Abstract
Mutations in the gene encoding leucine-rich repeat kinase 2 (LRRK2) are common genetic risk factors for both familial and sporadic Parkinson’s disease (PD). Pathogenic mutations in LRRK2 have been shown to induce changes in its activity, and abnormal increase in LRRK2 kinase activity is thought to contribute to PD pathology. The precise molecular mechanisms underlying LRRK2-associated PD pathology are far from clear, however the identification of LRRK2 substrates and the elucidation of cellular pathways involved suggest a role of LRRK2 in microtubule dynamics, vesicular trafficking, and synaptic transmission. Moreover, LRRK2 is associated with pathologies of α-synuclein, a major component of Lewy bodies (LBs). Evidence from various cellular and animal models supports a role of LRRK2 in the regulation of aggregation and propagation of α-synuclein. Here, we summarize our current understanding of how pathogenic mutations dysregulate LRRK2 and discuss the possible mechanisms leading to neurodegeneration.
1. Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease among the elderly population. The cardinal clinical symptoms of PD include resting tremors, muscular rigidity, bradykinesia, postural instability, and gait problems. Patients with PD often present unilateral motor symptoms that eventually become bilateral as the disease progresses [1]. Because motor symptoms predominantly result from the prominent loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc), current treatment strategies focus on dopamine replacement. Although the diagnosis of PD relies on clinical features derived from dopamine deficiency, it has long been recognized that pathology underlying PD involves several brain areas beyond the dopaminergic system, and that pathology also extends into the peripheral autonomous nervous system. Indeed, the majority of PD patients show a variety of non-motor symptoms, including hyposmia, sleep disruption, and constipation, and non-motor dysfunctions commonly precede motor symptoms by several years [2]. This has motivated clinical studies to assess olfactory dysfunction or rapid eye movement sleep behavior disorder as potential risk factors of developing PD [3]. Other non-motor symptoms, such as dementia, cognitive dysfunction, and hallucinations arise later in the disease. Collectively, the complicated clinical features imply that a broad spectrum of neurophysiological mechanisms are involved in PD pathology.
The majority of PD cases are idiopathic, while 5–10% of PD cases are familial and linked to mutations in multiple genes, such as α-synuclein (SNCA), Parkin (PRKN), PTEN-induced putative kinase 1 (PINK1), DJ-1, VPS35, glucocerebrosidase (GBA), and leucine-rich repeat kinase 2 (LRRK2) [4]. Among the PD genes, LRRK2 mutations comprise the most frequent cause of familial PD cases and are major risk factors for idiopathic PD cases. The most common mutation LRRK2-G2019S accounts for up to 6–40% of familial cases, depending on the ethnic group, and up to 2% of all sporadic cases [5,6,7,8]. Moreover, LRRK2-associated PD is generally indistinguishable from sporadic PD in terms of the age of onset, disease progression, and motor symptoms. Therefore, deciphering the role of LRRK2 might provide important insights into understanding molecular mechanisms of both familial and sporadic PD etiology and developing disease-modifying treatments for PD.
PD pathology is generally characterized by the preferential loss of DA neurons in SNpc and the presence of proteinaceous inclusions known as Lewy bodies (LBs). Enormous efforts have been made to understand the mechanisms by which each feature contributes to the pathology and progression of PD. Increasing lines of evidence suggest that the cardinal neuropathological features are not separate events. Molecular components, pathways, or mechanisms involved in each feature interact, and their interactions contribute to the pathogenesis of PD. In this review, we briefly discuss evidence regarding the role of LRRK2 in the regulation of key neuropathological features, and discuss a possibility that LRRK2 plays a role as an important modulator that mediates the interactions.
2. LRRK2 in Neurodegeneration
2.1. The Structure of LRRK2 and Regulation of Enzymatic Activities
LRRK2 is a large, multidomain protein (280 KDa) and belongs to the ROCO superfamily of proteins. It consists of multiple protein–protein interaction domains, such as N-terminal armadillo, ankyrin, and leucine-rich repeat domains, and a C-terminal WD40 domain [9]. An interesting feature of LRRK2 is that it contains two distinct enzymatic domains, a Ras of complex (ROC) GTPase domain and a serine/threonine kinase domain, separated by a C-terminal of ROC (COR) domain. Familial mutations tend to cluster within the catalytic domains of LRRK2. The kinase activity of LRRK2 has been assessed by measuring the autophosphorylation of LRRK2 [10], LRRK2 phosphorylation at Ser 1292, [11] and/or phosphorylation of an artificial peptide substrate—LRRKtide [12], or myelin basic protein (MBP) as a generic kinase substrate [13]. In addition to kinase activity, LRRK2 exhibits GTPase activity, presenting the capacity of GTP binding and hydrolysis in in vitro assay [14,15].
Molecular mechanisms that regulate the kinase and the GTPase activities of LRRK2 are not completely elucidated, but both inter- and intra-molecular regulation have been suggested to control enzymatic activities. LRRK2 forms a dimer in cells, and dimerization may be a critical step for the regulation of its enzymatic activities and membrane localization [16,17,18,19,20,21]. LRRK2 dimers show higher kinase activity compared to LRRK2 monomers, and the GTPase reaction also seems to depend on dimerization [22]. Furthermore, LRRK2 dimers are enriched in membrane fractions, and membrane LRRK2 shows enhanced kinase activity compared to cytosolic LRRK2 [20]. Biochemical analyses using truncated forms of LRRK2 and structural modeling studies with full length LRRK2 dimers suggest that the ROC and the COR domains function as primary dimerization interfaces [19]. It has been suggested that GTPase and kinase domains of LRRK2 could be regulated via an intramolecular mechanism. Missense mutations, such as K1347A and K1348N in the P-loop of the ROC domain of LRRK2, disrupt guanine nucleotide binding and impair LRRK2 autophosphorylation (although the caveat is that the mutations also inhibit dimerization) [23]. Conversely, LRRK2 kinase activity has been shown to regulate the GTPase activity through autophosphorylation of the ROC domain [24,25,26]. Kinase activity of LRRK2 can be regulated by guanine nucleotide exchange factor (GEF) or GTPase activating protein (GAP), such as ArfGAP1, RGS2, and ArhGEF7 [27,28,29,30].
2.2. Functions of Pathogenic LRRK2 Mutations in Neurodegeneration
Various genetic variants of LRRK2 have been identified and several missense mutations (N1437H, R1441C/G/H, Y1699C, G2019S and I2020T) are considered to be pathogenic. Those mutations segregate with disease in PD families, and pathophysiological roles have been suggested in a number of cellular and animal models [31]. Of note, pathogenic mutations are located in the catalytic domains: G2019S and I2020T mutations in the kinase domain; N1437H and R1441C/G/H in the ROC domain; and Y1699C in the COR domain. Thus, many studies that aim to understand the molecular and cellular mechanisms of pathogenic LRRK2 mutants have focused on elucidating whether and how the mutations change enzymatic activities, how they affect LRRK2 function, and how they contribute to the pathophysiology of PD. LRRK2 G2019S, the most common genetic determinant of PD, exhibits higher kinase activities compared to LRRK2 wild type (WT) [10,12,15,32,33,34]. The effect of I2020T mutation on LRRK2 kinase activity has been rather controversial, with some reporting increased activity [10,32], while others have shown decreased activity [12], and others have even described no changes in the kinase activity. ROC-COR domain mutations, LRRK2 R1441C/G/H and Y1699C show impaired GTP hydrolysis and enhanced GTP-binding [15,35] compared to LRRK2 WT. LRRK2 R1441C/G and Y1699C show increased kinase activity [15], whereas kinase domain mutants, LRRK2 G2019S and I2020T do not alter GTP binding [15,23].
The relationship between LRRK2 enzymatic activities and PD pathology has been and continues to be the subject of intensive investigation and has been tested in various PD model systems. In primary neuronal cultures, overexpression of LRRK2 G2019S, I2020T, R1441C, or Y1699C consistently induced neuronal toxicity, as evidenced by neurite shortening, cell death, or impaired functions of intracellular organelles [13,15,23,34,36,37,38,39]. Furthermore, many of these phenotypes were shown to be alleviated by introducing kinase-inactive or GTP binding-deficient mutations and/or treatment with chemical inhibitors of LRRK2 [13,40]. Several LRRK2 transgenic (Tg) animal models have been developed to recapitulate pathological phenotypes of PD. In Drosophila LRRK2 Tg models, Tg lines expressing LRRK2 G2019S, R1441C, Y1699C, or I2020T commonly exhibit age-dependent DA neuronal loss, disruption of dopamine homeostasis, locomotor defects or reduced survival [41,42,43,44,45]. Caenorhabditis elegans (C. elegans) LRRK2 Tg models expressing LRRK2 G2019S or R1441C also show neurodegeneration of DA neurons, reduced dopamine levels, and locomotor dysfunction [45,46]. These toxic gain-of-function effects of familial LRRK2 mutations are abolished by LRRK2 inhibitors. Liu et al. provided evidence that the administration of LRRK2 kinase inhibitors could rescue the toxic effects of LRRK2 G2019S in both Drosophila and C. elegans models [45]. In rodent models, herpes simplex viral- (HSV) or adenoviral-mediated expression of LRRK2 G2019S in the striatum of mice or rats induced robust degeneration of DA neurons in SNpc [13,47]. By contrast, the kinase-inactive variant LRRK2, G2019S/D1994A did not induce neurodegeneration, and neurodegeneration induced by HSV-LRRK2 G2019S was prevented by the administration of pharmacological inhibitors of LRRK2 kinase. Until now, several Tg rodent models overexpressing pathogenic LRRK2 mutants have been developed to mimic certain aspects of PD, such as selective DA neuronal loss, disruption of dopamine homeostasis, locomotor deficit or/and accumulation of tau and α-synuclein [48,49,50,51,52]. Knock-in (KI) models expressing a PD-associated LRRK2 mutation at endogenous levels did not show overt DA neuronal cell loss, but the mice at old age showed altered dopamine homeostasis, dysregulation of dopamine transport and accumulation into the synapse, and mild behavioral deficits, which are related to the prodromal phase of PD [53,54]. In fact, despite the fact that some models show impaired dopaminergic neurotransmission [55,56] and mild parkinsonian motor features at late stages [57], the majority of LRRK2 KI mice do not exhibit selective loss of DA neurons from the SNpc, the hallmark histopathological feature of PD [50,54,55,56,58,59]. In general, the pathological phenotypes in LRRK2 Tg or KI mice is highly dependent on aging and expression levels in DA neurons. Nonetheless, numerous cellular and animal LRRK2 models have repeatedly shown that LRRK2 exerts neuronal toxicity through kinase-dependent mechanisms. These observations attracted the interest of basic scientists and pharmacological companies to investigate how the kinase activity of LRRK2 in controlled.
