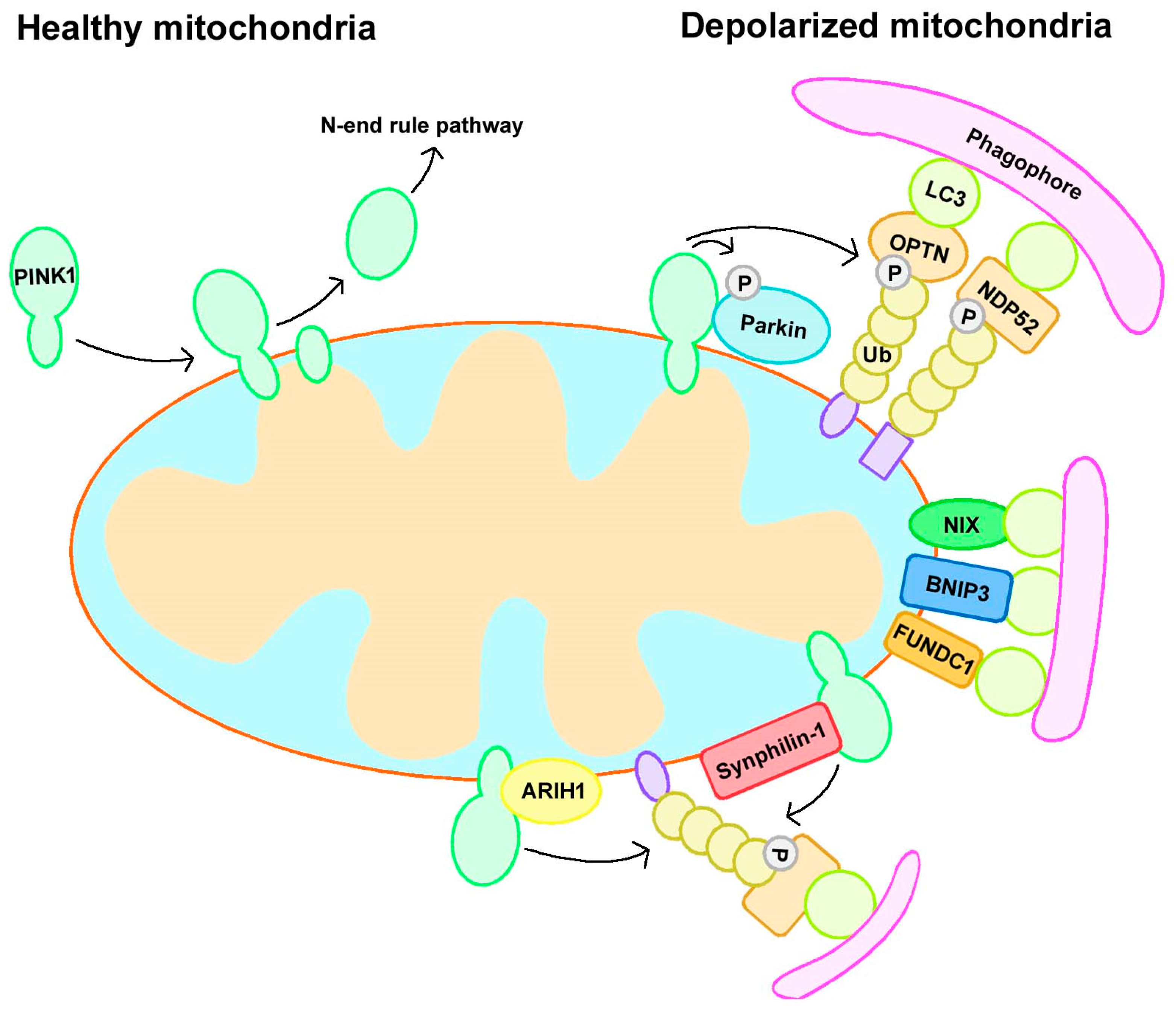
2.1. Parkin-Dependent Mitophagy
2.2. Parkin-Independent Mitophagy
2.2.1. Receptor-Mediated Mitophagy
2.2.2. Ubiquitin Ligase-Mediated Mitophagy
3. Mitophagy in PD Pathogenesis
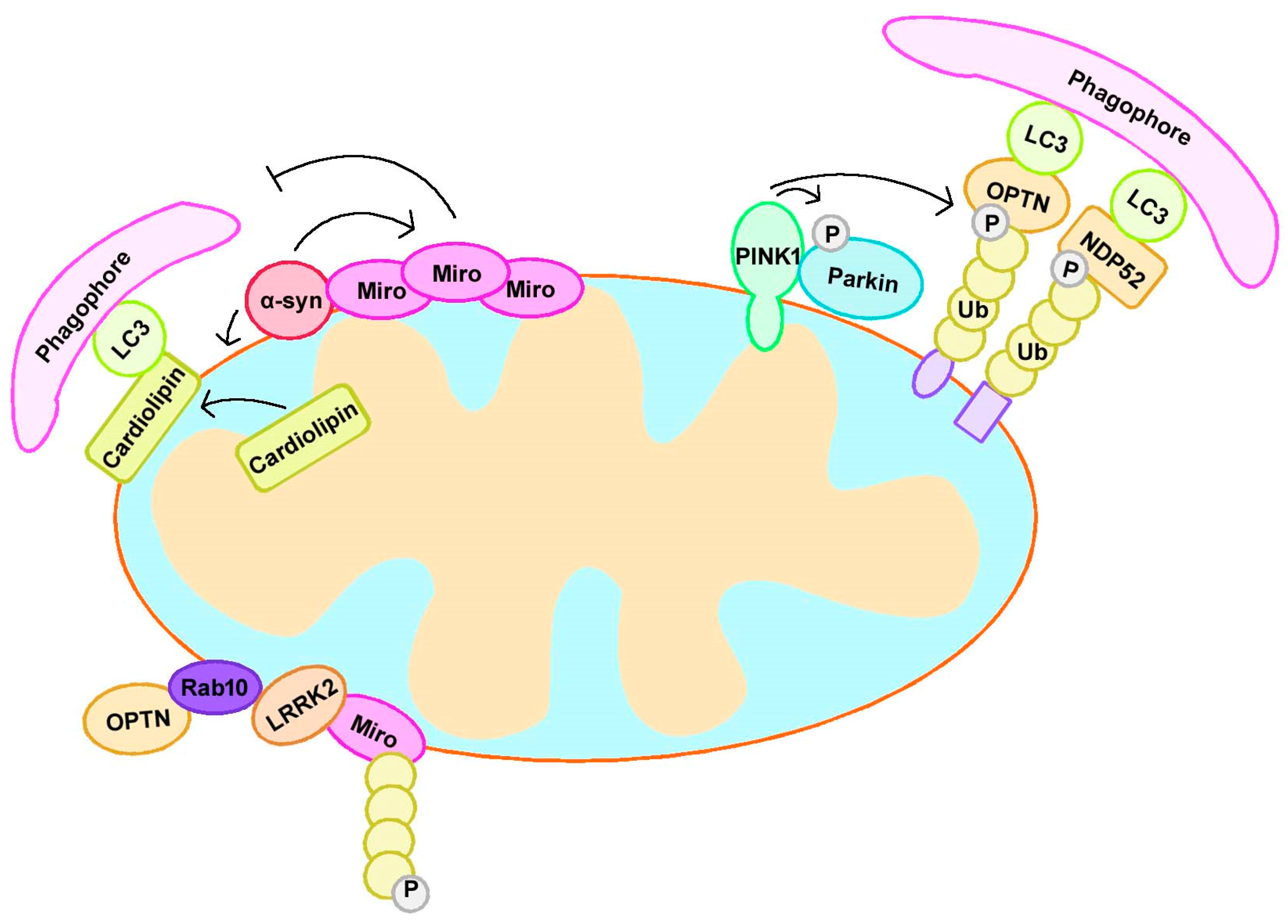
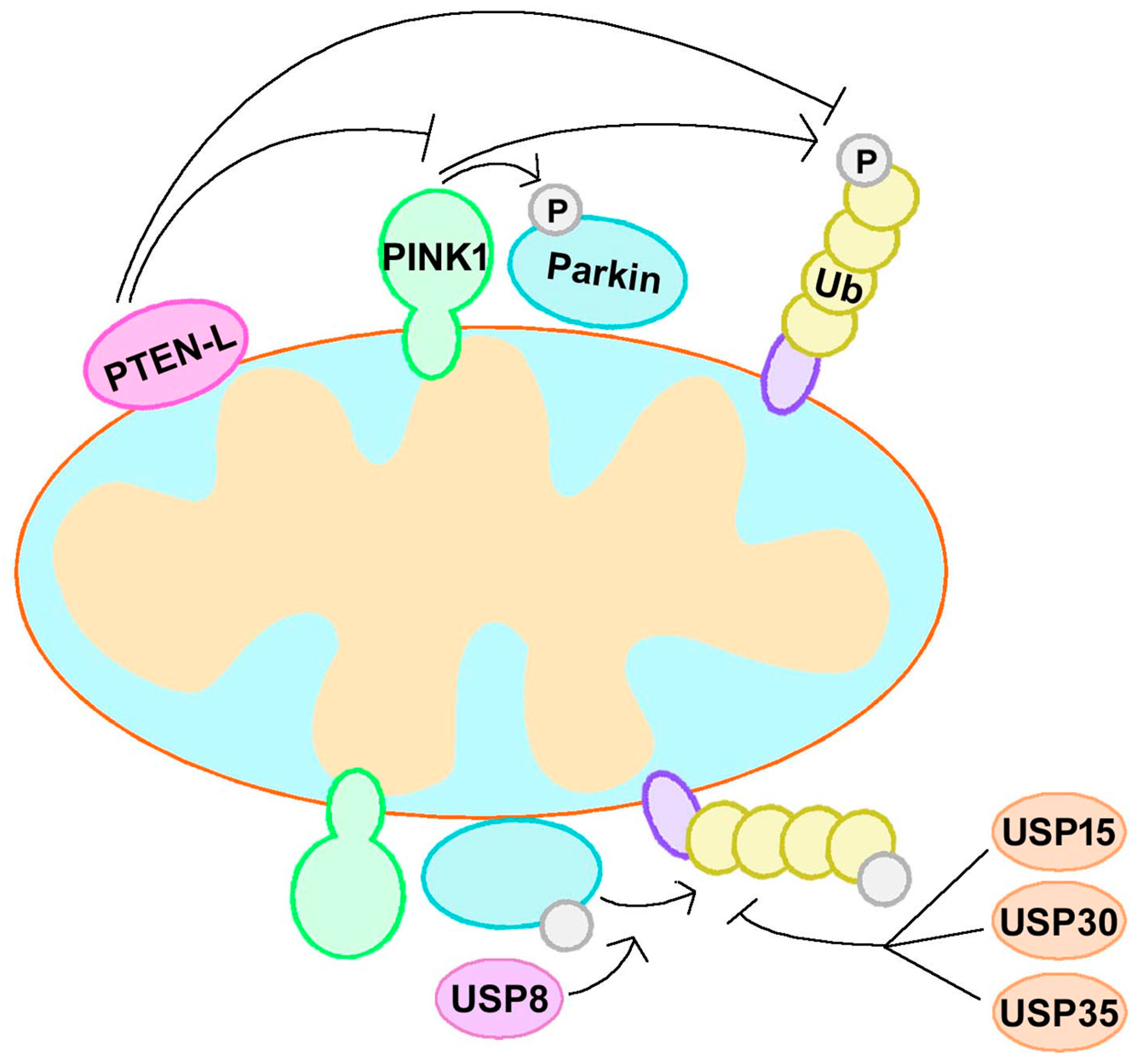
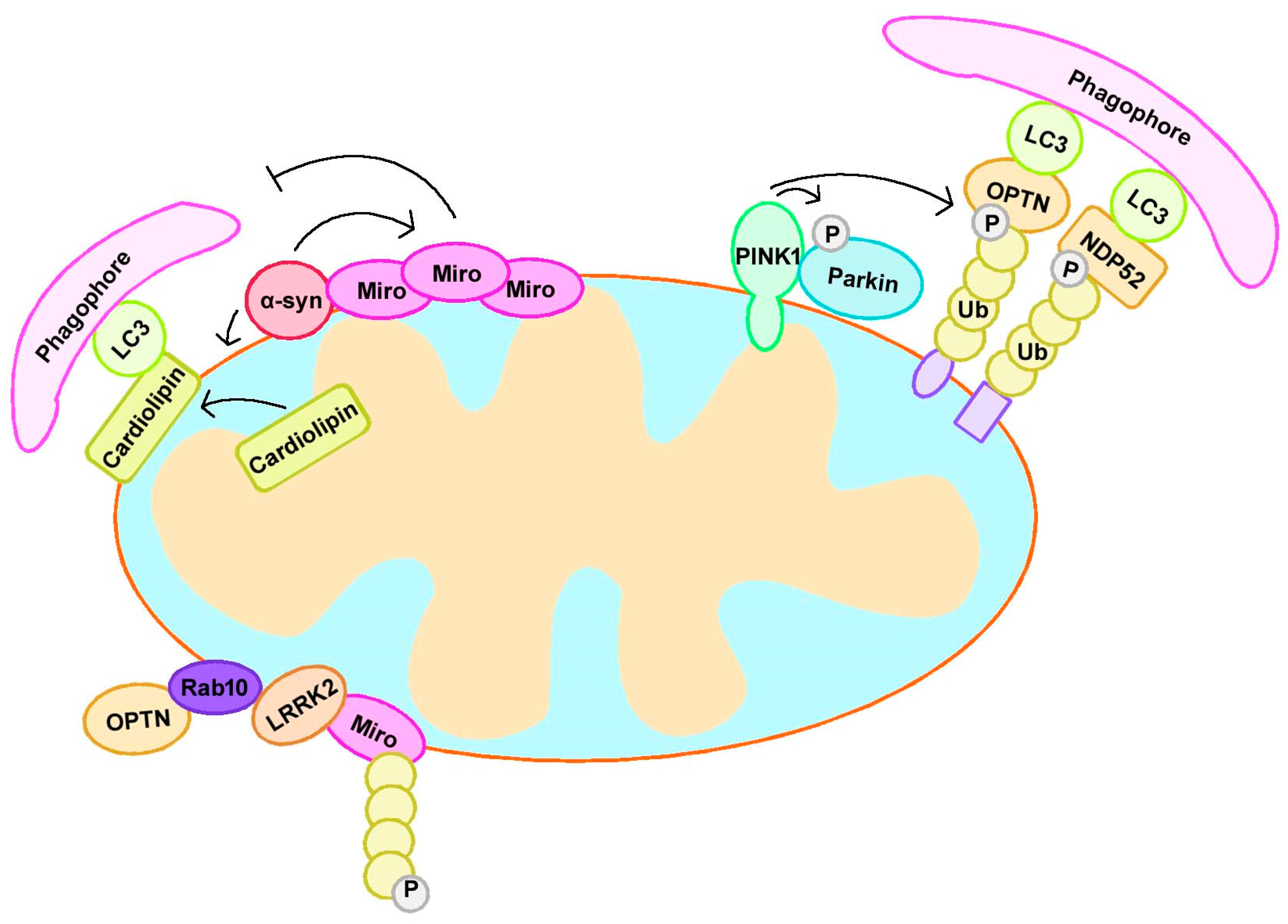
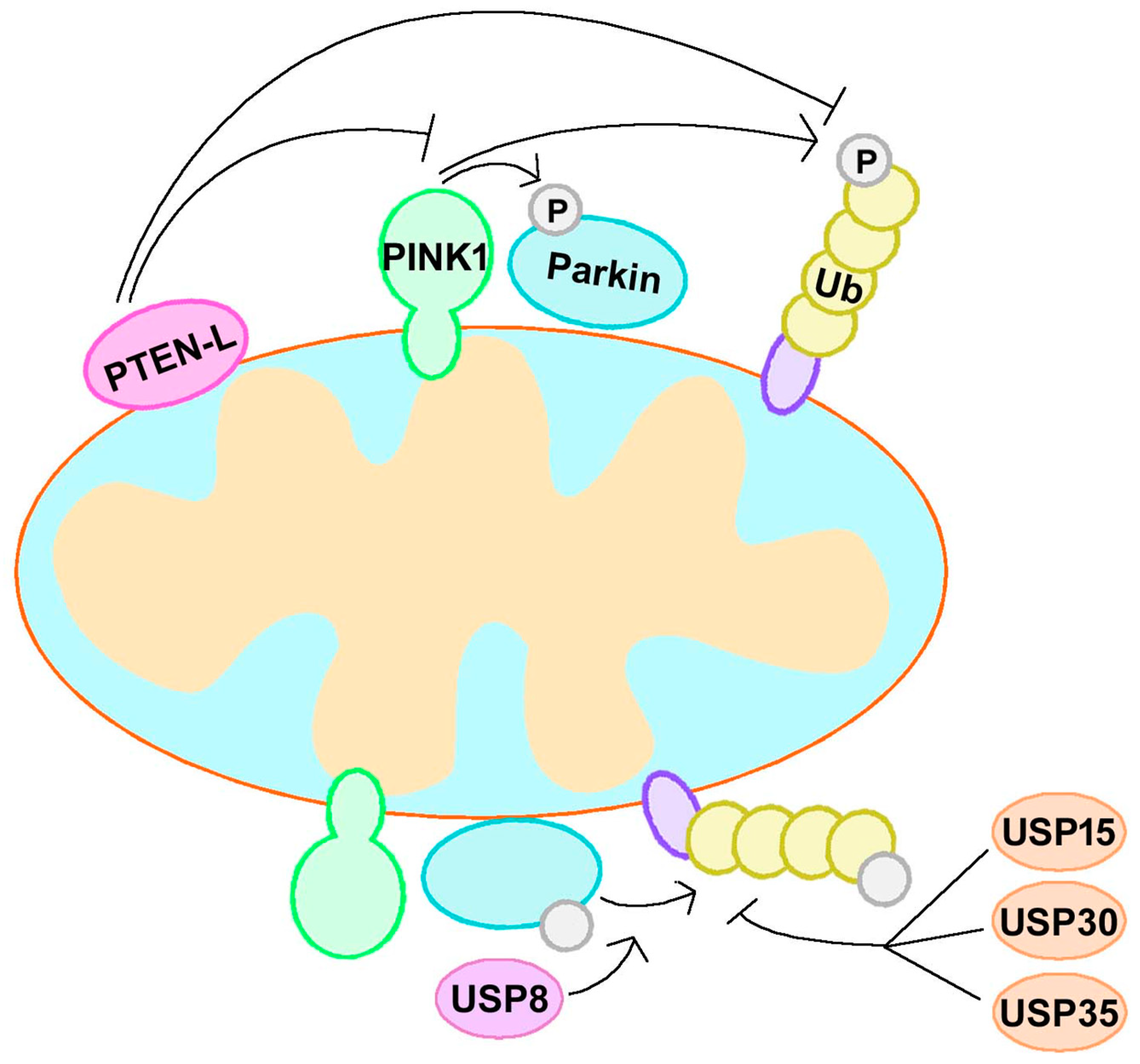
3.1. PINK1 and Parkin
3.1.1. Canonical Mitophagy Regulation
3.1.2. Non-Canonical Mitophagy Regulation
3.1.3. PINK1/Parkin-Mediated Mitophagy and Mitochondrial Dynamics
3.2. α-Synuclein
3.3. LRRK2
3.4. DJ-1
3.5. GBA1
3.6. Other PD-Related Proteins
4. Modulation of Mitophagy in PD Treatment
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Homayoun, H. Parkinson Disease. Ann. Intern. Med. 2018, 169, ITC33–ITC48. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Parkinson disease in 2015: Evolving basic, pathological and clinical concepts in PD. Nat. Rev. Neurol. 2016, 12, 65–66. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.H.; Min, J.; Jeon, S.R. Optogenetics to restore neural circuit function in Parkinson’s disease. J. Neurorestoratol. 2018, 6, 88–92. [Google Scholar] [CrossRef]
- Verstraeten, A.; Theuns, J.; Van Broeckhoven, C. Progress in unraveling the genetic etiology of Parkinson disease in a genomic era. Trends Genet. 2015, 31, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munishkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J.; et al. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J. Biol. Chem. 2011, 286, 20710–20726. [Google Scholar] [CrossRef] [PubMed]
- Papkovskaia, T.D.; Chau, K.Y.; Inesta-Vaquera, F.; Papkovsky, D.B.; Healy, D.G.; Nishio, K.; Staddon, J.; Duchen, M.R.; Hardy, J.; Schapira, A.H.; et al. G2019S leucine-rich repeat kinase 2 causes uncoupling protein-mediated mitochondrial depolarization. Hum. Mol. Genet. 2012, 21, 4201–4213. [Google Scholar] [CrossRef] [PubMed]
- Ramonet, D.; Podhajska, A.; Stafa, K.; Sonnay, S.; Trancikova, A.; Tsika, E.; Pletnikova, O.; Troncoso, J.C.; Glauser, L.; Moore, D.J. PARK9-associated ATP13A2 localizes to intracellular acidic vesicles and regulates cation homeostasis and neuronal integrity. Hum. Mol. Genet. 2012, 21, 1725–1743. [Google Scholar] [CrossRef]
- Wang, W.; Wang, X.; Fujioka, H.; Hoppel, C.; Whone, A.L.; Caldwell, M.A.; Cullen, P.J.; Liu, J.; Zhu, X. Parkinson’s disease-associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat. Med. 2016, 22, 54–63. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef]
- Palikaras, K.; Daskalaki, I.; Markaki, M.; Tavernarakis, N. Mitophagy and age-related pathologies: Development of new therapeutics by targeting mitochondrial turnover. Pharm. Ther. 2017, 178, 157–174. [Google Scholar] [CrossRef]
- Park, J.T.; Lee, Y.S.; Cho, K.A.; Park, S.C. Adjustment of the lysosomal-mitochondrial axis for control of cellular senescence. Ageing Res. Rev. 2018, 47, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.N.; Padman, B.S.; Lazarou, M. Deciphering the Molecular Signals of PINK1/Parkin Mitophagy. Trends Cell Biol. 2016, 26, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Villa, E.; Marchetti, S.; Ricci, J.E. No Parkin Zone: Mitophagy without Parkin. Trends Cell Biol. 2018, 28, 882–895. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. Plos Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Morais, V.A.; Haddad, D.; Craessaerts, K.; De Bock, P.J.; Swerts, J.; Vilain, S.; Aerts, L.; Overbergh, L.; Grunewald, A.; Seibler, P.; et al. PINK1 loss-of-function mutations affect mitochondrial complex I activity via NdufA10 ubiquinone uncoupling. Science 2014, 344, 203–207. [Google Scholar] [CrossRef]
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006, 441, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Khymenets, O.; Ortuno, J.; Fito, M.; Covas, M.A.; Farre, M.; de la Torre, R. Evaluation of RNA isolation procedures from human blood and its application for gene expression studies (Sod-1, Sod-2). Anal. Biochem. 2005, 347, 156–158. [Google Scholar] [CrossRef] [PubMed]
