Hypoxia-Driven Immune Escape in the Tumor Microenvironment



Abstract
1. Introduction
2. Hypoxia Signaling and Metabolism
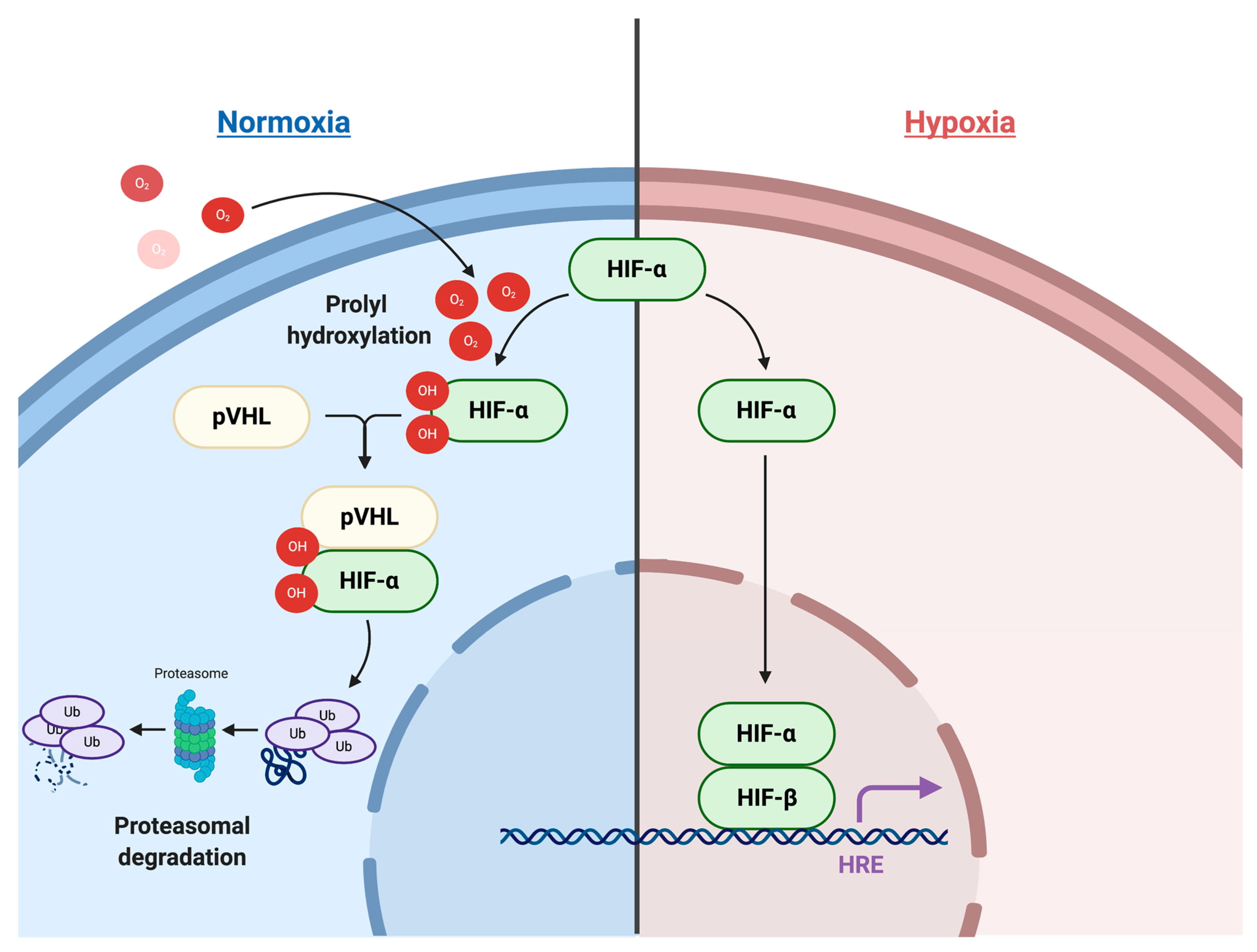
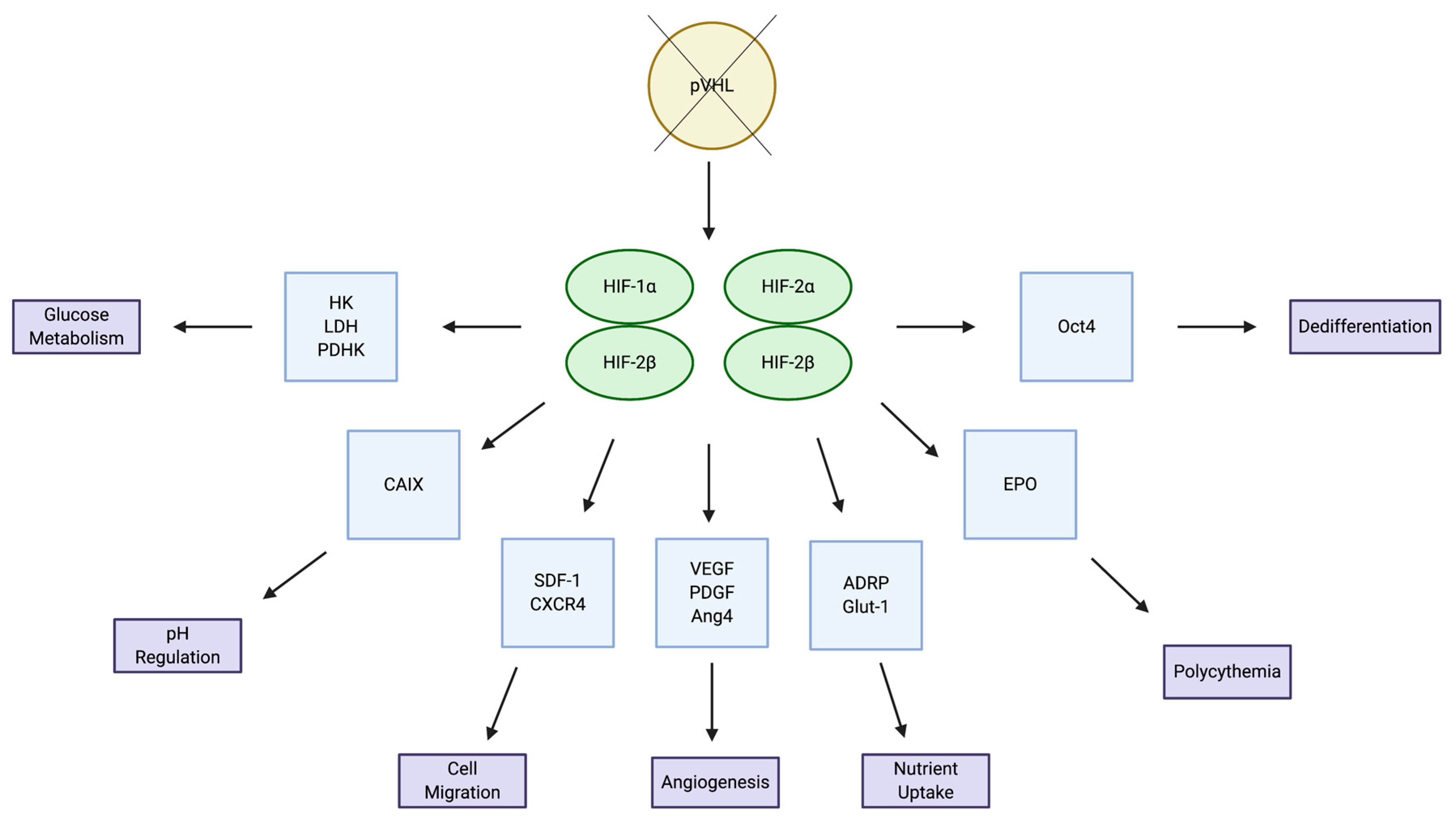
2.1. HIF Signaling Pathways
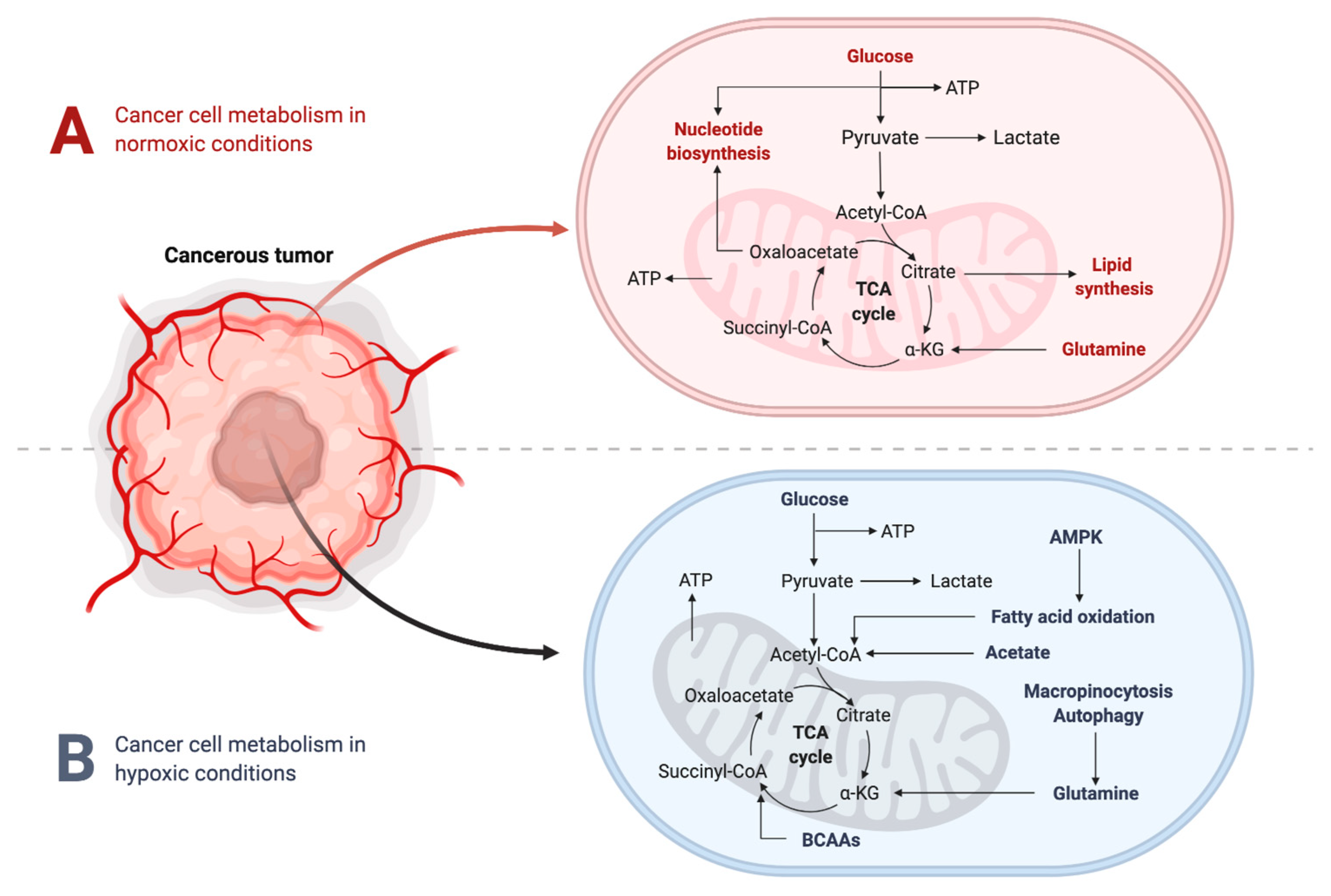
2.2. Metabolic Changes in the TME under Hypoxic Conditions
3. Hypoxia and the Immune System
3.1. Dendritic Cells
3.2. Macrophages
3.3. B Cells
3.4. T Cells
3.5. Natural Killer (NK) Cells
3.6. Myeloid-Derived Suppressor Cells (MDSCs)
4. Hypoxia and Immunogenic Cell Death
5. Hypoxia-Mediated Therapeutic Resistance
5.1. Hypoxia-Mediated Primary Resistance
5.2. Hypoxia-Mediated Acquired Resistance
6. Hypoxia-Targeted Immunotherapies
6.1. Strategies for Targeting Hypoxia-Induced Pathways
6.2. Considerations for Targeting Hypoxia-Induced Pathways
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Multhoff, G.; Vaupel, P. Hypoxia compromises anti-cancer immune responses. In Advances in Experimental Medicine and Biology; Springer: Berlin, Germany, 2020; Volume 1232, pp. 131–143. [Google Scholar]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-Induced Angiogenesis: Good and Evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Endler, A.; Shibasaki, F. Hypoxia and angiogenesis: Regulation of hypoxia-inducible factors via novel binding factors. Exp. Mol. Med. 2009, 41, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Engel, C.; Brügmann, G.; Lambing, S.; Mühlenbeck, L.H.; Marx, S.; Hagen, C.; Horv, D.; Goldeck, M.; Ludwig, J.; Herzne, A.M.; et al. RIG-I Resists Hypoxia-Induced Immunosuppression and Dedifferentiation. Cancer Immunol. Res. 2017, 5, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Ren, H.; Jia, L.; Chen, J.; Xin, W.; Yan, F.; Li, J.; Wang, X.; Gao, S.; Qian, D.; et al. Inhibition of HIF-1α by PX-478 enhances the anti-tumor effect of gemcitabine by inducing immunogenic cell death in pancreatic ductal adenocarcinoma. Oncotarget 2015, 6, 2250–2262. [Google Scholar] [CrossRef]
- Hatfield, S.; Veszeleiova, K.; Steingold, J.; Sethuraman, J.; Sitkovsky, M. Mechanistic justifications of systemic therapeutic oxygenation of tumors to weaken the hypoxia inducible factor 1α-mediated immunosuppression. Adv. Exp. Med. Biol. 2019, 1136, 113–121. [Google Scholar] [CrossRef]
- Han, Y.K.; Park, G.Y.; Bae, M.J.I.; Kim, J.S.; Jo, W.S.; Lee, C.G. Hypoxia induces immunogenic cell death of cancer cells by enhancing the exposure of cell surface calreticulin in an endoplasmic reticulum stress-dependent manner. Oncol. Lett. 2019, 18, 6269–6274. [Google Scholar] [CrossRef]
- Tamura, R.; Tanaka, T.; Akasaki, Y.; Murayama, Y.; Yoshida, K.; Sasaki, H. The role of vascular endothelial growth factor in the hypoxic and immunosuppressive tumor microenvironment: Perspectives for therapeutic implications. Med. Oncol. 2020, 37, 2. [Google Scholar] [CrossRef]
- Sormendi, S.; Wielockx, B. Hypoxia pathway proteins as central mediators of metabolism in the tumor cells and their microenvironment. Front. Immunol. 2018, 9, 40. [Google Scholar] [CrossRef]
- Koh, M.Y.; Powis, G. Passing the baton: The HIF switch. Trends Biochem. Sci. 2012, 37, 364–372. [Google Scholar] [CrossRef]
- Holmquist-Mengelbier, L.; Fredlund, E.; Löfstedt, T.; Noguera, R.; Navarro, S.; Nilsson, H.; Pietras, A.; Vallon-Christersson, J.; Borg, Å.; Gradin, K.; et al. Recruitment of HIF-1α and HIF-2α to common target genes is differentially regulated in neuroblastoma: HIF-2α promotes an aggressive phenotype. Cancer Cell 2006, 10, 413–423. [Google Scholar] [CrossRef]
- Peng, J.; Zhang, L.; Drysdale, L.; Fong, G.H. The transcription factor EPAS-1/hypoxia-inducible factor 2α plays an important role in vascular remodeling. Proc. Natl. Acad. Sci. USA 2000, 97, 8386–8391. [Google Scholar] [CrossRef]