2.3. Kinase Substrates of LRRK2 and Their Roles in Neurodegeneration
A considerable amount of work has been undertaken to identify the physiological substrates of LRRK2 kinase. LRRK2 has been suggested to play multiple functions, ranging from cytoskeletal remodeling to protein expression, to synaptic transmission, and to membrane trafficking, through the phosphorylation of diverse substrates (Table 1) [60,61,62,63,64,65,66], such as moesin [12], β-tubulin [67], tau [68], microtubule affinity-regulating kinase 1 (MARK1) [69], Futsch [70], FoxO1 [71], 4E-BP1 [72], ribosomal protein s15 (RPS15) [73], ArfGAP1 [27], endophilin A1 [74], snapin [75], RGS2 [29], N-ethylmaleimide sensitive fusion (NSF) [76], synaptojanin-1 [77], and a subset of Rab GTPases [78,79,80]. Although most of these substrates have been shown to be directly phosphorylated by LRRK2 in in vitro kinase assays, it is not clear if they are phosphorylated in a cellular context or in vivo. Phosphorylation of endogenous Rab GTPases (Rab3, Rab8, Rab10, Rab12, Rab35, and Rab43) by LRRK2 in cells and the mammalian brain has been validated by several groups [79,80].

Table 1.
The list of LRRK2 kinase substrates and their potential functions.
Early studies demonstrated that the expression of the pathogenic mutants of LRRK2 caused marked shortening of neurites, whereas depletion of LRRK2 increased neurite length and branching in primary cultures of embryonic cortical neurons [13,15,34,39]. Many LRRK2 substrates, such as moesin, β-tubulin, tau, and MARK1 have been suggested to play key roles in neurite extension via the regulation of cytoskeletal structures and dynamics. In cell-free assays, microtubule polymerization was enhanced by incubation with LRRK2 G2019S compared to LRRK2 WT or kinase-inactive LRRK2 D1994A [67]. It has been shown that tau, a microtubule-association protein (MAP), physically associates with and can be phosphorylated by LRRK2, in the presence of tubulin. In physiological conditions, tau associates with neuronal microtubules and is thought to play critical roles in the regulation of microtubule stability and dynamics [81,82,83]. In pathological conditions, tau detaches from microtubules and forms aggregates. Deposition of tau aggregates is a hallmark of Alzheimer’s disease, but is also found in a variety of other neurodegenerative diseases, including frontotemporal dementia with parkinsonism-17, Pick disease, progressive supranuclear palsy and corticobasal degeneration [84,85]. Phosphorylation induces tau to dissociate from microtubules, and hyperphosphorylation of tau has been proposed to contribute to neurodegeneration by the loss of microtubule stabilizing function of tau and by promoting tau aggregate formation. Increased phosphorylation of tau have been reported in multiple cellular and animal models expressing pathogenic LRRK2 mutants and in the postmortem brains from LRRK2-linked PD patients [39,86,87,88], although it is not clear whether the phosphorylation is directly mediated by LRRK2. It is plausible that hyperactivity of pathogenic LRRK2 plays a role in neurodegeneration by disturbing microtubule dynamics, but future studies are needed to investigate if LRRK2 phosphorylates tubulin and tau in vivo and whether and how the phosphorylation events contribute to PD pathology.
A group of LRRK2 substrates, such as endophilin A1, snapin, synaptojanin-1, and NSF, are key regulators of synaptic transmission. In neurons, synaptic transmission requires precisely controlled membrane trafficking at the presynaptic terminal in order to recover and recycle membranes that have fused with the plasma membrane during neurotransmitter release. Through postmortem brain studies of PD patients, synaptic dysfunction has been suggested to represent early events in PD [89]. Furthermore, multiple lines of evidences support the notion that LRRK2 plays a role as a regulator of clathrin-dependent endocytosis and recycling of synaptic vesicles [90]. In the course of synaptic vesicle endocytosis (SVE), endophilin promotes an early step by generating membrane curvature and a later step involved in vesicle release at the presynaptic membrane. Phosphorylation of endophilin by LRRK2 has been suggested to modulate the membrane deformation and release of endocytic vesicles [74]. In addition to SVE, LRRK2 can modulate synaptic vesicle fusion by regulating the dissociation of the SNARE complex via NSF phosphorylation [76]. NSF is an ATPase that catalyzes the release of SNARE complexes, thus allowing SVE and the next cycle of fusion. Phosphorylation of NSF by LRRK2 enhanced the catalytic activity of NSF and thus increased the rate of SNARE complex disassembly. These results imply that LRRK2 may tune the kinetics of synaptic vesicle recycling via NSF phosphorylation. Apart from endophilin and NSF, LRRK2 has been reported to bind and phosphorylate many other synaptic vesicle proteins [91], suggesting a broad role in synaptic transmission.
To date, it has been reported that LRRK2 can phosphorylate fourteen Rab GTPases (Rab3a/b/c/d, Rab5a/b/c, Rab8a/b, Rab10, Rab12, Rab29, Rab35, and Rab43) at a conserved residue, which is located in the switch II effector-binding motif [78,79,80]. The Rab GTPase family consists of more than 60 members in the human genome, and functions as a molecular switch in the regulation of intracellular vesicle trafficking. Rab GTPases cycle between the inactive GDP-bound and the active GTP-bound forms at specific membranous compartments. The rate of GDP/GTP cycling is regulated by specific GAPs and GEFs, which activate and inactivate the GTPase activity. GEF promotes GTP binding by inducing the dissociation of GDP from Rab GTPases and causes major conformational changes in two highly flexible regions, switch I and II. The conformational change enables the binding of γ-phosphate and interaction with regulatory proteins and effectors [92]. In the case of Rab8a, phosphorylation by different pathogenic LRRK2 mutants decreases the affinity for guanine dissociation inhibitor (GDI), which is required for membrane delivery and recycling of Rab, and the phosphorylation eventually disrupts the balanced membrane-cytosol distribution of Rab8a [78]. Steger et al. replaced the LRRK2 target site in all fourteen LRRK2 substrate Rabs with either a phosphomimetic glutamic acid or a non-phosphorylatable alanine residue and examined how the mutation affected partner protein binding. Interestingly, non-phosphorylatable mutants of Rabs stably bound to GDI1/2 and Rab escort proteins, CHM and CHML, whereas the phosphomimetic mutations strongly prevented partner binding [79]. These results imply that Rab phosphorylation by LRRK2 modulates the binding with regulators of the GDP/GTP cycle or downstream effector molecules, which may affect subcellular localization and function. Some studies have suggested that phosphorylation of Rab8a and Rab10 by pathogenic mutants of LRRK2 caused defects in primary cilia formation [79,93] and centrosomal cohesion [94,95]. Both ciliogenesis and centrosomal cohesion were regulated by phosphorylation-dependent recruitment of their effector, RILPL1. Rab8a/Rab10 phosphorylation by LRRK2 has also been suggested to be involved in endolysosomal trafficking [96] and lysosomal homeostasis [97]. Upon lysosomal overload stress, LRRK2 was activated and recruited onto lysosomes, where LRRK2 stabilized Rab8 and Rab10 through phosphorylation, and Rab8/10 further recruited their effectors, EH domain-binding protein 1 (EHBP1) and EHBP1-like 1 (EHBP1L1), to regulate stress-induced lysosomal enlargement and secretion [97]. Jeong et al. provided important clues regarding the pathological consequences of Rab phosphorylation by LRRK2 by using neuronal culture and in vivo models [80]. In primary cultures of cortical neuron, non-phosphorylatable and phosphomimetic mutants of Rab1a, 3c, and 35, but not the WTs, specifically induced neuronal toxicity. Furthermore, intracranial injection of adeno-associated viral (AAV) vectors expressing non-phosphorylatable or phosphomimetic mutants of Rab35 into SNpc of mouse brains caused profound DA neuronal loss. Of note, increased Rab35 expression was detected in the SN region of multiple PD animal models, including LRRK2 G2019S and R1441C Tg mice and MPTP and rotenone intoxication mice, as well as in the serums of PD patients when compared to age-matched subjects [98]. Rab35 has been reported to be localized in the plasma membrane and the endosome. Rab35 seems to play a role in delivering internalized cargos to late endosomes or multivesicular bodies (MVBs) directed either for lysosomal degradation or for secretion through exosomes [99,100]. Endolysosomal system dysfunctions have been repeatedly reported in a diverse range of neurodegenerative diseases, and defects in the endolysosomal trafficking may represent early events during the progression of PD [101]. Notably, LRRK2 is enriched in membrane-associated fractions, including the Golgi apparatus, endoplasmic reticulum, mitochondria, multivesicular bodies (MVBs), lysosomal and endosomal vesicles, and autophagic vacuoles, suggesting a role of LRRK2 in membrane trafficking [102]. Studies from LRRK2 knock-out (KO) animals support a role in autophagy and lysosomal function [103,104,105]. Investigating the interaction between LRRK2 and Rab GTPases in various membranous organelles and how Rab phosphorylation by LRRK2 affects the endolysosomal and autolysosmal pathways will enhance our understanding of LRRK2-mediated neuropathologies.