- McWilliams, T.G.; Muqit, M.M. PINK1 and Parkin: Emerging themes in mitochondrial homeostasis. Curr. Opin. Cell. Biol. 2017, 45, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W.; Ordureau, A.; Heo, J.M. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Yamano, K.; Youle, R.J. PINK1 is degraded through the N-end rule pathway. Autophagy 2013, 9, 1758–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meissner, C.; Lorenz, H.; Hehn, B.; Lemberg, M.K. Intramembrane protease PARL defines a negative regulator of PINK1- and PARK2/Parkin-dependent mitophagy. Autophagy 2015, 11, 1484–1498. [Google Scholar] [CrossRef] [Green Version]
- Trempe, J.F.; Sauve, V.; Grenier, K.; Seirafi, M.; Tang, M.Y.; Menade, M.; Al-Abdul-Wahid, S.; Krett, J.; Wong, K.; Kozlov, G.; et al. Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 2013, 340, 1451–1455. [Google Scholar] [CrossRef]
- Wong, Y.C.; Holzbaur, E.L. Temporal dynamics of PARK2/parkin and OPTN/optineurin recruitment during the mitophagy of damaged mitochondria. Autophagy 2015, 11, 422–424. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.C.; Holzbaur, E.L. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Sakakibara, K.; Chen, Q.; Okamoto, K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 2014, 24, 787–795. [Google Scholar] [CrossRef] [Green Version]
- Hanna, R.A.; Quinsay, M.N.; Orogo, A.M.; Giang, K.; Rikka, S.; Gustafsson, A.B. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J. Biol. Chem. 2012, 287, 19094–19104. [Google Scholar] [CrossRef] [PubMed]
- Rogov, V.V.; Suzuki, H.; Marinkovic, M.; Lang, V.; Kato, R.; Kawasaki, M.; Buljubasic, M.; Sprung, M.; Rogova, N.; Wakatsuki, S.; et al. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci. Rep. 2017, 7, 1131. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015, 22, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Van Humbeeck, C.; Cornelissen, T.; Hofkens, H.; Mandemakers, W.; Gevaert, K.; De Strooper, B.; Vandenberghe, W. Parkin interacts with Ambra1 to induce mitophagy. J. Neurosci. 2011, 31, 10249–10261. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 2013, 15, 1197–1205. [Google Scholar] [CrossRef] [Green Version]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Villa, E.; Proics, E.; Rubio-Patino, C.; Obba, S.; Zunino, B.; Bossowski, J.P.; Rozier, R.M.; Chiche, J.; Mondragon, L.; Riley, J.S.; et al. Parkin-Independent Mitophagy Controls Chemotherapeutic Response in Cancer Cells. Cell Rep. 2017, 20, 2846–2859. [Google Scholar] [CrossRef] [Green Version]
- Yun, J.; Puri, R.; Yang, H.; Lizzio, M.A.; Wu, C.; Sheng, Z.H.; Guo, M. MUL1 acts in parallel to the PINK1/parkin pathway in regulating mitofusin and compensates for loss of PINK1/parkin. Elife 2014, 3, e01958. [Google Scholar] [CrossRef]
- Szargel, R.; Shani, V.; Abd Elghani, F.; Mekies, L.N.; Liani, E.; Rott, R.; Engelender, S. The PINK1, synphilin-1 and SIAH-1 complex constitutes a novel mitophagy pathway. Hum. Mol. Genet. 2016, 25, 3476–3490. [Google Scholar] [CrossRef] [Green Version]
- Grunewald, A.; Kumar, K.R.; Sue, C.M. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Sharma, H.S.; Chen, L.; Saberi, H.; Mao, G. 2018 Yearbook of Neurorestoratology. J. Neurorestoratol. 2019, 7, 8–17. [Google Scholar] [Green Version]
- Parker, W.D., Jr.; Boyson, S.J.; Parks, J.K. Abnormalities of the electron transport chain in idiopathic Parkinson’s disease. Ann. Neurol. 1989, 26, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.H.; Guo, F.; Shelburne, J.; Watkins, S.; Chu, C.T. Localization of phosphorylated ERK/MAP kinases to mitochondria and autophagosomes in Lewy body diseases. Brain Pathol. 2003, 13, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Cherra, S.J., 3rd; Steer, E.; Gusdon, A.M.; Kiselyov, K.; Chu, C.T. Mutant LRRK2 elicits calcium imbalance and depletion of dendritic mitochondria in neurons. Am. J. Pathol. 2013, 182, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Mallajosyula, J.K.; Rane, A.; Andersen, J.K. Mitochondrial alpha-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci. Lett. 2010, 486, 235–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagda, R.K.; Cherra, S.J., 3rd; Kulich, S.M.; Tandon, A.; Park, D.; Chu, C.T. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. 2009, 284, 13843–13855. [Google Scholar] [CrossRef] [PubMed]