- Bangoura, G.; Yang, L.Y.; Huang, G.W.; Wang, W. Expression of HIF-2α/EPAS1 in hepatocellular carcinoma. World J. Gastroenterol. 2004, 10, 525–530. [Google Scholar] [CrossRef]
- Shestov, A.A.; Liu, X.; Ser, Z.; Cluntun, A.A.; Hung, Y.P.; Huang, L.; Kim, D.; Le, A.; Yellen, G.; Albeck, J.G.; et al. Quantitative determinants of aerobic glycolysis identify flux through the enzyme GAPDH as a limiting step. Elife 2014, 3, 1–18. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The Biology of Cancer: Metabolic Reprogramming Fuels Cell Growth and Proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef]
- Levine, A.J.; Puzio-Kuter, A.M. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 2010, 330, 1340–1344. [Google Scholar] [CrossRef]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Wielockx, B.; Meneses, A. PHD2: From hypoxia regulation to disease progression. Hypoxia 2016, 53. [Google Scholar] [CrossRef]
- Selfridge, A.C.; Cavadas, M.A.S.; Scholz, C.C.; Campbell, E.L.; Welch, L.C.; Lecuona, E.; Colgan, S.P.; Barrett, K.E.; Sporn, P.H.S.; Sznajder, J.I.; et al. Hypercapnia suppresses the HIF-dependent adaptive response to hypoxia. J. Biol. Chem. 2016, 291, 11800–11808. [Google Scholar] [CrossRef]
- Pezzuto, A.; Carico, E. Role of HIF-1 in Cancer Progression: Novel Insights. A Review. Curr. Mol. Med. 2018, 18, 343–351. [Google Scholar] [CrossRef]
- Elia, A.R.; Cappello, P.; Puppo, M.; Fraone, T.; Vanni, C.; Eva, A.; Musso, T.; Novelli, F.; Varesio, L.; Giovarelli, M. Human dendritic cells differentiated in hypoxia down-modulate antigen uptake and change their chemokine expression profile. J. Leukoc. Biol. 2008, 84, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Mancino, A.; Schioppa, T.; Larghi, P.; Pasqualini, F.; Nebuloni, M.; Chen, I.H.; Sozzani, S.; Austyn, J.M.; Mantovani, A.; Sica, A. Divergent effects of hypoxia on dendritic cell functions. Blood 2008, 112, 3723–3734. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Ostuni, R.; Kratochvill, F.; Murray, P.J.; Natoli, G. Macrophages and cancer: From mechanisms to therapeutic implications. Trends Immunol. 2015, 36, 229–239. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Henze, A.T.; Mazzone, M. The impact of hypoxia on tumor-associated macrophages. J. Clin. Investig. 2016, 126, 3672–3679. [Google Scholar] [CrossRef]
- Hughes, R.; Qian, B.Z.; Rowan, C.; Muthana, M.; Keklikoglou, I.; Olson, O.C.; Tazzyman, S.; Danson, S.; Addison, C.; Clemons, M.; et al. Perivascular M2 macrophages stimulate tumor relapse after chemotherapy. Cancer Res. 2015, 75, 3479–3491. [Google Scholar] [CrossRef]
- Ohno, S.; Ohno, Y.; Suzuki, N.; Kamei, T.; Koike, K.; Inagawa, H.; Kohchi, C.; Soma, G.I.; Inoue, M. Correlation of histological localization of tumor-associated macrophages with clinicopathological features in endometrial cancer. Anticancer Res. 2004, 24, 3335–3342. [Google Scholar]
- Hoffman, W.; Lakkis, F.G.; Chalasani, G. B cells, antibodies, and more. Clin. J. Am. Soc. Nephrol. 2016, 11, 137–154. [Google Scholar] [CrossRef]
- Mauri, C.; Bosma, A. Immune Regulatory Function of B Cells. Annu. Rev. Immunol. 2012, 30, 221–241. [Google Scholar] [CrossRef]
- Krzywinska, E.; Stockmann, C. Hypoxia, metabolism and immune cell function. Biomedicines 2018, 6, 56. [Google Scholar] [CrossRef]
- Kojima, H.; Gu, H.; Nomura, S.; Caldwell, C.C.; Kobata, T.; Carmeliet, P.; Semenza, G.L.; Sitkovsky, M.V. Abnormal B lymphocyte development and autoimmunity in hypoxia-inducible factor 1α-deficient chimeric mice. Proc. Natl. Acad. Sci. USA 2002, 99, 2170–2174. [Google Scholar] [CrossRef]