3. Functions of LRRK2 in Lewy Pathology and Synucleinopathies
3.1. Lewy Pathology in PD
The presence of LBs, the proteinaceous inclusions characterized by the accumulation of misfolded and aggregated α-synuclein, is the main histopathological hallmark of PD. LBs are widely distributed in multiple brain regions, including the mesostriatal system, cortex, thalamus, hypothalamus, olfactory bulb and brain stem [106]. The morphology of LBs varies depending on the location in the brain (brainstem, limbic, or neocortical), and the heterogeneity may reflect the maturation stage and/or the biochemical variability of Lewy pathology [106,107]. LBs are also found in neurites, mainly in axons, referred to as intraneurite LBs or Lewy neurites. Misfolded and aggregated forms of α-synuclein are main protein components of LBs. It has been proposed that α-synuclein exists in a dynamic equilibrium of the unfolded monomers and helically folded tetramers, and that chronically shifting the physiological tetramers to excess monomers is associated with PD-like states [108]. Under pathological conditions, α-synuclein monomers become aggregated and initiate the formation of protofibrils and insoluble fibrils [109]. WT and disease-linked mutants of α-synuclein spontaneously form amyloid-like fibrils during prolonged incubation in vitro [110], but few inclusion bodies are found in various Tg mice overexpressing α-synuclein. Molecular mechanisms that trigger the fibrillization of α-synuclein and the formation of LBs in the brain remain poorly understood. Identification of the molecular components of LBs may provide important clues regarding the mechanisms of LB formation and disease progression. Proteomic analyses have identified more than 300 proteins in LBs, and about a hundred proteins have been validated by immunohistochemical analyses in various postmortem studies [106,111,112]. Notably, LRRK2 [36,113,114,115] has also been detected in LBs together with other PD-linked gene products, such as DJ-1 [116,117], Parkin [118], and PINK-1 [119]. In addition to proteinaceous components, LBs also contain non-proteinaceous (lipid) materials. Using correlative light and electron microscopy and tomography on postmortem human brain tissue from PD brain donors, one study [107] has revealed that α-synuclein-positive LBs and Lewy neurites contained a crowded mix-up of dysmorphic organellar and membranous features. Detection of LRRK2 and distorted membranous organelles in α-synuclein-positive Lewy pathology not only suggests a molecular and functional interaction between LRRK2 and α-synuclein but also provides support for the hypothesis that LRRK2 dysfunction may disrupt organelle trafficking and contribute to the pathogenesis of PD.
3.2. Roles of LRRK2 in Synucleinopathy
PD is now thought of as a complex multisystem disease with premotor and nonmotor symptoms, however, clinical-pathological diagnosis related to motor symptoms still remains the gold standard for the diagnosis of PD [120]. The current pathological criteria for PD require both DA neuronal loss in SNpc and LB pathology. Most LRRK2-assocaited PD patients are indistinguishable from idiopathic PD, both pathologically and clinically [121]. Pathological analyses revealed typical PD-type LB in most patients, but atypical neuropathology was also observed in some LRRK2 mutation carriers [122]. A subset of PD patients carrying a highly penetrant LRRK2 mutation, such as G2019S, R1441C/G, Y1699C, or I2020T, did not show LBs or α-synuclein pathology [120,123,124,125,126,127], despite the loss of DA neurons in SNpc [120]. However, it should be noted that the absence of LBs does not necessarily indicate that there is no α-synuclein-linked pathology. Native α-synuclein monomers can give rise to heterogeneous soluble oligomeric forms, which are not readily detectable in histological sections. Several lines of evidence suggest that LB themselves may be innocent bystanders in PD pathogenesis and that the neurotoxic species are in fact oligomers [128,129,130]. In this aspect, formation of LBs can represent a protective mechanism whereby insoluble fibrils function to sequester toxic oligomers [131]. It is a matter of debate whether LBs are neurotoxic or neuroprotective. Monitoring the dynamic process of α-synuclein aggregation rather than detecting the mere existence of LBs may be more informative to understand α-synuclein pathology. Levels of α-synuclein oligomers were significantly higher in the cerebrospinal fluid (CSF) of asymptomatic LRRK2 mutant carriers relative to healthy age-matched controls, suggesting a possibility that oligomeric α-synuclein is formed during the early stages of the disease prior to any major clinical manifestation and that LRRK2 might play a role in this pre-symptomatic stage by contributing to the formation of the toxic α-synuclein oligomers [132]. Supporting this notion, another study showed greater age-dependent accumulation of oligomeric α-synuclein in the striatum and cortex of LRRK2 R1441G KI mice at 15 and 18 months of age, compared to age-matched LRRK2 WT mice [133].
Attempts have been made to recapitulate Lewy pathology or synucleinopathy in rodent models and mimic the increased expression or phosphorylation of α-synuclein and/or the formation of insoluble α-synuclein aggregates or LB-like inclusions. In Tg mice overexpressing LRRK2 G2019S in catecholaminergic neurons under the control of TH promoter, DA and norepinephrine neurons degenerated in an age-dependent manner [52]. Moreover, elevated phosphorylation (pS129) of α-synuclein and high molecular weight species of α-synuclein were detected in the striatum and ventral midbrain of the LRRK2 G2019S Tg mice, whereas none of the pathogenic forms of α-synuclein were detected in nonTg or the LRRK2 G2019S/D1994A mice at 15 and 24 months of age [52]. LRRK2 G2019S KI mice showed age-dependent increases in phospho-α-synuclein (pS129) in SN and striatum [134]. However, most other Tg or KI mouse lines expressing pathogenic LRRK2 mutants under different promoter systems do not exhibit synucleinopathy, which may be in part explained by the insufficient amount of seeds required for α-synuclein aggregation. Tg mice expressing LRRK2 alone did not cause noticeable neurodegeneration, but the presence of excess LRRK2 accelerated the progression of neuropathological abnormalities developed in PD-related A53T α-synuclein Tg mice [135]. Co-expression of WT or LRRK2 G2019S with α-synuclein A53T caused synergistic toxicity to neurons that accelerated the progression of α-synuclein-mediated neuropathology. In another study, aged LRRK2 G2019S KI mice were more prone to develop α-synuclein toxicity than WT mice, and larger amounts of α-synuclein aggregates were present in aged LRRK2 G2019S KI mice [134,135]. Conversely, deletion of LRRK2 suppressed the aggregation and somatic accumulation of α-synuclein, and thereby delayed the progression of neuropathology developed in A53T Tg mice [135]. Preformed fibrils (PFFs) are artificially generated short fragments of fibrils that can trigger the conversion of endogenous α-synuclein into pathogenic fibril forms, and PFFs can function as seeds for the generation of insoluble fibrils or LB-like inclusions. In the α-synuclein PFF model, LRRK2 G2019S Tg mice showed accelerated α-synuclein aggregation, degeneration of DA neurons in SNpc, and neuroinflammation [136]. These lines of evidence suggest that LRRK2 may regulate α-synuclein-induced pathogenesis in PD. However, the link between LRRK2 and synuclein aggregation is far from clear and the literature is still controversial.
3.3. Roles of LRRK2 in α-Synuclein Propagation
The Braak model suggests that PD pathology spreads in a stereotypical fashion. According to the Braak model, PD pathology ascends caudo-rostrally from the lower brainstem and olfactory bulbs at the prodromal stage of disease, spreads through the midbrain and forebrain regions at disease diagnosis, and eventually propagates into the cerebral cortex at later stages [137]. Studies have suggested that α-synuclein exhibits prion-like properties and self-propagates via templating endogenous α-synuclein to form polymers [138]. The spreading of α-synuclein is considered to be an underlying molecular mechanism for the Braak hypothesis. Currently, cell-to-cell transmission of α-synuclein has been recapitulated in both cell culture and animal models [139,140,141,142]. The initial step of α-synuclein transmission may be the transformation of an innocuous, physiological form of α-synuclein into a toxic fibril, which may be caused by the misfolding of α-synuclein [128,129,130]. Infectious cells release α-synuclein oligomers or fibrils through undefined nonclassical secretory pathways, and α-synuclein then gains entry to nearby naïve cells [143], presumably by utilizing cell-to-cell communication mechanisms, such as passive diffusion, endocytosis, or transsynaptic transport [144,145]. Finally, the internalized pathogenic forms of α-synuclein act as seeds for de novo formation of α-synuclein fibrils, and thereby converts a naïve cell into an infectious cell.
Multiple lines of studies have demonstrated that LRRK2 plays a role in the regulation of cell-to-cell transmission of α-synuclein. Kondo et al. [146] provided the first clues regarding a role of LRRK2 in α-synuclein transmission. Co-expression of LRRK2 G2019S and α-synuclein in human neuroblastoma SH-SY5Y cells increased α-synuclein aggregation and secretion when compared to cells expressing α-synuclein alone. Furthermore, more α-synuclein+ cells were detected after treatment with conditioned media collected from cells co-expressing LRRK2 G2019S and α-synuclein, when compared to cells treated with conditioned media collected from cells expressing α-synuclein alone. These results suggest that LRRK2 may be involved in the process of α-synuclein secretion and/or transmission. In an α-synuclein PFF-based transmission assay, where human α-synuclein PFFs trigger the aggregation of endogenous α-synuclein, α-synuclein aggregation was enhanced by LRRK2 G2019S compared to WT, but was decreased by the loss of LRRK2 in PD patient-derived neurons differentiated from induced pluripotent stem cells [136]. LRRK2 dependent α-synuclein transmission has also been demonstrated in multiple animal models. Bae et al. [147] generated C. elegans Tg models that expressed the N- and the C-terminal halves of the Venus fluorescent protein fused to α-synuclein in the pharyngeal muscles and the connected neurons, respectively, to assess intercellular α-synuclein transmission between pharyngeal muscles and neurons. Age-dependent increases in Venus fluorescence were detected in the WT background, but not in worms lacking lrk1 (Lrrk1 and Lrrk2 ortholog in C. elegans). Bae et al. also injected AAV vectors encoding human α-synuclein into the vagus nerve of rats and tested neuron-to-neuron transmission of α-synuclein and long-distance protein spreading. The number of immunoreactive axons against α-synuclein were significantly decreased in the pons, the caudal and rostral midbrain, and the forebrain in rats lacking Lrrk2 compared to WT. These studies imply that LRRK2 plays a role in cell-to-cell transmission and long distance spreading of α-synuclein, presumably through regulation of the release, uptake and/or lysosomal/proteosomal degradation of α-synuclein. The exact molecular mechanisms by which LRRK2 regulates α-synuclein transmission remain to be determined.