- Osellame, L.D.; Duchen, M.R. Defective quality control mechanisms and accumulation of damaged mitochondria link Gaucher and Parkinson diseases. Autophagy 2013, 9, 1633–1635. [Google Scholar] [CrossRef] [Green Version]
- Dagda, R.K.; Zhu, J.; Kulich, S.M.; Chu, C.T. Mitochondrially localized ERK2 regulates mitophagy and autophagic cell stress: Implications for Parkinson’s disease. Autophagy 2008, 4, 770–782. [Google Scholar] [CrossRef]
- Zhu, J.H.; Horbinski, C.; Guo, F.; Watkins, S.; Uchiyama, Y.; Chu, C.T. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am. J. Pathol. 2007, 170, 75–86. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [PubMed]
- Ordureau, A.; Sarraf, S.A.; Duda, D.M.; Heo, J.M.; Jedrychowski, M.P.; Sviderskiy, V.O.; Olszewski, J.L.; Koerber, J.T.; Xie, T.; Beausoleil, S.A.; et al. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol. Cell 2014, 56, 360–375. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Fiesel, F.C.; Truban, D.; Castanedes Casey, M.; Lin, W.L.; Soto, A.I.; Tacik, P.; Rousseau, L.G.; Diehl, N.N.; Heckman, M.G.; et al. Age- and disease-dependent increase of the mitophagy marker phospho-ubiquitin in normal aging and Lewy body disease. Autophagy 2018, 14, 1404–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ando, M.; Fiesel, F.C.; Hudec, R.; Caulfield, T.R.; Ogaki, K.; Gorka-Skoczylas, P.; Koziorowski, D.; Friedman, A.; Chen, L.; Dawson, V.L.; et al. The PINK1 p.I368N mutation affects protein stability and ubiquitin kinase activity. Mol. Neurodegener. 2017, 12, 32. [Google Scholar] [CrossRef] [PubMed]
- Yamano, K.; Queliconi, B.B.; Koyano, F.; Saeki, Y.; Hirokawa, T.; Tanaka, K.; Matsuda, N. Site-specific Interaction Mapping of Phosphorylated Ubiquitin to Uncover Parkin Activation. J. Biol. Chem. 2015, 290, 25199–25211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauve, V.; Lilov, A.; Seirafi, M.; Vranas, M.; Rasool, S.; Kozlov, G.; Sprules, T.; Wang, J.; Trempe, J.F.; Gehring, K. A Ubl/ubiquitin switch in the activation of Parkin. EMBO J. 2015, 34, 2492–2505. [Google Scholar] [CrossRef] [PubMed]
- Bingol, B.; Tea, J.S.; Phu, L.; Reichelt, M.; Bakalarski, C.E.; Song, Q.; Foreman, O.; Kirkpatrick, D.S.; Sheng, M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 2014, 510, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Serricchio, M.; Jauregui, M.; Shanbhag, R.; Stoltz, T.; Di Paolo, C.T.; Kim, P.K.; McQuibban, G.A. Deubiquitinating enzymes regulate PARK2-mediated mitophagy. Autophagy 2015, 11, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Durcan, T.M.; Fon, E.A. USP8 and PARK2/parkin-mediated mitophagy. Autophagy 2015, 11, 428–429. [Google Scholar] [CrossRef] [Green Version]
- Durcan, T.M.; Tang, M.Y.; Perusse, J.R.; Dashti, E.A.; Aguileta, M.A.; McLelland, G.L.; Gros, P.; Shaler, T.A.; Faubert, D.; Coulombe, B.; et al. USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from parkin. EMBO J. 2014, 33, 2473–2491. [Google Scholar] [CrossRef]
- Cornelissen, T.; Haddad, D.; Wauters, F.; Van Humbeeck, C.; Mandemakers, W.; Koentjoro, B.; Sue, C.; Gevaert, K.; De Strooper, B.; Verstreken, P.; et al. The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum. Mol. Genet. 2014, 23, 5227–5242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Wang, J.; Tang, Y.; Shen, H.M. PTEN-L puts a brake on mitophagy. Autophagy 2018, 14, 2023–2025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Cho, Y.L.; Tang, Y.; Wang, J.; Park, J.E.; Wu, Y.; Wang, C.; Tong, Y.; Chawla, R.; Zhang, J.; et al. PTEN-L is a novel protein phosphatase for ubiquitin dephosphorylation to inhibit PINK1-Parkin-mediated mitophagy. Cell Res. 2018, 28, 787–802. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, J.D.; Dunkerley, K.M.; Mercier, P.; Shaw, G.S. Structure of phosphorylated UBL domain and insights into PINK1-orchestrated parkin activation. Proc. Natl. Acad. Sci. USA 2017, 114, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, J.D.; Dunkerley, K.M.; Lam, R.; Rusal, M.; Shaw, G.S. Impact of altered phosphorylation on loss of function of juvenile Parkinsonism-associated genetic variants of the E3 ligase parkin. J. Biol. Chem. 2018, 293, 6337–6348. [Google Scholar] [CrossRef] [PubMed]