- Goda, N.; Ryan, H.E.; Khadivi, B.; McNulty, W.; Rickert, R.C.; Johnson, R.S. Hypoxia-Inducible Factor 1 Is Essential for Cell Cycle Arrest during Hypoxia. Mol. Cell. Biol. 2003, 23, 359–369. [Google Scholar] [CrossRef]
- Meng, X.; Grötsch, B.; Luo, Y.; Knaup, K.X.; Wiesener, M.S.; Chen, X.X.; Jantsch, J.; Fillatreau, S.; Schett, G.; Bozec, A. Hypoxia-inducible factor-1α is a critical transcription factor for IL-10-producing B cells in autoimmune disease. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Zhang, Y.; Garcia-Ibanez, L.; Toellner, K.M. Regulation of germinal center B-cell differentiation. Immunol. Rev. 2016, 270, 8–19. [Google Scholar] [CrossRef]
- Jellusova, J.; Cato, M.H.; Apgar, J.R.; Ramezani-Rad, P.; Leung, C.R.; Chen, C.; Richardson, A.D.; Conner, E.M.; Benschop, R.J.; Woodgett, J.R.; et al. Gsk3 is a metabolic checkpoint regulator in B cells. Nat. Immunol. 2017, 18, 303–312. [Google Scholar] [CrossRef]
- Caro-Maldonado, A.; Wang, R.; Nichols, A.G.; Kuraoka, M.; Milasta, S.; Sun, L.D.; Gavin, A.L.; Abel, E.D.; Kelsoe, G.; Green, D.R.; et al. Metabolic Reprogramming Is Required for Antibody Production That Is Suppressed in Anergic but Exaggerated in Chronically BAFF-Exposed B Cells. J. Immunol. 2014, 192, 3626–3636. [Google Scholar] [CrossRef]
- Griss, J.; Bauer, W.; Wagner, C.; Simon, M.; Chen, M.; Grabmeier-Pfistershammer, K.; Maurer-Granofszky, M.; Roka, F.; Penz, T.; Bock, C.; et al. B cells sustain inflammation and predict response to immune checkpoint blockade in human melanoma. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Masopust, D.; Schenkel, J.M. The integration of T cell migration, differentiation and function. Nat. Rev. Immunol. 2013, 13, 309–320. [Google Scholar] [CrossRef]
- Cho, S.H.; Raybuck, A.L.; Blagih, J.; Kemboi, E.; Haase, V.H.; Jones, R.G.; Boothby, M.R. Hypoxia-inducible factors in CD4+ T cells promote metabolism, switch cytokine secretion, and T cell help in humoral immunity. Proc. Natl. Acad. Sci. USA 2019, 116, 8975–8984. [Google Scholar] [CrossRef]
- Tao, J.H.; Barbi, J.; Pan, F. Hypoxia-inducible factors in T lymphocyte differentiation and function. A review in the theme: Cellular responses to hypoxia. Am. J. Physiol. - Cell Physiol. 2015, 309, C580–C589. [Google Scholar] [CrossRef]
- Waickman, A.T.; Powell, J.D. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol. Rev. 2012, 249, 43–58. [Google Scholar] [CrossRef]
- Frauwirth, K.A.; Thompson, C.B. Regulation of T Lymphocyte Metabolism. J. Immunol. 2004, 172, 4661–4665. [Google Scholar] [CrossRef]
- Chen, J.L.Y.; Lucas, J.E.; Schroeder, T.; Mori, S.; Wu, J.; Nevins, J.; Dewhirst, M.; West, M.; Chi, J.T. The genomic analysis of lactic acidosis and acidosis response in human cancers. PLoS Genet. 2008, 4, e100293. [Google Scholar] [CrossRef]
- Pauken, K.E.; Wherry, E.J. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015, 36, 265–276. [Google Scholar] [CrossRef]
- Caldwell, C.C.; Kojima, H.; Lukashev, D.; Armstrong, J.; Farber, M.; Apasov, S.G.; Sitkovsky, M.V. Differential Effects of Physiologically Relevant Hypoxic Conditions on T Lymphocyte Development and Effector Functions. J. Immunol. 2001, 167, 6140–6149. [Google Scholar] [CrossRef]
- Sukumar, M.; Liu, J.; Ji, Y.; Subramanian, M.; Crompton, J.G.; Yu, Z.; Roychoudhuri, R.; Palmer, D.C.; Muranski, P.; Karoly, E.D.; et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Investig. 2013, 123, 4479–4488. [Google Scholar] [CrossRef]