The identification of multiple Rab GTPases as physiological substrates of LRRK2 has raised an interesting hypothesis that links LRRK2 and α-synuclein transmission (Figure 1). In particular, Rab35, which has been suggested to mediate neurodegeneration induced by LRRK2 [80], has also been pointed out to mediate α-synuclein transmission stimulated by LRRK2 [147]. Rab35 could enhance the secretion and aggregation of A53T α-synuclein in SH-SY5Y cells [98]. How phosphorylation of Rab35 regulates α-synuclein propagation is unclear at this point. Considering the role of LRRK2 and Rab35 in endosomal recycling, it is plausible to suggest that activation of the LRRK2–Rab35 pathway would hijack the internalized α-synuclein aggregates from the endolysosomal degradation pathway, causing amplification of aggregates and continuous propagation [147]. Interestingly, several Rab GTPases that have been identified as modulators of α-synuclein toxicity, such as Rab1, 3, and 8 [148,149], are also LRRK2 substrates [80]. It is possible that more Rab GTPases are involved in LRRK2-mediated α-synuclein transmission. Rab8, 10, and 29 have been suggested as upstream and downstream molecules of LRRK2 in the regulation of endolysosomal pathway [97,150]. More studies are needed to define the link between LRRK2 and α-synuclein, but the LRRK2–Rab axis may have a critical role in α-synuclein transmission through the regulation of lysosomal degradation and secretory pathways.
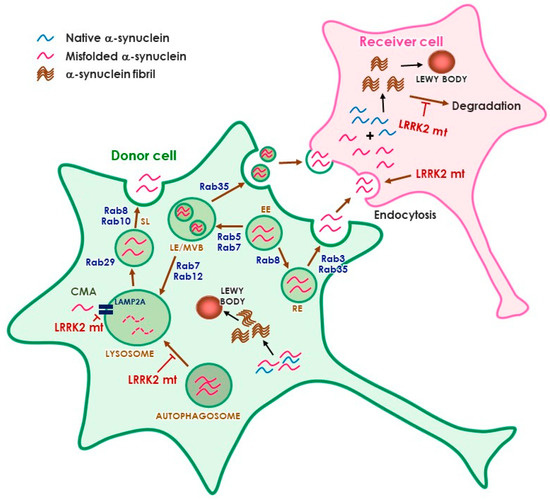
Figure 1.
Potential role of LRRK2 in α-synuclein propagation. The cell may refer to neurons and/or glia. Abbreviations: EE, early endosome; LE, late endosome, MVB, multivesicular body, RE, recycling endosome, SL, stress lysosome, CMA, chaperone-mediated autophagy.
4. Conclusions and Future Directions
Since the first descriptions of pathogenic mutations of LRRK2 in 2004 [151], remarkable advances have been made in our understanding of the function and dysfunction of LRRK2. Evidence so far suggests that LRRK2 plays multiple roles to control neuronal survival and death through cell-autonomous and non-cell-autonomous mechanisms. Apparently, the aberrant kinase activity of pathogenic LRRK2 mutants induces neurodegeneration by disturbing various intracellular processes, such as protein translation, endolysosomal pathway, autophagy, synaptic functions, and cytoskeleton dynamics, which may be mediated by the phosphorylation of several distinct putative substrates (Table 1). Although future studies are needed to identify the mechanistic link between LRRK2 and α-synuclein, pathogenic LRRK2 could exacerbate α-synuclein-mediated neurotoxicity, and LRRK2 has been suggested to interact with α-synuclein through intra- and inter-neuronal mechanisms. The available data suggest that targeting LRRK2 might be beneficial not only for patients with LRRK2 mutations but also for idiopathic PD patients. Therefore, LRRK2 has emerged as a promising target for potential disease-modifying therapies for PD, and clinical trials with small-molecule LRRK2 kinase inhibitors have already commenced. Until now, to understand LRRK2 pathology in PD, research interests have largely focused on the neurotoxic effects of pathogenic LRRK2 in the brain. Of note, accumulating lines of evidence suggest that LRRK2 plays a fundamental role in the regulation of inflammation in both the central and peripheral immune system [152,153]. Moreover, LRRK2 protein is also highly expressed in the kidney and the respiratory system. Therefore, increased knowledge of the role of LRRK2 in the immune system and in the periphery is needed and should be taken into consideration to develop effective and safe treatments for PD.
Author Contributions
Conceptualization, B.D.L; Writing and Editing, G.R.J. and B.D.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Research Foundation of Korea (NRF) funded by the Korea Government (MSIT) (NRF-2016M3C7A1905386, 2017M3C7A1043848, 2019R1A2C1088975).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Clarke, C.E. Parkinson’s disease. BMJ 2007, 335, 441–445. [Google Scholar] [CrossRef]
- Poewe, W. Non-motor symptoms in Parkinson’s disease. Eur. J. Neurol. 2008, 15 (Suppl. 1), 14–20. [Google Scholar] [CrossRef] [PubMed]
- Ponsen, M.M.; Stoffers, D.; Booij, J.; van Eck-Smit, B.L.; Wolters, E.; Berendse, H.W. Idiopathic hyposmia as a preclinical sign of Parkinson’s disease. Ann. Neurol. 2004, 56, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Bandres-Ciga, S.; Diez-Fairen, M.; Kim, J.J.; Singleton, A.B. Genetics of Parkinson’s disease: An introspection of its journey towards precision medicine. Neurobiol. Dis. 2020, 137, 104782. [Google Scholar] [CrossRef] [PubMed]
- Aasly, J.O.; Toft, M.; Fernandez-Mata, I.; Kachergus, J.; Hulihan, M.; White, L.R.; Farrer, M. Clinical features of LRRK2-associated Parkinson’s disease in central Norway. Ann. Neurol. 2005, 57, 762–765. [Google Scholar] [CrossRef]
- Bras, J.M.; Guerreiro, R.J.; Ribeiro, M.H.; Januario, C.; Morgadinho, A.; Oliveira, C.R.; Cunha, L.; Hardy, J.; Singleton, A. G2019S dardarin substitution is a common cause of Parkinson’s disease in a Portuguese cohort. Mov. Disord. 2005, 20, 1653–1655. [Google Scholar] [CrossRef]
- Infante, J.; Rodriguez, E.; Combarros, O.; Mateo, I.; Fontalba, A.; Pascual, J.; Oterino, A.; Polo, J.M.; Leno, C.; Berciano, J. LRRK2 G2019S is a common mutation in Spanish patients with late-onset Parkinson’s disease. Neurosci. Lett. 2006, 395, 224–226. [Google Scholar] [CrossRef]
- Kachergus, J.; Mata, I.F.; Hulihan, M.; Taylor, J.P.; Lincoln, S.; Aasly, J.; Gibson, J.M.; Ross, O.A.; Lynch, T.; Wiley, J.; et al. Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: Evidence of a common founder across European populations. Am. J. Hum. Genet. 2005, 76, 672–680. [Google Scholar] [CrossRef]
- Lee, B.D.; Dawson, V.L.; Dawson, T.M. Leucine-rich repeat kinase 2 (LRRK2) as a potential therapeutic target in Parkinson’s disease. Trends Pharmacol. Sci. 2012, 33, 365–373. [Google Scholar] [CrossRef]
- West, A.B.; Moore, D.J.; Biskup, S.; Bugayenko, A.; Smith, W.W.; Ross, C.A.; Dawson, V.L.; Dawson, T.M. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. USA 2005, 102, 16842–16847. [Google Scholar] [CrossRef]
- Sheng, Z.; Zhang, S.; Bustos, D.; Kleinheinz, T.; Le Pichon, C.E.; Dominguez, S.L.; Solanoy, H.O.; Drummond, J.; Zhang, X.; Ding, X.; et al. Ser1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of PD mutations. Sci. Transl. Med. 2012, 4, 164ra161. [Google Scholar] [CrossRef] [PubMed]
- Jaleel, M.; Nichols, R.J.; Deak, M.; Campbell, D.G.; Gillardon, F.; Knebel, A.; Alessi, D.R. LRRK2 phosphorylates moesin at threonine-558: Characterization of how Parkinson’s disease mutants affect kinase activity. Biochem. J. 2007, 405, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.D.; Shin, J.H.; VanKampen, J.; Petrucelli, L.; West, A.B.; Ko, H.S.; Lee, Y.I.; Maguire-Zeiss, K.A.; Bowers, W.J.; Federoff, H.J.; et al. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson’s disease. Nat. Med. 2010, 16, 998–1000. [Google Scholar] [CrossRef] [PubMed]
- Ito, G.; Okai, T.; Fujino, G.; Takeda, K.; Ichijo, H.; Katada, T.; Iwatsubo, T. GTP binding is essential to the protein kinase activity of LRRK2, a causative gene product for familial Parkinson’s disease. Biochemistry 2007, 46, 1380–1388. [Google Scholar] [CrossRef]
- West, A.B.; Moore, D.J.; Choi, C.; Andrabi, S.A.; Li, X.; Dikeman, D.; Biskup, S.; Zhang, Z.; Lim, K.L.; Dawson, V.L.; et al. Parkinson’s disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum. Mol. Genet. 2007, 16, 223–232. [Google Scholar] [CrossRef]
- Greggio, E.; Zambrano, I.; Kaganovich, A.; Beilina, A.; Taymans, J.M.; Daniels, V.; Lewis, P.; Jain, S.; Ding, J.; Syed, A.; et al. The Parkinson disease-associated leucine-rich repeat kinase 2 (LRRK2) is a dimer that undergoes intramolecular autophosphorylation. J. Biol. Chem. 2008, 283, 16906–16914. [Google Scholar] [CrossRef]
- Daniels, V.; Vancraenenbroeck, R.; Law, B.M.; Greggio, E.; Lobbestael, E.; Gao, F.; De Maeyer, M.; Cookson, M.R.; Harvey, K.; Baekelandt, V.; et al. Insight into the mode of action of the LRRK2 Y1699C pathogenic mutant. J. Neurochem. 2011, 116, 304–315. [Google Scholar] [CrossRef]
- Civiero, L.; Vancraenenbroeck, R.; Belluzzi, E.; Beilina, A.; Lobbestael, E.; Reyniers, L.; Gao, F.; Micetic, I.; De Maeyer, M.; Bubacco, L.; et al. Biochemical characterization of highly purified leucine-rich repeat kinases 1 and 2 demonstrates formation of homodimers. PLoS ONE 2012, 7, e43472. [Google Scholar] [CrossRef]