- McWilliams, T.G.; Barini, E.; Pohjolan-Pirhonen, R.; Brooks, S.P.; Singh, F.; Burel, S.; Balk, K.; Kumar, A.; Montava-Garriga, L.; Prescott, A.R.; et al. Phosphorylation of Parkin at serine 65 is essential for its activation in vivo. Open Biol. 2018, 8, 180108. [Google Scholar] [CrossRef] [PubMed]
- Shiba-Fukushima, K.; Ishikawa, K.I.; Inoshita, T.; Izawa, N.; Takanashi, M.; Sato, S.; Onodera, O.; Akamatsu, W.; Okano, H.; Imai, Y.; et al. Evidence that phosphorylated ubiquitin signaling is involved in the etiology of Parkinson’s disease. Hum. Mol. Genet. 2017, 26, 3172–3185. [Google Scholar] [CrossRef]
- Aerts, L.; Craessaerts, K.; De Strooper, B.; Morais, V.A. PINK1 kinase catalytic activity is regulated by phosphorylation on serines 228 and 402. J. Biol. Chem. 2015, 290, 2798–2811. [Google Scholar] [CrossRef]
- Oh, C.K.; Sultan, A.; Platzer, J.; Dolatabadi, N.; Soldner, F.; McClatchy, D.B.; Diedrich, J.K.; Yates, J.R., 3rd; Ambasudhan, R.; Nakamura, T.; et al. S-Nitrosylation of PINK1 Attenuates PINK1/Parkin-Dependent Mitophagy in hiPSC-Based Parkinson’s Disease Models. Cell Rep. 2017, 21, 2171–2182. [Google Scholar] [CrossRef]
- Da Costa, C.A.; Sunyach, C.; Giaime, E.; West, A.; Corti, O.; Brice, A.; Safe, S.; Abou-Sleiman, P.M.; Wood, N.W.; Takahashi, H.; et al. Transcriptional repression of p53 by parkin and impairment by mutations associated with autosomal recessive juvenile Parkinson’s disease. Nat. Cell Biol. 2009, 11, 1370–1375. [Google Scholar] [CrossRef]
- Zhang, C.; Lin, M.; Wu, R.; Wang, X.; Yang, B.; Levine, A.J.; Hu, W.; Feng, Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc. Natl. Acad. Sci. USA 2011, 108, 16259–16264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viotti, J.; Duplan, E.; Caillava, C.; Condat, J.; Goiran, T.; Giordano, C.; Marie, Y.; Idbaih, A.; Delattre, J.Y.; Honnorat, J.; et al. Glioma tumor grade correlates with parkin depletion in mutant p53-linked tumors and results from loss of function of p53 transcriptional activity. Oncogene 2014, 33, 1764–1775. [Google Scholar] [CrossRef] [PubMed]
- Goiran, T.; Duplan, E.; Rouland, L.; El Manaa, W.; Lauritzen, I.; Dunys, J.; You, H.; Checler, F.; Alves da Costa, C. Nuclear p53-mediated repression of autophagy involves PINK1 transcriptional down-regulation. Cell Death Differ. 2018, 25, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Goiran, T.; Duplan, E.; Chami, M.; Bourgeois, A.; El Manaa, W.; Rouland, L.; Dunys, J.; Lauritzen, I.; You, H.; Stambolic, V.; et al. beta-Amyloid Precursor Protein Intracellular Domain Controls Mitochondrial Function by Modulating Phosphatase and Tensin Homolog-Induced Kinase 1 Transcription in Cells and in Alzheimer Mice Models. Biol. Psychiatry 2018, 83, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Choubey, V.; Cagalinec, M.; Liiv, J.; Safiulina, D.; Hickey, M.A.; Kuum, M.; Liiv, M.; Anwar, T.; Eskelinen, E.L.; Kaasik, A. BECN1 is involved in the initiation of mitophagy: It facilitates PARK2 translocation to mitochondria. Autophagy 2014, 10, 1105–1119. [Google Scholar] [CrossRef] [PubMed]
- Michiorri, S.; Gelmetti, V.; Giarda, E.; Lombardi, F.; Romano, F.; Marongiu, R.; Nerini-Molteni, S.; Sale, P.; Vago, R.; Arena, G.; et al. The Parkinson-associated protein PINK1 interacts with Beclin1 and promotes autophagy. Cell Death Differ. 2010, 17, 962–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelmetti, V.; De Rosa, P.; Torosantucci, L.; Marini, E.S.; Romagnoli, A.; Di Rienzo, M.; Arena, G.; Vignone, D.; Fimia, G.M.; Valente, E.M. PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy 2017, 13, 654–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McWilliams, T.G.; Prescott, A.R.; Montava-Garriga, L.; Ball, G.; Singh, F.; Barini, E.; Muqit, M.M.K.; Brooks, S.P.; Ganley, I.G. Basal Mitophagy Occurs Independently of PINK1 in Mouse Tissues of High Metabolic Demand. Cell Metab. 2018, 27, 439–449.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birsa, N.; Norkett, R.; Wauer, T.; Mevissen, T.E.; Wu, H.C.; Foltynie, T.; Bhatia, K.; Hirst, W.D.; Komander, D.; Plun-Favreau, H.; et al. Lysine 27 ubiquitination of the mitochondrial transport protein Miro is dependent on serine 65 of the Parkin ubiquitin ligase. J. Biol. Chem. 2014, 289, 14569–14582. [Google Scholar] [CrossRef]