- Parodi, M.; Raggi, F.; Cangelosi, D.; Manzini, C.; Balsamo, M.; Blengio, F.; Eva, A.; Varesio, L.; Pietra, G.; Moretta, L.; et al. Hypoxia modifies the transcriptome of human NK cells, modulates their immunoregulatory profile, and influences NK cell subset migration. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Balsamo, M.; Manzini, C.; Pietra, G.; Raggi, F.; Blengio, F.; Mingari, M.C.; Varesio, L.; Moretta, L.; Bosco, M.C.; Vitale, M. Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC. Eur. J. Immunol. 2013, 43, 2756–2764. [Google Scholar] [CrossRef]
- Krzywinska, E.; Kantari-Mimoun, C.; Kerdiles, Y.; Sobecki, M.; Isagawa, T.; Gotthardt, D.; Castells, M.; Haubold, J.; Millien, C.; Viel, T.; et al. Loss of HIF-1α in natural killer cells inhibits tumour growth by stimulating non-productive angiogenesis. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Cluff, E.R.; Nolan, J.; Collins, C.; Varadaraj, A.; Rajasekaran, N. Hypoxia-Inducible Factor-1α is upregulated in Natural Killer cells by Interleukin-2 and hypoxia via PI3K/mTOR signaling pathway. J. Immunol. 2019, 202, 194.37. [Google Scholar]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.; Muthana, M.; Coffelt, S.B.; Lewis, C.E. The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer 2008, 8, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.I.; Cheng, P.; Cho, H.I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef]
- Ye, X.Z.; Yu, S.C.; Bian, X.W. Contribution of myeloid-derived suppressor cells to tumor-induced immune suppression, angiogenesis, invasion and metastasis. J. Genet. Genomics 2010, 37, 423–430. [Google Scholar] [CrossRef]
- Chiu, D.K.C.; Tse, A.P.W.; Xu, I.M.J.; Di Cui, J.; Lai, R.K.H.; Li, L.L.; Koh, H.Y.; Tsang, F.H.C.; Wei, L.L.; Wong, C.M.; et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Chiu, D.K.C.; Xu, I.M.J.; Lai, R.K.H.; Tse, A.P.W.; Wei, L.L.; Koh, H.Y.; Li, L.L.; Lee, D.; Lo, R.C.L.; Wong, C.M.; et al. Hypoxia induces myeloid-derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C-C motif) ligand 26. Hepatology 2016, 64, 797–813. [Google Scholar] [CrossRef]
- Erler, J.T.; Bennewith, K.L.; Nicolau, M.; Dornhöfer, N.; Kong, C.; Le, Q.T.; Chi, J.T.A.; Jeffrey, S.S.; Giaccia, A.J. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 2006, 440, 1222–1226. [Google Scholar] [CrossRef]
- Kepp, O.; Tartour, E.; Vitale, I.; Vacchelli, E.; Adjemian, S.; Agostinis, P.; Apetoh, L.; Aranda, F.; Barnaba, V.; Bloy, N.; et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014, 3, e955691. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abeliovich, H.; Agostinis, P.; Agrawal, D.K.; Aliev, G.; Askew, D.S.; Baba, M.; Baehrecke, E.H.; Bahr, B.A.; Ballabio, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008, 4, 151–175. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.G.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic Cell Death in Cancer Therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Cirone, M.; Renzo, L.D.; Lotti, L.V.; Conte, V.; Trivedi, P.; Santarelli, R.; Gonnella, R.; Frati, L.; Faggioni, A. Activation of dendritic cells by tumor cell death. Oncoimmunology 2012, 1, 1218–1219. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, G.; Chen, Y.; Wang, H.; Hua, Y.; Cai, Z. Immunogenic cell death in cancer therapy: Present and emerging inducers. J. Cell. Mol. Med. 2019, 23, 4854–4865. [Google Scholar] [CrossRef]
- Olin, M.R.; Andersen, B.M.; Zellmer, D.M.; Grogan, P.T.; Popescu, F.E.; Xiong, Z.; Forster, C.L.; Seiler, C.; SantaCruz, K.S.; Chen, W.; et al. Superior efficacy of tumor cell vaccines grown in physiologic oxygen. Clin. Cancer Res. 2010, 16, 4800–4808. [Google Scholar] [CrossRef]
- Olin, M.R.; Andersen, B.M.; Litterman, A.J.; Grogan, P.T.; Sarver, A.L.; Robertson, P.T.; Liang, X.; Chen, W.; Parney, I.F.; Hunt, M.A.; et al. Oxygen is a master regulator of the immunogenicity of primary human glioma cells. Cancer Res. 2011, 71, 6583–6589. [Google Scholar] [CrossRef]