- Guaitoli, G.; Raimondi, F.; Gilsbach, B.K.; Gomez-Llorente, Y.; Deyaert, E.; Renzi, F.; Li, X.; Schaffner, A.; Jagtap, P.K.; Boldt, K.; et al. Structural model of the dimeric Parkinson’s protein LRRK2 reveals a compact architecture involving distant interdomain contacts. Proc. Natl. Acad. Sci. USA 2016, 113, 4357–4366. [Google Scholar] [CrossRef]
- Berger, Z.; Smith, K.A.; Lavoie, M.J. Membrane localization of LRRK2 is associated with increased formation of the highly active LRRK2 dimer and changes in its phosphorylation. Biochemistry 2010, 49, 5511–5523. [Google Scholar] [CrossRef]
- Sen, S.; Webber, P.J.; West, A.B. Dependence of leucine-rich repeat kinase 2 (LRRK2) kinase activity on dimerization. J. Biol. Chem. 2009, 284, 36346–36356. [Google Scholar] [CrossRef] [PubMed]
- Deyaert, E.; Wauters, L.; Guaitoli, G.; Konijnenberg, A.; Leemans, M.; Terheyden, S.; Petrovic, A.; Gallardo, R.; Nederveen-Schippers, L.M.; Athanasopoulos, P.S.; et al. A homologue of the Parkinson’s disease-associated protein LRRK2 undergoes a monomer-dimer transition during GTP turnover. Nat. Commun. 2017, 8, 1008. [Google Scholar] [CrossRef] [PubMed]
- Biosa, A.; Trancikova, A.; Civiero, L.; Glauser, L.; Bubacco, L.; Greggio, E.; Moore, D.J. GTPase activity regulates kinase activity and cellular phenotypes of Parkinson’s disease-associated LRRK2. Hum. Mol. Genet. 2013, 22, 1140–1156. [Google Scholar] [CrossRef] [PubMed]
- Greggio, E.; Taymans, J.M.; Zhen, E.Y.; Ryder, J.; Vancraenenbroeck, R.; Beilina, A.; Sun, P.; Deng, J.; Jaffe, H.; Baekelandt, V.; et al. The Parkinson’s disease kinase LRRK2 autophosphorylates its GTPase domain at multiple sites. Biochem. Biophys. Res. Commun. 2009, 389, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Webber, P.J.; Smith, A.D.; Sen, S.; Renfrow, M.B.; Mobley, J.A.; West, A.B. Autophosphorylation in the leucine-rich repeat kinase 2 (LRRK2) GTPase domain modifies kinase and GTP-binding activities. J. Mol. Biol. 2011, 412, 94–110. [Google Scholar] [CrossRef]
- Gloeckner, C.J.; Boldt, K.; von Zweydorf, F.; Helm, S.; Wiesent, L.; Sarioglu, H.; Ueffing, M. Phosphopeptide analysis reveals two discrete clusters of phosphorylation in the N-terminus and the Roc domain of the Parkinson-disease associated protein kinase LRRK2. J. Proteome. Res. 2010, 9, 1738–1745. [Google Scholar] [CrossRef]
- Xiong, Y.; Yuan, C.; Chen, R.; Dawson, T.M.; Dawson, V.L. ArfGAP1 is a GTPase activating protein for LRRK2: Reciprocal regulation of ArfGAP1 by LRRK2. J. Neurosci. 2012, 32, 3877–3886. [Google Scholar] [CrossRef]
- Stafa, K.; Trancikova, A.; Webber, P.J.; Glauser, L.; West, A.B.; Moore, D.J. GTPase activity and neuronal toxicity of Parkinson’s disease-associated LRRK2 is regulated by ArfGAP1. PLoS Genet. 2012, 8, e1002526. [Google Scholar] [CrossRef]
- Dusonchet, J.; Li, H.; Guillily, M.; Liu, M.; Stafa, K.; Derada Troletti, C.; Boon, J.Y.; Saha, S.; Glauser, L.; Mamais, A.; et al. A Parkinson’s disease gene regulatory network identifies the signaling protein RGS2 as a modulator of LRRK2 activity and neuronal toxicity. Hum. Mol. Genet. 2014, 23, 4887–4905. [Google Scholar] [CrossRef]
- Haebig, K.; Gloeckner, C.J.; Miralles, M.G.; Gillardon, F.; Schulte, C.; Riess, O.; Ueffing, M.; Biskup, S.; Bonin, M. ARHGEF7 (Beta-PIX) acts as guanine nucleotide exchange factor for leucine-rich repeat kinase 2. PLoS ONE 2010, 5, e13762. [Google Scholar] [CrossRef]
- Paisan-Ruiz, C.; Lewis, P.A.; Singleton, A.B. LRRK2: Cause, risk, and mechanism. J. Parkinsons Dis. 2013, 3, 85–103. [Google Scholar] [CrossRef] [PubMed]
- Gloeckner, C.J.; Kinkl, N.; Schumacher, A.; Braun, R.J.; O’Neill, E.; Meitinger, T.; Kolch, W.; Prokisch, H.; Ueffing, M. The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum. Mol. Genet. 2006, 15, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Bender, S.; Kang, S.; Lin, R.; Glicksman, M.A.; Liu, M. The Parkinson disease-linked LRRK2 protein mutation I2020T stabilizes an active state conformation leading to increased kinase activity. J. Biol. Chem. 2014, 289, 13042–13053. [Google Scholar] [CrossRef]
- Smith, W.W.; Pei, Z.; Jiang, H.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat. Neurosci. 2006, 9, 1231–1233. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Wu, C.X.; Burlak, C.; Zhang, S.; Sahm, H.; Wang, M.; Zhang, Z.Y.; Vogel, K.W.; Federici, M.; Riddle, S.M.; et al. Parkinson disease-associated mutation R1441H in LRRK2 prolongs the “active state” of its GTPase domain. Proc. Natl. Acad. Sci. USA 2014, 111, 4055–4060. [Google Scholar] [CrossRef] [PubMed]
- Greggio, E.; Jain, S.; Kingsbury, A.; Bandopadhyay, R.; Lewis, P.; Kaganovich, A.; van der Brug, M.P.; Beilina, A.; Blackinton, J.; Thomas, K.J.; et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis. 2006, 23, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Yu, M.; Wang, C.; Xu, Z. Leucine-rich repeat kinase 2 disturbs mitochondrial dynamics via Dynamin-like protein. J. Neurochem. 2012, 122, 650–658. [Google Scholar] [CrossRef]
- Wang, X.; Yan, M.H.; Fujioka, H.; Liu, J.; Wilson-Delfosse, A.; Chen, S.G.; Perry, G.; Casadesus, G.; Zhu, X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 2012, 21, 1931–1944. [Google Scholar] [CrossRef]
- MacLeod, D.; Dowman, J.; Hammond, R.; Leete, T.; Inoue, K.; Abeliovich, A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron 2006, 52, 587–593. [Google Scholar] [CrossRef]
- Ramsden, N.; Perrin, J.; Ren, Z.; Lee, B.D.; Zinn, N.; Dawson, V.L.; Tam, D.; Bova, M.; Lang, M.; Drewes, G.; et al. Chemoproteomics-based design of potent LRRK2-selective lead compounds that attenuate Parkinson’s disease-related toxicity in human neurons. ACS Chem. Biol. 2011, 6, 1021–1028. [Google Scholar] [CrossRef]
- Lin, C.H.; Tsai, P.I.; Wu, R.M.; Chien, C.T. LRRK2 G2019S mutation induces dendrite degeneration through mislocalization and phosphorylation of tau by recruiting autoactivated GSK3ss. J. Neurosci. 2010, 30, 13138–13149. [Google Scholar] [CrossRef] [PubMed]
- Venderova, K.; Kabbach, G.; Abdel-Messih, E.; Zhang, Y.; Parks, R.J.; Imai, Y.; Gehrke, S.; Ngsee, J.; Lavoie, M.J.; Slack, R.S.; et al. Leucine-Rich Repeat Kinase 2 interacts with Parkin, DJ-1 and PINK-1 in a Drosophila melanogaster model of Parkinson’s disease. Hum. Mol. Genet. 2009, 18, 4390–4404. [Google Scholar] [CrossRef] [PubMed]
- Hindle, S.; Afsari, F.; Stark, M.; Middleton, C.A.; Evans, G.J.; Sweeney, S.T.; Elliott, C.J. Dopaminergic expression of the Parkinsonian gene LRRK2-G2019S leads to non-autonomous visual neurodegeneration, accelerated by increased neural demands for energy. Hum. Mol. Genet. 2013, 22, 2129–2140. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.H.; Mok, S.Z.; Koh, C.; Ouyang, X.; Fivaz, M.L.; Tan, E.K.; Dawson, V.L.; Dawson, T.M.; Yu, F.; Lim, K.L. Parkin protects against LRRK2 G2019S mutant-induced dopaminergic neurodegeneration in Drosophila. J. Neurosci. 2009, 29, 11257–11262. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Hamamichi, S.; Lee, B.D.; Yang, D.; Ray, A.; Caldwell, G.A.; Caldwell, K.A.; Dawson, T.M.; Smith, W.W.; Dawson, V.L. Inhibitors of LRRK2 kinase attenuate neurodegeneration and Parkinson-like phenotypes in Caenorhabditis elegans and Drosophila Parkinson’s disease models. Hum. Mol. Genet. 2011, 20, 3933–3942. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; El Khoury, R.; Wang, W.; Byrd, T.A.; Pehek, E.A.; Thacker, C.; Zhu, X.; Smith, M.A.; Wilson-Delfosse, A.L.; Chen, S.G. LRRK2-mediated neurodegeneration and dysfunction of dopaminergic neurons in a Caenorhabditis elegans model of Parkinson’s disease. Neurobiol. Dis. 2010, 40, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Tsika, E.; Nguyen, A.P.; Dusonchet, J.; Colin, P.; Schneider, B.L.; Moore, D.J. Adenoviral-mediated expression of G2019S LRRK2 induces striatal pathology in a kinase-dependent manner in a rat model of Parkinson’s disease. Neurobiol. Dis. 2015, 77, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Weng, Y.H.; Chien, K.Y.; Lin, K.J.; Yeh, T.H.; Cheng, Y.P.; Lu, C.S.; Wang, H.L. (G2019S) LRRK2 activates MKK4-JNK pathway and causes degeneration of SN dopaminergic neurons in a transgenic mouse model of PD. Cell Death Differ. 2012, 19, 1623–1633. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.S.; Chen, C.Y.; Chen, Y.L.; Weng, Y.H.; Yeh, T.H.; Lu, C.S.; Chang, Y.M.; Wang, H.L. (G2019S) LRRK2 causes early-phase dysfunction of SNpc dopaminergic neurons and impairment of corticostriatal long-term depression in the PD transgenic mouse. Neurobiol. Dis. 2014, 68, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, T.; Mori, S.; Sasaki, Y.; Miyajima, T.; Azuma, S.; Ohta, E.; Obata, F. The I2020T Leucine-rich repeat kinase 2 transgenic mouse exhibits impaired locomotive ability accompanied by dopaminergic neuron abnormalities. Mol. Neurodegener. 2012, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Weng, Y.H.; Chen, C.Y.; Lin, K.J.; Chen, Y.L.; Yeh, T.H.; Hsiao, I.T.; Chen, I.J.; Lu, C.S.; Wang, H.L. (R1441C) LRRK2 induces the degeneration of SN dopaminergic neurons and alters the expression of genes regulating neuronal survival in a transgenic mouse model. Exp. Neurol. 2016, 275, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Neifert, S.; Karuppagounder, S.S.; Liu, Q.; Stankowski, J.N.; Lee, B.D.; Ko, H.S.; Lee, Y.; Grima, J.C.; Mao, X.; et al. Robust kinase- and age-dependent dopaminergic and norepinephrine neurodegeneration in LRRK2 G2019S transgenic mice. Proc. Natl. Acad. Sci. USA 2018, 115, 1635–1640. [Google Scholar] [CrossRef] [PubMed]