- Shlevkov, E.; Kramer, T.; Schapansky, J.; LaVoie, M.J.; Schwarz, T.L. Miro phosphorylation sites regulate Parkin recruitment and mitochondrial motility. Proc. Natl. Acad. Sci. USA 2016, 113, E6097–E6106. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; LaVoie, M.J.; Schwarz, T.L. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef] [PubMed]
- McLelland, G.L.; Fon, E.A. MFN2 retrotranslocation boosts mitophagy by uncoupling mitochondria from the ER. Autophagy 2018, 14, 1658–1660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Dorn, G.W., 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Ziviani, E.; Tao, R.N.; Whitworth, A.J. Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc. Natl. Acad. Sci. USA 2010, 107, 5018–5023. [Google Scholar] [CrossRef] [PubMed]
- Rakovic, A.; Grunewald, A.; Kottwitz, J.; Bruggemann, N.; Pramstaller, P.P.; Lohmann, K.; Klein, C. Mutations in PINK1 and Parkin impair ubiquitination of Mitofusins in human fibroblasts. PLoS ONE 2011, 6, e16746. [Google Scholar] [CrossRef] [PubMed]
- Karbowski, M.; Youle, R.J. Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr. Opin. Cell Biol. 2011, 23, 476–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, S.T.; Bain, A.M.; Bedimo, R.G.; Hall, R.G.; Busti, A.J. Acute elevation of triglycerides after initiation of fosamprinavir/ritonavir in an HIV-negative patient with baseline hypertriglyceridemia. J. Clin. Lipidol. 2008, 2, 398–400. [Google Scholar] [CrossRef]
- McLelland, G.L.; Goiran, T.; Yi, W.; Dorval, G.; Chen, C.X.; Lauinger, N.D.; Krahn, A.I.; Valimehr, S.; Rakovic, A.; Rouiller, I.; et al. Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. Elife 2018, 7, e32866. [Google Scholar] [CrossRef]
- Gautier, C.A.; Erpapazoglou, Z.; Mouton-Liger, F.; Muriel, M.P.; Cormier, F.; Bigou, S.; Duffaure, S.; Girard, M.; Foret, B.; Iannielli, A.; et al. The endoplasmic reticulum-mitochondria interface is perturbed in PARK2 knockout mice and patients with PARK2 mutations. Hum. Mol. Genet. 2016, 25, 2972–2984. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, J.; Wang, X.; Duan, C.; Wang, X.; Yang, H. V63 and N65 of overexpressed alpha-synuclein are involved in mitochondrial dysfunction. Brain Res. 2016, 1642, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Du, T.; Wang, X.; Duan, C.; Gao, G.; Zhang, J.; Lu, L.; Yang, H. alpha-Synuclein amino terminus regulates mitochondrial membrane permeability. Brain Res. 2014, 1591, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.H.; Alaimo, A.; Gorojod, R.M.; Porte Alcon, S.; Fuentes, F.; Coluccio Leskow, F.; Kotler, M.L. Drp-1 dependent mitochondrial fragmentation and protective autophagy in dopaminergic SH-SY5Y cells overexpressing alpha-synuclein. Mol. Cell Neurosci. 2018, 88, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, X.; Lu, Y.; Duan, C.; Gao, G.; Lu, L.; Yang, H. Pink1 interacts with alpha-synuclein and abrogates alpha-synuclein-induced neurotoxicity by activating autophagy. Cell Death Dis. 2017, 8, e3056. [Google Scholar] [CrossRef] [PubMed]
- Shaltouki, A.; Hsieh, C.H.; Kim, M.J.; Wang, X. Alpha-synuclein delays mitophagy and targeting Miro rescues neuron loss in Parkinson’s models. Acta Neuropathol. 2018, 136, 607–620. [Google Scholar] [CrossRef]
- Chen, J.; Ren, Y.; Gui, C.; Zhao, M.; Wu, X.; Mao, K.; Li, W.; Zou, F. Phosphorylation of Parkin at serine 131 by p38 MAPK promotes mitochondrial dysfunction and neuronal death in mutant A53T alpha-synuclein model of Parkinson’s disease. Cell Death Dis. 2018, 9, 700. [Google Scholar] [CrossRef] [PubMed]
- Choubey, V.; Safiulina, D.; Vaarmann, A.; Cagalinec, M.; Wareski, P.; Kuum, M.; Zharkovsky, A.; Kaasik, A. Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J. Biol. Chem. 2011, 286, 10814–10824. [Google Scholar] [CrossRef]
- Ryan, T.; Bamm, V.V.; Stykel, M.G.; Coackley, C.L.; Humphries, K.M.; Jamieson-Williams, R.; Ambasudhan, R.; Mosser, D.D.; Lipton, S.A.; Harauz, G.; et al. Cardiolipin exposure on the outer mitochondrial membrane modulates alpha-synuclein. Nat. Commun 2018, 9, 817. [Google Scholar] [CrossRef]
- Sampaio-Marques, B.; Felgueiras, C.; Silva, A.; Rodrigues, M.; Tenreiro, S.; Franssens, V.; Reichert, A.S.; Outeiro, T.F.; Winderickx, J.; Ludovico, P. SNCA (alpha-synuclein)-induced toxicity in yeast cells is dependent on sirtuin 2 (Sir2)-mediated mitophagy. Autophagy 2012, 8, 1494–1509. [Google Scholar] [CrossRef]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef]