- Garg, A.D.; Krysko, D.V.; Verfaillie, T.; Kaczmarek, A.; Ferreira, G.B.; Marysael, T.; Rubio, N.; Firczuk, M.; Mathieu, C.; Roebroek, A.J.M.; et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012, 31, 1062–1079. [Google Scholar] [CrossRef]
- Garg, A.D.; Krysko, D.V.; Vandenabeele, P.; Agostinis, P. Hypericin-based photodynamic therapy induces surface exposure of damage-associated molecular patterns like HSP70 and calreticulin. Cancer Immunol. Immunother. 2012, 61, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Enlightening the impact of immunogenic cell death in photodynamic cancer therapy. EMBO J. 2012, 31, 1055–1057. [Google Scholar] [CrossRef] [PubMed]
- Lucky, S.S.; Soo, K.C.; Zhang, Y. Nanoparticles in photodynamic therapy. Chem. Rev. 2015, 115, 1990–2042. [Google Scholar] [CrossRef] [PubMed]
- Chouaib, S.; Noman, M.Z.; Kosmatopoulos, K.; Curran, M.A. Hypoxic stress: Obstacles and opportunities for innovative immunotherapy of cancer. Oncogene 2017, 36, 439–445. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, L.; Liang, R.; Luo, Z.; He, H.; Wu, Z.; Tian, H.; Zheng, M.; Ma, Y.; Cai, L. Bioinspired hybrid protein oxygen nanocarrier amplified photodynamic therapy for eliciting anti-tumor immunity and abscopal effect. ACS Nano 2018, 12, 8633–8645. [Google Scholar] [CrossRef]
- Hou, W.; Zhang, Q.; Yan, Z.; Chen, R.; Zeh, H.J.; Kang, R.; Lotze, M.T.; Tang, D. Strange attractors: DAMPs and autophagy link tumor cell death and immunity. Cell Death Dis. 2013, 4, e966. [Google Scholar] [CrossRef]
- Garg, A.D.; Dudek, A.M.; Agostinis, P. Calreticulin surface exposure is abrogated in cells lacking, chaperone-mediated autophagy-essential gene, LAMP2A. Cell Death Dis. 2013, 4, e826. [Google Scholar] [CrossRef]
- Papandreou, I.; Lim, A.L.; Laderoute, K.; Denko, N.C. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ. 2008, 15, 1572–1581. [Google Scholar] [CrossRef]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-Induced Autophagy Is Mediated through Hypoxia-Inducible Factor Induction of BNIP3 and BNIP3L via Their BH3 Domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Y.; Kim, E.; Beemiller, P.; Wang, C.Y.; Swanson, J.; You, M.; Guan, K.L. Bnip3 mediates the hypoxia-induced inhibition on mammalian target of rapamycin by interacting with Rheb. J. Biol. Chem. 2007, 282, 35803–35813. [Google Scholar] [CrossRef]
- Cam, H.; Easton, J.B.; High, A.; Houghton, P.J. mTORC1 signaling under hypoxic conditions is controlled by atm-dependent phosphorylation of HIF-1α. Mol. Cell 2010, 40, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Li, D.D.; Xie, B.; Wu, X.J.; Li, J.J.; Ding, Y.; Wen, X.Z.; Zhang, X.; Zhu, S.G.; Liu, W.; Zhang, X.S.; et al. Late-stage inhibition of autophagy enhances calreticulin surface exposure. Oncotarget 2016, 7, 80842–80854. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F.; Aptsiauri, N.; Doorduijn, E.M.; Garcia Lora, A.M.; van Hall, T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 2016, 39, 44–51. [Google Scholar] [CrossRef]
- Garrido, F.; Algarra, I. MHC antigens and tumor escape from immune surveillance. Adv. Cancer Res. 2001, 83, 117–158. [Google Scholar]
- Seliger, B.; Cabrera, T.; Garrido, F.; Ferrone, S. HLA class I antigen abnormalities and immune escape by malignant cells. Semin. Cancer Biol. 2002, 12, 3–13. [Google Scholar] [CrossRef]
- Sethumadhavan, S.; Silva, M.; Philbrook, P.; Nguyen, T.; Hatfield, S.M.; Ohta, A.; Sitkovsky, M.V. Hypoxia and hypoxia-inducible factor (HIF) downregulate antigen-presenting MHC class I molecules limiting tumor cell recognition by T cells. PLoS ONE 2017, 12, e0187314. [Google Scholar] [CrossRef]