- Giesert, F.; Glasl, L.; Zimprich, A.; Ernst, L.; Piccoli, G.; Stautner, C.; Zerle, J.; Holter, S.M.; Vogt Weisenhorn, D.M.; Wurst, W. The pathogenic LRRK2 R1441C mutation induces specific deficits modeling the prodromal phase of Parkinson’s disease in the mouse. Neurobiol. Dis. 2017, 105, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Longo, F.; Mercatelli, D.; Novello, S.; Arcuri, L.; Brugnoli, A.; Vincenzi, F.; Russo, I.; Berti, G.; Mabrouk, O.S.; Kennedy, R.T.; et al. Age-dependent dopamine transporter dysfunction and Serine129 phospho-alpha-synuclein overload in G2019S LRRK2 mice. Acta Neuropathol. Commun. 2017, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Pisani, A.; Martella, G.; Karouani, M.; Yamaguchi, H.; Pothos, E.N.; Shen, J. R1441C mutation in LRRK2 impairs dopaminergic neurotransmission in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 14622–14627. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Patel, J.C.; Wang, J.; Avshalumov, M.V.; Nicholson, C.; Buxbaum, J.D.; Elder, G.A.; Rice, M.E.; Yue, Z. Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson’s disease mutation G2019S. J. Neurosci. 2010, 30, 1788–1797. [Google Scholar] [CrossRef]
- Bichler, Z.; Lim, H.C.; Zeng, L.; Tan, E.K. Non-motor and motor features in LRRK2 transgenic mice. PLoS ONE 2013, 8, e70249. [Google Scholar] [CrossRef]
- Herzig, M.C.; Kolly, C.; Persohn, E.; Theil, D.; Schweizer, T.; Hafner, T.; Stemmelen, C.; Troxler, T.J.; Schmid, P.; Danner, S.; et al. LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Hum. Mol. Genet. 2011, 20, 4209–4223. [Google Scholar] [CrossRef]
- Yue, M.; Hinkle, K.M.; Davies, P.; Trushina, E.; Fiesel, F.C.; Christenson, T.A.; Schroeder, A.S.; Zhang, L.; Bowles, E.; Behrouz, B.; et al. Progressive dopaminergic alterations and mitochondrial abnormalities in LRRK2 G2019S knock-in mice. Neurobiol. Dis. 2015, 78, 172–195. [Google Scholar] [CrossRef]
- Kalogeropulou, A.F.; Zhao, J.; Bolliger, M.F.; Memou, A.; Narasimha, S.; Molitor, T.P.; Wilson, W.H.; Rideout, H.J.; Nichols, R.J. P62/SQSTM1 is a novel leucine-rich repeat kinase 2 (LRRK2) substrate that enhances neuronal toxicity. Biochem. J. 2018, 475, 1271–1293. [Google Scholar] [CrossRef]
- Kiral, F.R.; Kohrs, F.E.; Jin, E.J.; Hiesinger, P.R. Rab GTPases and Membrane Trafficking in Neurodegeneration. Curr. Biol. 2018, 28, R471–R486. [Google Scholar] [CrossRef] [PubMed]
- Hutagalung, A.H.; Novick, P.J. Role of Rab GTPases in membrane traffic and cell physiology. Physiol. Rev. 2011, 91, 119–149. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Fukuda, M. Small GTPase Rab12 regulates transferrin receptor degradation: Implications for a novel membrane trafficking pathway from recycling endosomes to lysosomes. Cell Logist. 2011, 1, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wei, Z.; Fan, Y.; Huang, W.; Su, Y.; Li, H.; Dong, Z.; Fukuda, M.; Khater, M.; Wu, G. The GTPase Rab43 Controls the Anterograde ER-Golgi Trafficking and Sorting of GPCRs. Cell Rep. 2017, 21, 1089–1101. [Google Scholar] [CrossRef]
- Yun, H.J.; Kim, H.; Ga, I.; Oh, H.; Ho, D.H.; Kim, J.; Seo, H.; Son, I.; Seol, W. An early endosome regulator, Rab5b, is an LRRK2 kinase substrate. J. Biochem. 2015, 157, 485–495. [Google Scholar] [CrossRef]
- Parisiadou, L.; Xie, C.; Cho, H.J.; Lin, X.; Gu, X.L.; Long, C.X.; Lobbestael, E.; Baekelandt, V.; Taymans, J.M.; Sun, L.; et al. Phosphorylation of ezrin/radixin/moesin proteins by LRRK2 promotes the rearrangement of actin cytoskeleton in neuronal morphogenesis. J. Neurosci. 2009, 29, 13971–13980. [Google Scholar] [CrossRef]
- Gillardon, F. Leucine-rich repeat kinase 2 phosphorylates brain tubulin-beta isoforms and modulates microtubule stability—A point of convergence in parkinsonian neurodegeneration? J. Neurochem. 2009, 110, 1514–1522. [Google Scholar] [CrossRef]
- Kawakami, F.; Yabata, T.; Ohta, E.; Maekawa, T.; Shimada, N.; Suzuki, M.; Maruyama, H.; Ichikawa, T.; Obata, F. LRRK2 phosphorylates tubulin-associated tau but not the free molecule: LRRK2-mediated regulation of the tau-tubulin association and neurite outgrowth. PLoS ONE 2012, 7, e30834. [Google Scholar] [CrossRef]
- Krumova, P.; Reyniers, L.; Meyer, M.; Lobbestael, E.; Stauffer, D.; Gerrits, B.; Muller, L.; Hoving, S.; Kaupmann, K.; Voshol, J.; et al. Chemical genetic approach identifies microtubule affinity-regulating kinase 1 as a leucine-rich repeat kinase 2 substrate. FASEB J. 2015, 29, 2980–2992. [Google Scholar] [CrossRef]
- Lee, S.; Liu, H.P.; Lin, W.Y.; Guo, H.; Lu, B. LRRK2 kinase regulates synaptic morphology through distinct substrates at the presynaptic and postsynaptic compartments of the Drosophila neuromuscular junction. J. Neurosci. 2010, 30, 16959–16969. [Google Scholar] [CrossRef]
- Kanao, T.; Venderova, K.; Park, D.S.; Unterman, T.; Lu, B.; Imai, Y. Activation of FoxO by LRRK2 induces expression of proapoptotic proteins and alters survival of postmitotic dopaminergic neuron in Drosophila. Hum. Mol. Genet. 2010, 19, 3747–3758. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Gehrke, S.; Wang, H.Q.; Takahashi, R.; Hasegawa, K.; Oota, E.; Lu, B. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J. 2008, 27, 2432–2443. [Google Scholar] [CrossRef] [PubMed]
- Martin, I.; Kim, J.W.; Lee, B.D.; Kang, H.C.; Xu, J.C.; Jia, H.; Stankowski, J.; Kim, M.S.; Zhong, J.; Kumar, M.; et al. Ribosomal protein s15 phosphorylation mediates LRRK2 neurodegeneration in Parkinson’s disease. Cell 2014, 157, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Matta, S.; Van Kolen, K.; da Cunha, R.; van den Bogaart, G.; Mandemakers, W.; Miskiewicz, K.; De Bock, P.J.; Morais, V.A.; Vilain, S.; Haddad, D.; et al. LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron 2012, 75, 1008–1021. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.J.; Park, J.; Ho, D.H.; Kim, H.; Kim, C.H.; Oh, H.; Ga, I.; Seo, H.; Chang, S.; Son, I.; et al. LRRK2 phosphorylates Snapin and inhibits interaction of Snapin with SNAP-25. Exp. Mol. Med. 2013, 45, e36. [Google Scholar] [CrossRef][Green Version]
- Belluzzi, E.; Gonnelli, A.; Cirnaru, M.D.; Marte, A.; Plotegher, N.; Russo, I.; Civiero, L.; Cogo, S.; Carrion, M.P.; Franchin, C.; et al. LRRK2 phosphorylates pre-synaptic N-ethylmaleimide sensitive fusion (NSF) protein enhancing its ATPase activity and SNARE complex disassembling rate. Mol. Neurodegener. 2016, 11, 1. [Google Scholar] [CrossRef]
- Islam, M.S.; Nolte, H.; Jacob, W.; Ziegler, A.B.; Putz, S.; Grosjean, Y.; Szczepanowska, K.; Trifunovic, A.; Braun, T.; Heumann, H.; et al. Human R1441C LRRK2 regulates the synaptic vesicle proteome and phosphoproteome in a Drosophila model of Parkinson’s disease. Hum. Mol. Genet. 2016, 25, 5365–5382. [Google Scholar] [CrossRef]
- Steger, M.; Tonelli, F.; Ito, G.; Davies, P.; Trost, M.; Vetter, M.; Wachter, S.; Lorentzen, E.; Duddy, G.; Wilson, S.; et al. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. eLife 2016, 5. [Google Scholar] [CrossRef]
- Steger, M.; Diez, F.; Dhekne, H.S.; Lis, P.; Nirujogi, R.S.; Karayel, O.; Tonelli, F.; Martinez, T.N.; Lorentzen, E.; Pfeffer, S.R.; et al. Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. eLife 2017, 6. [Google Scholar] [CrossRef]