- Schwab, A.J.; Sison, S.L.; Meade, M.R.; Broniowska, K.A.; Corbett, J.A.; Ebert, A.D. Decreased Sirtuin Deacetylase Activity in LRRK2 G2019S iPSC-Derived Dopaminergic Neurons. Stem Cell Rep. 2017, 9, 1839–1852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, J.; Bolognin, S.; Antony, P.M.A.; Nickels, S.L.; Poovathingal, S.K.; Salamanca, L.; Magni, S.; Perfeito, R.; Hoel, F.; Qing, X.; et al. Neural Stem Cells of Parkinson’s Disease Patients Exhibit Aberrant Mitochondrial Morphology and Functionality. Stem Cell Rep. 2019, 12, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Yakhine-Diop, S.M.S.; Niso-Santano, M.; Rodriguez-Arribas, M.; Gomez-Sanchez, R.; Martinez-Chacon, G.; Uribe-Carretero, E.; Navarro-Garcia, J.A.; Ruiz-Hurtado, G.; Aiastui, A.; Cooper, J.M.; et al. Impaired Mitophagy and Protein Acetylation Levels in Fibroblasts from Parkinson’s Disease Patients. Mol. Neurobiol. 2019, 56, 2466–2481. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.H.; Shaltouki, A.; Gonzalez, A.E.; Bettencourt da Cruz, A.; Burbulla, L.F.; St Lawrence, E.; Schule, B.; Krainc, D.; Palmer, T.D.; Wang, X. Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson’s Disease. Cell Stem Cell 2016, 19, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. Destructive cellular paths underlying familial and sporadic Parkinson disease converge on mitophagy. Autophagy 2017, 13, 1998–1999. [Google Scholar] [CrossRef] [Green Version]
- Wauters, F.; Cornelissen, T.; Imberechts, D.; Martin, S.; Koentjoro, B.; Sue, C.; Vangheluwe, P.; Vandenberghe, W. LRRK2 mutations impair depolarization-induced mitophagy through inhibition of mitochondrial accumulation of RAB10. Autophagy 2019, 1–20. [Google Scholar] [CrossRef]
- Bonello, F.; Hassoun, S.M.; Mouton-Liger, F.; Shin, Y.S.; Muscat, A.; Tesson, C.; Lesage, S.; Beart, P.M.; Brice, A.; Krupp, J.; et al. LRRK2 impairs PINK1/Parkin-dependent mitophagy via its kinase activity: Pathologic insights into Parkinson’s disease. Hum. Mol. Genet. 2019. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef]
- Andres-Mateos, E.; Perier, C.; Zhang, L.; Blanchard-Fillion, B.; Greco, T.M.; Thomas, B.; Ko, H.S.; Sasaki, M.; Ischiropoulos, H.; Przedborski, S.; et al. DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proc. Natl. Acad. Sci. USA 2007, 104, 14807–14812. [Google Scholar] [CrossRef] [Green Version]
- Meulener, M.C.; Xu, K.; Thomson, L.; Ischiropoulos, H.; Bonini, N.M. Mutational analysis of DJ-1 in Drosophila implicates functional inactivation by oxidative damage and aging. Proc. Natl. Acad. Sci. USA 2006, 103, 12517–12522. [Google Scholar] [CrossRef]
- Krebiehl, G.; Ruckerbauer, S.; Burbulla, L.F.; Kieper, N.; Maurer, B.; Waak, J.; Wolburg, H.; Gizatullina, Z.; Gellerich, F.N.; Woitalla, D.; et al. Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson’s disease-associated protein DJ-1. PLoS ONE 2010, 5, e9367. [Google Scholar] [CrossRef] [PubMed]
- Joselin, A.P.; Hewitt, S.J.; Callaghan, S.M.; Kim, R.H.; Chung, Y.H.; Mak, T.W.; Shen, J.; Slack, R.S.; Park, D.S. ROS-dependent regulation of Parkin and DJ-1 localization during oxidative stress in neurons. Hum. Mol. Genet. 2012, 21, 4888–4903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duplan, E.; Giaime, E.; Viotti, J.; Sevalle, J.; Corti, O.; Brice, A.; Ariga, H.; Qi, L.; Checler, F.; Alves da Costa, C. ER-stress-associated functional link between Parkin and DJ-1 via a transcriptional cascade involving the tumor suppressor p53 and the spliced X-box binding protein XBP-1. J. Cell Sci. 2013, 126, 2124–2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, K.J.; McCoy, M.K.; Blackinton, J.; Beilina, A.; van der Brug, M.; Sandebring, A.; Miller, D.; Maric, D.; Cedazo-Minguez, A.; Cookson, M.R. DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum. Mol. Genet. 2011, 20, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.Y.; Giasson, B.I.; Bonini, N.M. DJ-1 is critical for mitochondrial function and rescues PINK1 loss of function. Proc. Natl. Acad. Sci. USA 2010, 107, 9747–9752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [PubMed]