- Murthy, A.; Gerber, S.A.; Koch, C.J.; Lord, E.M. Intratumoral Hypoxia Reduces IFN-γ–Mediated Immunity and MHC Class I Induction in a Preclinical Tumor Model. ImmunoHorizons 2019, 3, 149–160. [Google Scholar] [CrossRef]
- Siemens, D.R.; Hu, N.; Sheikhi, A.K.; Chung, E.; Frederiksen, L.J.; Pross, H.; Graham, C.H. Hypoxia increases tumor cell shedding of MHC class I chain-related molecule: Role of nitric oxide. Cancer Res. 2008, 68, 4746–4753. [Google Scholar] [CrossRef]
- Marijt, K.A.; Sluijter, M.; Blijleven, L.; Tolmeijer, S.H.; Scheeren, F.A.; Van Der Burg, S.H.; Van Hall, T. Metabolic stress in cancer cells induces immune escape through a PI3K-dependent blockade of IFNγreceptor signaling. J. Immunother. Cancer 2019, 7, 152. [Google Scholar] [CrossRef]
- Qin, W.; Hu, L.; Zhang, X.; Jiang, S.; Li, J.; Zhang, Z.; Wang, X. The Diverse Function of PD-1/PD-L Pathway Beyond Cancer. Front. Immunol. 2019, 10, 152. [Google Scholar] [CrossRef] [PubMed]
- Tawadros, A.I.F.; Khalafalla, M.M.M. Expression of programmed death-ligand 1 and hypoxia-inducible factor-1α proteins in endometrial carcinoma. J. Cancer Res. Ther. 2018, 14, S1063–S1069. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced: MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Zapata, C.; Avendaño-Ortiz, J.; Hernandez-Jimenez, E.; Toledano, V.; Casas-Martin, J.; Varela-Serrano, A.; Torres, M.; Almendros, I.; Casitas, R.; Fernández-Navarro, I.; et al. Hypoxia-induced PD-L1/PD-1 crosstalk impairs T-cell function in sleep apnoea. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 2019, 18, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Messai, Y.; Gad, S.; Noman, M.Z.; Le Teuff, G.; Couve, S.; Janji, B.; Kammerer, S.F.; Rioux-Leclerc, N.; Hasmim, M.; Ferlicot, S.; et al. Renal Cell Carcinoma Programmed Death-ligand 1, a New Direct Target of Hypoxia-inducible Factor-2 Alpha, is Regulated by von Hippel–Lindau Gene Mutation Status. Eur. Urol. 2016, 70, 623–632. [Google Scholar] [CrossRef]
- Sansom, D.M. CD28, CTLA-4 and their ligands: Who does what and to whom? Immunology 2000, 101, 169–177. [Google Scholar] [CrossRef]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef]
- Köhler, T.; Reizis, B.; Johnson, R.S.; Weighardt, H.; Förster, I. Influence of hypoxia-inducible factor 1α on dendritic cell differentiation and migration. Eur. J. Immunol. 2012, 42, 1226–1236. [Google Scholar] [CrossRef]
- Ho, P.-C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.-C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef]
- Chang, C.-H.; Qiu, J.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; Tonc, E.; et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression Graphical Abstract HHS Public Access. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Vyas, M.; Müller, R.; Pogge von Strandmann, E. Antigen Loss Variants: Catching Hold of Escaping Foes. Front. Immunol. 2017, 8, 175. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018, 173, 879–893.e13. [Google Scholar] [CrossRef] [PubMed]
- Verdegaal, E.M.E.; de Miranda, N.F.C.C.; Visser, M.; Harryvan, T.; van Buuren, M.M.; Andersen, R.S.; Hadrup, S.R.; van der Minne, C.E.; Schotte, R.; Spits, H.; et al. Neoantigen landscape dynamics during human melanoma–T cell interactions. Nature 2016, 536, 91–95. [Google Scholar] [CrossRef]
- Sanchez-Perez, L.; Kottke, T.; Diaz, R.M.; Ahmed, A.; Thompson, J.; Chong, H.; Melcher, A.; Holmen, S.; Daniels, G.; Vile, R.G. Potent Selection of Antigen Loss Variants of B16 Melanoma following Inflammatory Killing of Melanocytes In vivo. Cancer Res. 2005, 65, 2009–2017. [Google Scholar] [CrossRef]