- Jeong, G.R.; Jang, E.H.; Bae, J.R.; Jun, S.; Kang, H.C.; Park, C.H.; Shin, J.H.; Yamamoto, Y.; Tanaka-Yamamoto, K.; Dawson, V.L.; et al. Dysregulated phosphorylation of Rab GTPases by LRRK2 induces neurodegeneration. Mol. Neurodegener. 2018, 13, 8. [Google Scholar] [CrossRef]
- Barbier, P.; Zejneli, O.; Martinho, M.; Lasorsa, A.; Belle, V.; Smet-Nocca, C.; Tsvetkov, P.O.; Devred, F.; Landrieu, I. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Front. Aging Neurosci. 2019, 11, 204. [Google Scholar] [CrossRef] [PubMed]
- Avila, J.; Lucas, J.J.; Perez, M.; Hernandez, F. Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 2004, 84, 361–384. [Google Scholar] [CrossRef] [PubMed]
- Drechsel, D.N.; Hyman, A.A.; Cobb, M.H.; Kirschner, M.W. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol. Biol. Cell 1992, 3, 1141–1154. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Liu, F.; Gong, C.X. Tau and neurodegenerative disease: The story so far. Nat. Rev. Neurol. 2016, 12, 15–27. [Google Scholar] [CrossRef]
- Gao, Y.L.; Wang, N.; Sun, F.R.; Cao, X.P.; Zhang, W.; Yu, J.T. Tau in neurodegenerative disease. Ann. Transl. Med. 2018, 6, 175. [Google Scholar] [CrossRef]
- Li, Y.; Liu, W.; Oo, T.F.; Wang, L.; Tang, Y.; Jackson-Lewis, V.; Zhou, C.; Geghman, K.; Bogdanov, M.; Przedborski, S.; et al. Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson’s disease. Nat. Neurosci. 2009, 12, 826–828. [Google Scholar] [CrossRef]
- Melrose, H.L.; Dachsel, J.C.; Behrouz, B.; Lincoln, S.J.; Yue, M.; Hinkle, K.M.; Kent, C.B.; Korvatska, E.; Taylor, J.P.; Witten, L.; et al. Impaired dopaminergic neurotransmission and microtubule-associated protein tau alterations in human LRRK2 transgenic mice. Neurobiol. Dis. 2010, 40, 503–517. [Google Scholar] [CrossRef]
- Rajput, A.; Dickson, D.W.; Robinson, C.A.; Ross, O.A.; Dachsel, J.C.; Lincoln, S.J.; Cobb, S.A.; Rajput, M.L.; Farrer, M.J. Parkinsonism, Lrrk2 G2019S, and tau neuropathology. Neurology 2006, 67, 1506–1508. [Google Scholar] [CrossRef]
- Picconi, B.; Piccoli, G.; Calabresi, P. Synaptic dysfunction in Parkinson’s disease. Adv. Exp. Med. Biol. 2012, 970, 553–572. [Google Scholar] [CrossRef]
- Arranz, A.M.; Delbroek, L.; Van Kolen, K.; Guimaraes, M.R.; Mandemakers, W.; Daneels, G.; Matta, S.; Calafate, S.; Shaban, H.; Baatsen, P.; et al. LRRK2 functions in synaptic vesicle endocytosis through a kinase-dependent mechanism. J. Cell Sci. 2015, 128, 541–552. [Google Scholar] [CrossRef]
- Nguyen, M.; Krainc, D. LRRK2 phosphorylation of auxilin mediates synaptic defects in dopaminergic neurons from patients with Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, 5576–5581. [Google Scholar] [CrossRef] [PubMed]
- Toma-Fukai, S.; Shimizu, T. Structural Insights into the Regulation Mechanism of Small GTPases by GEFs. Molecules 2019, 24, 3308. [Google Scholar] [CrossRef] [PubMed]
- Dhekne, H.S.; Yanatori, I.; Gomez, R.C.; Tonelli, F.; Diez, F.; Schule, B.; Steger, M.; Alessi, D.R.; Pfeffer, S.R. A pathway for Parkinson’s Disease LRRK2 kinase to block primary cilia and Sonic hedgehog signaling in the brain. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Madero-Perez, J.; Fdez, E.; Fernandez, B.; Lara Ordonez, A.J.; Blanca Ramirez, M.; Gomez-Suaga, P.; Waschbusch, D.; Lobbestael, E.; Baekelandt, V.; Nairn, A.C.; et al. Parkinson disease-associated mutations in LRRK2 cause centrosomal defects via Rab8a phosphorylation. Mol. Neurodegener. 2018, 13, 3. [Google Scholar] [CrossRef]
- Fernandez, B.; Lara Ordonez, A.J.; Fdez, E.; Mutez, E.; Comptdaer, T.; Leghay, C.; Kreisler, A.; Simonin, C.; Vandewynckel, L.; Defebvre, L.; et al. Centrosomal cohesion deficits as cellular biomarker in lymphoblastoid cell lines from LRRK2 Parkinson’s disease patients. Biochem. J. 2019, 476, 2797–2813. [Google Scholar] [CrossRef]
- Rivero-Rios, P.; Romo-Lozano, M.; Madero-Perez, J.; Thomas, A.P.; Biosa, A.; Greggio, E.; Hilfiker, S. The G2019S variant of leucine-rich repeat kinase 2 (LRRK2) alters endolysosomal trafficking by impairing the function of the GTPase RAB8A. J. Biol. Chem. 2019, 294, 4738–4758. [Google Scholar] [CrossRef]
- Eguchi, T.; Kuwahara, T.; Sakurai, M.; Komori, T.; Fujimoto, T.; Ito, G.; Yoshimura, S.I.; Harada, A.; Fukuda, M.; Koike, M.; et al. LRRK2 and its substrate Rab GTPases are sequentially targeted onto stressed lysosomes and maintain their homeostasis. Proc. Natl. Acad. Sci. USA 2018, 115, E9115–E9124. [Google Scholar] [CrossRef]
- Chiu, C.C.; Yeh, T.H.; Lai, S.C.; Weng, Y.H.; Huang, Y.C.; Cheng, Y.C.; Chen, R.S.; Huang, Y.Z.; Hung, J.; Chen, C.C.; et al. Increased Rab35 expression is a potential biomarker and implicated in the pathogenesis of Parkinson’s disease. Oncotarget 2016, 7, 54215–54227. [Google Scholar] [CrossRef]
- Hsu, C.; Morohashi, Y.; Yoshimura, S.; Manrique-Hoyos, N.; Jung, S.; Lauterbach, M.A.; Bakhti, M.; Gronborg, M.; Mobius, W.; Rhee, J.; et al. Regulation of exosome secretion by Rab35 and its GTPase-activating proteins TBC1D10A-C. J. Cell Biol. 2010, 189, 223–232. [Google Scholar] [CrossRef]
- Kouranti, I.; Sachse, M.; Arouche, N.; Goud, B.; Echard, A. Rab35 regulates an endocytic recycling pathway essential for the terminal steps of cytokinesis. Curr. Biol. 2006, 16, 1719–1725. [Google Scholar] [CrossRef]
- Malik, B.R.; Maddison, D.C.; Smith, G.A.; Peters, O.M. Autophagic and endo-lysosomal dysfunction in neurodegenerative disease. Mol. Brain. 2019, 12, 100. [Google Scholar] [CrossRef] [PubMed]
- Hur, E.M.; Jang, E.H.; Jeong, G.R.; Lee, B.D. LRRK2 and membrane trafficking: Nexus of Parkinson’s disease. BMB Rep. 2019, 52, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Yamaguchi, H.; Giaime, E.; Boyle, S.; Kopan, R.; Kelleher, R.J., 3rd; Shen, J. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc. Natl. Acad. Sci. USA 2010, 107, 9879–9884. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.W.; Zhang, T.; Jiang, C.; Chen, S.; Guo, M. Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Hum. Mol. Genet. 2012, 21, 1350–1363. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Giaime, E.; Yamaguchi, H.; Ichimura, T.; Liu, Y.; Si, H.; Cai, H.; Bonventre, J.V.; Shen, J. Loss of leucine-rich repeat kinase 2 causes age-dependent bi-phasic alterations of the autophagy pathway. Mol. Neurodegener. 2012, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Tanji, K.; Odagiri, S.; Miki, Y.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease and related neurodegenerative disorders. Mol. Neurobiol. 2013, 47, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castano-Diez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef]
- Nuber, S.; Rajsombath, M.; Minakaki, G.; Winkler, J.; Muller, C.P.; Ericsson, M.; Caldarone, B.; Dettmer, U.; Selkoe, D.J. Abrogating Native alpha-Synuclein Tetramers in Mice Causes a L-DOPA-Responsive Motor Syndrome Closely Resembling Parkinson’s Disease. Neuron 2018, 100, 75–90.e75. [Google Scholar] [CrossRef]
- Dettmer, U.; Newman, A.J.; Soldner, F.; Luth, E.S.; Kim, N.C.; von Saucken, V.E.; Sanderson, J.B.; Jaenisch, R.; Bartels, T.; Selkoe, D. Parkinson-causing alpha-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat. Commun. 2015, 6, 7314. [Google Scholar] [CrossRef]
- Conway, K.A.; Lee, S.J.; Rochet, J.C.; Ding, T.T.; Harper, J.D.; Williamson, R.E.; Lansbury, P.T., Jr. Accelerated oligomerization by Parkinson’s disease linked alpha-synuclein mutants. Ann. N. Y. Acad. Sci. 2000, 920, 42–45. [Google Scholar] [CrossRef]
- Leverenz, J.B.; Umar, I.; Wang, Q.; Montine, T.J.; McMillan, P.J.; Tsuang, D.W.; Jin, J.; Pan, C.; Shin, J.; Zhu, D.; et al. Proteomic identification of novel proteins in cortical lewy bodies. Brain Pathol. 2007, 17, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Liao, L.; Cheng, D.; Duong, D.M.; Gearing, M.; Lah, J.J.; Levey, A.I.; Peng, J. Proteomic identification of novel proteins associated with Lewy bodies. Front. Biosci. 2008, 13, 3850–3856. [Google Scholar] [CrossRef] [PubMed]
- Miklossy, J.; Arai, T.; Guo, J.P.; Klegeris, A.; Yu, S.; McGeer, E.G.; McGeer, P.L. LRRK2 expression in normal and pathologic human brain and in human cell lines. J. Neuropathol. Exp. Neurol. 2006, 65, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Siedlak, S.L.; Smith, M.A.; Perry, G.; Chen, S.G. LRRK2 protein is a component of Lewy bodies. Ann. Neurol. 2006, 60, 617–618. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Babar, A.; Siedlak, S.L.; Yang, Q.; Ito, G.; Iwatsubo, T.; Smith, M.A.; Perry, G.; Chen, S.G. LRRK2 in Parkinson’s disease and dementia with Lewy bodies. Mol. Neurodegener. 2006, 1, 17. [Google Scholar] [CrossRef]