- Tayebi, N.; Callahan, M.; Madike, V.; Stubblefield, B.K.; Orvisky, E.; Krasnewich, D.; Fillano, J.J.; Sidransky, E. Gaucher disease and parkinsonism: A phenotypic and genotypic characterization. Mol. Genet. Metab. 2001, 73, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Migdalska-Richards, A.; Schapira, A.H. The relationship between glucocerebrosidase mutations and Parkinson disease. J. Neurochem. 2016, 139 (Suppl. 1), 77–90. [Google Scholar] [CrossRef] [Green Version]
- Valley, N.A.; Blower, P.G.; Wood, S.R.; Plath, K.L.; McWilliams, L.E.; Richmond, G.L. Doubling down: Delving into the details of diacid adsorption at aqueous surfaces. J. Phys. Chem. A 2014, 118, 4778–4789. [Google Scholar] [CrossRef]
- De la Mata, M.; Cotan, D.; Oropesa-Avila, M.; Garrido-Maraver, J.; Cordero, M.D.; Villanueva Paz, M.; Delgado Pavon, A.; Alcocer-Gomez, E.; de Lavera, I.; Ybot-Gonzalez, P.; et al. Pharmacological Chaperones and Coenzyme Q10 Treatment Improves Mutant beta-Glucocerebrosidase Activity and Mitochondrial Function in Neuronopathic Forms of Gaucher Disease. Sci. Rep. 2015, 5, 10903. [Google Scholar] [CrossRef]
- Choi, J.H.; Stubblefield, B.; Cookson, M.R.; Goldin, E.; Velayati, A.; Tayebi, N.; Sidransky, E. Aggregation of alpha-synuclein in brain samples from subjects with glucocerebrosidase mutations. Mol. Genet. Metab. 2011, 104, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Cullen, V.; Sardi, S.P.; Ng, J.; Xu, Y.H.; Sun, Y.; Tomlinson, J.J.; Kolodziej, P.; Kahn, I.; Saftig, P.; Woulfe, J.; et al. Acid beta-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter alpha-synuclein processing. Ann. Neurol. 2011, 69, 940–953. [Google Scholar] [CrossRef] [PubMed]
- Osellame, L.D.; Rahim, A.A.; Hargreaves, I.P.; Gegg, M.E.; Richard-Londt, A.; Brandner, S.; Waddington, S.N.; Schapira, A.H.; Duchen, M.R. Mitochondria and quality control defects in a mouse model of Gaucher disease--links to Parkinson’s disease. Cell Metab. 2013, 17, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ham, A.; Ma, T.C.; Kuo, S.H.; Kanter, E.; Kim, D.; Ko, H.S.; Quan, Y.; Sardi, S.P.; Li, A.; et al. Mitochondrial dysfunction and mitophagy defect triggered by heterozygous GBA mutations. Autophagy 2019, 15, 113–130. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.L.; Liu, W.; Hu, J.X.; Erion, J.R.; Ye, J.; Mei, L.; Xiong, W.C. VPS35 Deficiency or Mutation Causes Dopaminergic Neuronal Loss by Impairing Mitochondrial Fusion and Function. Cell Rep. 2015, 12, 1631–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, B.R.; Godena, V.K.; Whitworth, A.J. VPS35 pathogenic mutations confer no dominant toxicity but partial loss of function in Drosophila and genetically interact with parkin. Hum. Mol. Genet. 2015, 24, 6106–6117. [Google Scholar] [CrossRef]
- Delgado-Camprubi, M.; Esteras, N.; Soutar, M.P.; Plun-Favreau, H.; Abramov, A.Y. Deficiency of Parkinson’s disease-related gene Fbxo7 is associated with impaired mitochondrial metabolism by PARP activation. Cell Death Differ. 2017, 24, 120–131. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Xie, S.P.; Sathiyamoorthy, S.; Saw, W.T.; Sing, T.Y.; Ng, S.H.; Chua, H.P.; Tang, A.M.; Shaffra, F.; Li, Z.; et al. F-box protein 7 mutations promote protein aggregation in mitochondria and inhibit mitophagy. Hum. Mol. Genet. 2015, 24, 6314–6330. [Google Scholar] [CrossRef] [Green Version]
- Burchell, V.S.; Nelson, D.E.; Sanchez-Martinez, A.; Delgado-Camprubi, M.; Ivatt, R.M.; Pogson, J.H.; Randle, S.J.; Wray, S.; Lewis, P.A.; Houlden, H.; et al. The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat. Neurosci. 2013, 16, 1257–1265. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Sathiyamoorthy, S.; Angeles, D.C.; Tan, E.K. Linking F-box protein 7 and parkin to neuronal degeneration in Parkinson’s disease (PD). Mol. Brain 2016, 9, 41. [Google Scholar] [CrossRef]
- Moors, T.E.; Hoozemans, J.J.; Ingrassia, A.; Beccari, T.; Parnetti, L.; Chartier-Harlin, M.C.; van de Berg, W.D. Therapeutic potential of autophagy-enhancing agents in Parkinson’s disease. Mol. Neurodegener. 2017, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- East, D.A.; Campanella, M. Mitophagy and the therapeutic clearance of damaged mitochondria for neuroprotection. Int. J. Biochem. Cell. Biol. 2016, 79, 382–387. [Google Scholar] [CrossRef] [Green Version]