- Rodríguez-Jiménez, F.J.; Moreno-Manzano, V.; Lucas-Dominguez, R.; Sánchez-Puelles, J.-M. Hypoxia Causes Downregulation of Mismatch Repair System and Genomic Instability in Stem Cells. Stem Cells 2008, 26, 2052–2062. [Google Scholar] [CrossRef]
- Cowman, S.; Pizer, B.; See, V. Downregulation of both mismatch repair and non-homologous end-joining pathways in hypoxic brain tumour cell lines. bioRxiv Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Mihaylova, V.T.; Bindra, R.S.; Yuan, J.; Campisi, D.; Narayanan, L.; Jensen, R.; Giordano, F.; Johnson, R.S.; Rockwell, S.; Glazer, P.M. Decreased Expression of the DNA Mismatch Repair Gene Mlh1 under Hypoxic Stress in Mammalian Cells. Mol. Cell. Biol. 2003, 23, 3265–3273. [Google Scholar] [CrossRef]
- Kondo, A.; Safaei, R.; Mishima, M.; Niedner, H.; Lin, X.; Howell, S.B. Hypoxia-induced enrichment and mutagenesis of cells that have lost DNA mismatch repair. Cancer Res. 2001, 61, 7603–7607. [Google Scholar]
- Luoto, K.R.; Kumareswaran, R.; Bristow, R.G. Tumor hypoxia as a driving force in genetic instability. Genome Integr. 2013, 4, 5. [Google Scholar] [CrossRef]
- Kumareswaran, R.; Ludkovski, O.; Meng, A.; Sykes, J.; Pintilie, M.; Bristow, R.G. Chronic hypoxia compromises repair of DNA double-strand breaks to drive genetic instability. J. Cell Sci. 2012, 125, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.J.; Semenza, G.L.; Zhang, H. feng Hypoxia-inducible factor 1 and breast cancer metastasis. J. Zhejiang Univ. Sci. B 2015, 16, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Jing, S.W.; Wang, Y.D.; Kuroda, M.; Su, J.W.; Sun, G.G.; Liu, Q.; Cheng, Y.J.; Yang, C.R. HIF-1α contributes to hypoxia-induced invasion and metastasis of esophageal carcinoma via inibiting E-cadherin and promoting MMP-2 expression. Acta Med. Okayama 2012, 66, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Role of angiogenesis in tumor growth and metastasis. Semin. Oncol. 2002, 29, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Bielenberg, D.R.; Zetter, B.R. The Contribution of Angiogenesis to the Process of Metastasis. Cancer J. (USA) 2015, 21, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Büchler, P.; Reber, H.A.; Büchler, M.; Shrinkante, S.; Büchler, M.W.; Friess, H.; Semenza, G.L.; Hines, O.J. Hypoxia-inducible factor 1 regulates vascular endothelial growth factor expression in human pancreatic cancer. Pancreas 2003, 26, 56–64. [Google Scholar] [CrossRef]
- Sivridis, E.; Giatromanolaki, A.; Gatter, K.C.; Harris, A.L.; Koukourakis, M.I. Association of hypoxia-inducible factors 1α and 2α with activated angiogenic pathways and prognosis in patients with endometrial carcinoma. Cancer 2002, 95, 1055–1063. [Google Scholar] [CrossRef]
- Oladipupo, S.; Hu, S.; Kovalski, J.; Yao, J.; Santeford, A.; Sohn, R.E.; Shohet, R.; Maslov, K.; Wang, L.V.; Arbeit, J.M. VEGF is essential for hypoxia-inducible factor-mediated neovascularization but dispensable for endothelial sprouting. Proc. Natl. Acad. Sci. USA 2011, 108, 13264–13269. [Google Scholar] [CrossRef]
- Skinner, H.D.; Zheng, J.Z.; Fang, J.; Agani, F.; Jiang, B.H. Vascular endothelial growth factor transcriptional activation is mediated by hypoxia-inducible factor 1α, HDM2, and p70S6K1 in response to phosphatidylinositol 3-kinase/AKT signaling. J. Biol. Chem. 2004, 279, 45643–45651. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Lappano, R.; Santolla, M.F.; Marsico, S.; Caruso, A.; Maggiolini, M. HIF-1α/GPER signaling mediates the expression of VEGF induced by hypoxia in breast cancer associated fibroblasts (CAFs). Breast Cancer Res. 2013, 15, R64. [Google Scholar] [CrossRef]