- Bandopadhyay, R.; Kingsbury, A.E.; Cookson, M.R.; Reid, A.R.; Evans, I.M.; Hope, A.D.; Pittman, A.M.; Lashley, T.; Canet-Aviles, R.; Miller, D.W.; et al. The expression of DJ-1 (PARK7) in normal human CNS and idiopathic Parkinson’s disease. Brain 2004, 127, 420–430. [Google Scholar] [CrossRef]
- Jin, J.; Meredith, G.E.; Chen, L.; Zhou, Y.; Xu, J.; Shie, F.S.; Lockhart, P.; Zhang, J. Quantitative proteomic analysis of mitochondrial proteins: Relevance to Lewy body formation and Parkinson’s disease. Brain Res. Mol. Brain. Res. 2005, 134, 119–138. [Google Scholar] [CrossRef]
- Schlossmacher, M.G.; Frosch, M.P.; Gai, W.P.; Medina, M.; Sharma, N.; Forno, L.; Ochiishi, T.; Shimura, H.; Sharon, R.; Hattori, N.; et al. Parkin localizes to the Lewy bodies of Parkinson disease and dementia with Lewy bodies. Am. J. Pathol. 2002, 160, 1655–1667. [Google Scholar] [CrossRef]
- Gandhi, S.; Muqit, M.M.; Stanyer, L.; Healy, D.G.; Abou-Sleiman, P.M.; Hargreaves, I.; Heales, S.; Ganguly, M.; Parsons, L.; Lees, A.J.; et al. PINK1 protein in normal human brain and Parkinson’s disease. Brain 2006, 129, 1720–1731. [Google Scholar] [CrossRef]
- Schneider, S.A.; Alcalay, R.N. Neuropathology of genetic synucleinopathies with parkinsonism: Review of the literature. Mov. Disord. 2017, 32, 1504–1523. [Google Scholar] [CrossRef]
- Mehta, S.H.; Sethi, K.D. Learning PD from the lark. Mov. Disord. 2011, 26, 2178. [Google Scholar] [CrossRef] [PubMed]
- Wider, C.; Dickson, D.W.; Wszolek, Z.K. Leucine-rich repeat kinase 2 gene-associated disease: Redefining genotype-phenotype correlation. Neurodegener. Dis. 2010, 7, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Poulopoulos, M.; Cortes, E.; Vonsattel, J.P.; Fahn, S.; Waters, C.; Cote, L.J.; Moskowitz, C.; Honig, L.S.; Clark, L.N.; Marder, K.S.; et al. Clinical and pathological characteristics of LRRK2 G2019S patients with PD. J. Mol. Neurosci. 2012, 47, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E.; Hazrati, L.N.; Fujioka, S.; Wszolek, Z.K.; Dickson, D.W.; Ross, O.A.; Van Deerlin, V.M.; Trojanowski, J.Q.; Hurtig, H.I.; et al. Clinical correlations with Lewy body pathology in LRRK2-related Parkinson disease. JAMA Neurol. 2015, 72, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Agin-Liebes, J.; Cortes, E.; Vonsattel, J.P.; Marder, K.; Alcalay, R.N. Movement disorders rounds: A case of missing pathology in a patient with LRRK2 Parkinson’s disease. Parkinsonism. Relat. Disord. 2020, 74, 76–77. [Google Scholar] [CrossRef]
- Henderson, M.X.; Sengupta, M.; Trojanowski, J.Q.; Lee, V.M.Y. Alzheimer’s disease tau is a prominent pathology in LRRK2 Parkinson’s disease. Acta Neuropathol. Commun. 2019, 7, 183. [Google Scholar] [CrossRef]
- Kalia, L.V. Expert comment: “A case of missing pathology in a patient with LRRK2 Parkinson’s disease”. Parkinsonism Relat. Disord. 2020, 74, 78–79. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Petre, B.M.; Wall, J.; Simon, M.; Nowak, R.J.; Walz, T.; Lansbury, P.T., Jr. Alpha-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J. Mol. Biol. 2002, 322, 1089–1102. [Google Scholar] [CrossRef]
- Ding, T.T.; Lee, S.J.; Rochet, J.C.; Lansbury, P.T., Jr. Annular alpha-synuclein protofibrils are produced when spherical protofibrils are incubated in solution or bound to brain-derived membranes. Biochemistry 2002, 41, 10209–10217. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Hartley, D.; Petre, B.M.; Walz, T.; Lansbury, P.T., Jr. Neurodegenerative disease: Amyloid pores from pathogenic mutations. Nature 2002, 418, 291. [Google Scholar] [CrossRef]
- Chen, L.; Feany, M.B. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci. 2005, 8, 657–663. [Google Scholar] [CrossRef]
- Aasly, J.O.; Johansen, K.K.; Bronstad, G.; Waro, B.J.; Majbour, N.K.; Varghese, S.; Alzahmi, F.; Paleologou, K.E.; Amer, D.A.; Al-Hayani, A.; et al. Elevated levels of cerebrospinal fluid alpha-synuclein oligomers in healthy asymptomatic LRRK2 mutation carriers. Front. Aging Neurosci. 2014, 6, 248. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.W.; Leung, C.T.; Liu, H.; Pang, S.Y.; Lam, C.S.; Xian, J.; Li, L.; Kung, M.H.; Ramsden, D.B.; Ho, S.L. Age-dependent accumulation of oligomeric SNCA/alpha-synuclein from impaired degradation in mutant LRRK2 knockin mouse model of Parkinson disease: Role for therapeutic activation of chaperone-mediated autophagy (CMA). Autophagy 2020, 16, 347–370. [Google Scholar] [CrossRef] [PubMed]
- Novello, S.; Arcuri, L.; Dovero, S.; Dutheil, N.; Shimshek, D.R.; Bezard, E.; Morari, M. G2019S LRRK2 mutation facilitates alpha-synuclein neuropathology in aged mice. Neurobiol. Dis. 2018, 120, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Parisiadou, L.; Gu, X.L.; Wang, L.; Shim, H.; Sun, L.; Xie, C.; Long, C.X.; Yang, W.J.; Ding, J.; et al. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant alpha-synuclein. Neuron 2009, 64, 807–827. [Google Scholar] [CrossRef] [PubMed]
- Bieri, G.; Brahic, M.; Bousset, L.; Couthouis, J.; Kramer, N.J.; Ma, R.; Nakayama, L.; Monbureau, M.; Defensor, E.; Schule, B.; et al. LRRK2 modifies alpha-syn pathology and spread in mouse models and human neurons. Acta Neuropathol. 2019, 137, 961–980. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K.; Rub, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Hijaz, B.A.; Volpicelli-Daley, L.A. Initiation and propagation of alpha-synuclein aggregation in the nervous system. Mol. Neurodegener. 2020, 15, 19. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef]
- Luk, K.C.; Song, C.; O’Brien, P.; Stieber, A.; Branch, J.R.; Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. USA 2009, 106, 20051–20056. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.M.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J. Exp. Med. 2012, 209, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Karampetsou, M.; Ardah, M.T.; Semitekolou, M.; Polissidis, A.; Samiotaki, M.; Kalomoiri, M.; Majbour, N.; Xanthou, G.; El-Agnaf, O.M.A.; Vekrellis, K. Phosphorylated exogenous alpha-synuclein fibrils exacerbate pathology and induce neuronal dysfunction in mice. Sci. Rep. 2017, 7, 16533. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Patel, S.; Lee, S.J. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J. Neurosci. 2005, 25, 6016–6024. [Google Scholar] [CrossRef] [PubMed]
- Freundt, E.C.; Maynard, N.; Clancy, E.K.; Roy, S.; Bousset, L.; Sourigues, Y.; Covert, M.; Melki, R.; Kirkegaard, K.; Brahic, M. Neuron-to-neuron transmission of alpha-synuclein fibrils through axonal transport. Ann. Neurol. 2012, 72, 517–524. [Google Scholar] [CrossRef]
- Lee, H.J.; Suk, J.E.; Bae, E.J.; Lee, J.H.; Paik, S.R.; Lee, S.J. Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int. J. Biochem. Cell Biol. 2008, 40, 1835–1849. [Google Scholar] [CrossRef]
- Kondo, K.; Obitsu, S.; Teshima, R. alpha-Synuclein aggregation and transmission are enhanced by leucine-rich repeat kinase 2 in human neuroblastoma SH-SY5Y cells. Biol. Pharm. Bull. 2011, 34, 1078–1083. [Google Scholar] [CrossRef]
- Bae, E.J.; Kim, D.K.; Kim, C.; Mante, M.; Adame, A.; Rockenstein, E.; Ulusoy, A.; Klinkenberg, M.; Jeong, G.R.; Bae, J.R.; et al. LRRK2 kinase regulates alpha-synuclein propagation via RAB35 phosphorylation. Nat. Commun. 2018, 9, 3465. [Google Scholar] [CrossRef]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef]
- Gitler, A.D.; Bevis, B.J.; Shorter, J.; Strathearn, K.E.; Hamamichi, S.; Su, L.J.; Caldwell, K.A.; Caldwell, G.A.; Rochet, J.C.; McCaffery, J.M.; et al. The Parkinson’s disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 145–150. [Google Scholar] [CrossRef]
- Kuwahara, T.; Iwatsubo, T. The Emerging Functions of LRRK2 and Rab GTPases in the Endolysosomal System. Front. Neurosci. 2020, 14, 227. [Google Scholar] [CrossRef]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Kozina, E.; Sadasivan, S.; Jiao, Y.; Dou, Y.; Ma, Z.; Tan, H.; Kodali, K.; Shaw, T.; Peng, J.; Smeyne, R.J. Mutant LRRK2 mediates peripheral and central immune responses leading to neurodegeneration in vivo. Brain 2018, 141, 1753–1769. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; James, W.S.; Cowley, S.A. LRRK2 in peripheral and central nervous system innate immunity: Its link to Parkinson’s disease. Biochem. Soc. Trans. 2017, 45, 131–139. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).