- East, D.A.; Fagiani, F.; Crosby, J.; Georgakopoulos, N.D.; Bertrand, H.; Schaap, M.; Fowkes, A.; Wells, G.; Campanella, M. PMI: A DeltaPsim independent pharmacological regulator of mitophagy. Chem. Biol. 2014, 21, 1585–1596. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; MacDougall, E.J.; Tang, M.Y.; Krahn, A.I.; Gan-Or, Z.; Trempe, J.F.; Fon, E.A. The Landscape of Parkin Variants Reveals Pathogenic Mechanisms and Therapeutic Targets in Parkinson’s Disease. Hum. Mol. Genet. 2019. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulos, N.D.; Wells, G.; Campanella, M. The pharmacological regulation of cellular mitophagy. Nat. Chem. Bio.l 2017, 13, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Koentjoro, B.; Park, J.S.; Sue, C.M. Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Sci. Rep. 2017, 7, 44373. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Koentjoro, B.; Sue, C.M. Commentary: Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Front. Mol. Neurosci. 2017, 10, 297. [Google Scholar] [CrossRef]
- Olszewska, D.A.; Lynch, T. Nature’s Parkin experiment: Nix-a novel protective mechanism in Parkinson’s disease. Mov. Disord. 2017, 32, 992. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Loci | Gene | Protein | Position | Inheritance | Onset |
|---|---|---|---|---|---|
| PARK1 | SNCA | Alpha-synuclein | 4q21 | AD, sporadic | Early or late |
| PARK2 | PRKN | Parkin | 6q25–q27 | AR, sporadic | Early |
| PARK3 | Unknown | Unknown | 2p13 | AD | Late |
| PARK5 | UCHL1 | Ubiquitin C-Terminal Hydrolase L1 | 4p14 | AD | Late |
| PARK6 | PINK1 | PTEN-induced putative kinase 1 | 1p35–p36 | AR | Early |
| PARK7 | DJ-1 | Protein DJ-1 | 1p36 | AR | Early |
| PARK8 | LRRK2 | Leucine-rich repeat kinase 2 | 12q12 | AD, sporadic | Early or late |
| PARK9 | ATP13A2 | ATPase 13A2 | 1p36 | AR | Early |
| PARK10 | Unknown | Unknown | 1p32 | Unknown | Unknown |
| PARK11 | GIGYF2 | GRB10 interacting GYF protein 2 | 2q36–q37 | AD | Late |
| PARK12 | Unknown | Unknown | Xq21–q25 | Unknown | Unknown |
| PARK13 | HTRA2 | Serine peptidase 2 | 2p13 | AD | Late |
| PARK14 | PLA2G6 | Phospholipase A2 Group VI | 22q12–q13 | AR | Early |
| PARK15 | FBX07 | F-Box protein 7 | 22q12–q13 | AR | Early |
| PARK17 | VPS35 | Vacuolar protein sorting 35 | 16q11.2 | AD | Late |
| PARK18 | EIF4G1 | Eukaryotic translation initiation factor 4 gamma, 1 | 3q27.1 | AD | Late |
| PARK19 | DNAJC6 | DNAJ subfamily C member 6 | 1p31.3 | AR | Early |
| PARK20 | SYNJ1 | Synaptojanin-1 | 21q22.11 | AR | Early |
| PARK21 | DNAJC13 | DNAJ subfamily C member 13 | 3q22.1 | AD | Early |
| PARK22 | CHCHD2 | Coiled coil-helix-coiled coil-helix domain 2 | 7p11.2 | AD | Late |
| PARK23 | VPS13C | Vacuolar protein sorting 13 homolog C | 15q22.2 | AR | Early |
| - | GBA | Glucocerebrosidase | 1q21 | AD | Unknown |
| Gene | Model | Motor Deficits | Loss of DA Neurons | α-syn Pathology | Mitophagy Defect | Mitochondrial Dynamics Defect |
|---|---|---|---|---|---|---|
| SNCA | Wide-type α-syn overexpression | + | + | + | + | + |
| A53T α-syn overexpression | + | + | + | + | + | |
| A30P α-syn overexpression | + | + | + | - | - | |
| PRKN | Parkin deficiency | - | - | - | + | + |
| PINK1 | PINK1 deficiency | - | - | - | + | + |
| DJ-1 | DJ-1 deficiency | + | - | - | + | + |
| LRRK2 | G2019S LRRK2 overexpression | + | + | - | + | + |
| GBA | L1444P GBA overexpression | + | unknown | + | + | + |
| GBA deficiency | + | unknown | + | + | + |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Liu, W.; Li, R.; Yang, H. Mitophagy in Parkinson’s Disease: From Pathogenesis to Treatment. Cells 2019, 8, 712. https://doi.org/10.3390/cells8070712
Liu J, Liu W, Li R, Yang H. Mitophagy in Parkinson’s Disease: From Pathogenesis to Treatment. Cells. 2019; 8(7):712. https://doi.org/10.3390/cells8070712
Chicago/Turabian StyleLiu, Jia, Weijin Liu, Ruolin Li, and Hui Yang. 2019. "Mitophagy in Parkinson’s Disease: From Pathogenesis to Treatment" Cells 8, no. 7: 712. https://doi.org/10.3390/cells8070712
APA StyleLiu, J., Liu, W., Li, R., & Yang, H. (2019). Mitophagy in Parkinson’s Disease: From Pathogenesis to Treatment. Cells, 8(7), 712. https://doi.org/10.3390/cells8070712