- Han, S.; Huang, T.; Li, W.; Liu, S.; Yang, W.; Shi, Q.; Li, H.; Ren, J.; Hou, F. Association between Hypoxia-Inducible Factor-2α (HIF-2α) Expression and Colorectal Cancer and Its Prognostic Role: A Systematic Analysis. Cell. Physiol. Biochem. 2018, 48, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Wierzbicki, P.M.; Klacz, J.; Kotulak-Chrzaszcz, A.; Wronska, A.; Stanislawowski, M.; Rybarczyk, A.; Ludziejewska, A.; Kmiec, Z.; Matuszewski, M. Prognostic significance of VHL, HIF1A, HIF2A, VEGFA and p53 expression in patients with clear-cell renal cell carcinoma treated with sunitinib as first-line treatment. Int. J. Oncol. 2019, 55, 371–390. [Google Scholar] [CrossRef] [PubMed]
- Baba, Y.; Nosho, K.; Shima, K.; Irahara, N.; Chan, A.T.; Meyerhardt, J.A.; Chung, D.C.; Giovannucci, E.L.; Fuchs, C.S.; Ogino, S. HIF1A overexpression is associated with poor prognosis in a cohort of 731 colorectal cancers. Am. J. Pathol. 2010, 176, 2292–2301. [Google Scholar] [CrossRef] [PubMed]
- Burroughs, S.K.; Kaluz, S.; Wang, D.; Wang, K.; Van Meir, E.G.; Wang, B. Hypoxia inducible factor pathway inhibitors as anticancer therapeutics. Future Med. Chem. 2013, 5, 553–572. [Google Scholar] [CrossRef]
- Wigerup, C.; Påhlman, S.; Bexell, D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol. Ther. 2016, 164, 152–169. [Google Scholar] [CrossRef]
- Onnis, B.; Rapisarda, A.; Melillo, G. Development of HIF-1 inhibitors for cancer therapy. J. Cell. Mol. Med. 2009, 13, 2780–2786. [Google Scholar] [CrossRef]
- Li, Y.; Patel, S.P.; Roszik, J.; Qin, Y. Hypoxia-driven immunosuppressive metabolites in the tumor microenvironment: New approaches for combinational immunotherapy. Front. Immunol. 2018, 9, 1. [Google Scholar] [CrossRef]
- Yu, T.; Tang, B.; Sun, X. Development of Inhibitors Targeting Hypoxia-Inducible Factor 1 and 2 for Cancer Therapy. Yonsei Med. J. 2017, 58, 489–496. [Google Scholar] [CrossRef]
- Fallah, J.; Rini, B.I. HIF Inhibitors: Status of Current Clinical Development. Curr. Oncol. Rep. 2019, 21, 1–10. [Google Scholar] [CrossRef]
- Carbajo-Pescador, S.; Ordoñez, R.; Benet, M.; Jover, R.; García-Palomo, A.; Mauriz, J.L.; González-Gallego, J. Inhibition of VEGF expression through blockade of Hif1α and STAT3 signalling mediates the anti-angiogenic effect of melatonin in HepG2 liver cancer cells. Br. J. Cancer 2013, 109, 83–91. [Google Scholar] [CrossRef]
- Ban, H.S.; Kim, B.K.; Lee, H.; Kim, H.M.; Harmalkar, D.; Nam, M.; Park, S.K.; Lee, K.; Park, J.T.; Kim, I.; et al. The novel hypoxia-inducible factor-1α inhibitor IDF-11774 regulates cancer metabolism, thereby suppressing tumor growth. Cell Death Dis. 2017, 8, e2843. [Google Scholar] [CrossRef] [PubMed]
- Laughner, E.; Taghavi, P.; Chiles, K.; Mahon, P.C.; Semenza, G.L. HER2 (neu) Signaling Increases the Rate of Hypoxia-Inducible Factor 1 (HIF-1) Synthesis: Novel Mechanism for HIF-1-Mediated Vascular Endothelial Growth Factor Expression. Mol. Cell. Biol. 2001, 21, 3995–4004. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J.B.; Vazquez, F.; Reddy, A.; Sellers, W.R.; Kaelin, W.G. TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell 2003, 4, 147–158. [Google Scholar] [CrossRef]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of Hypoxia-Inducible Factor 1 Expression and Function by the Mammalian Target of Rapamycin. Mol. Cell. Biol. 2002, 22, 7004–7014. [Google Scholar] [CrossRef]
- Lastwika, K.J.; Wilson, W.; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res. 2016, 76, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Chen, D.; Lu, B.; Wang, C.; Zhang, J.; Huang, L.; Wang, X.; Timmons, C.L.; Hu, J.; Liu, B.; et al. PTEN Loss Increases PD-L1 Protein Expression and Affects the Correlation between PD-L1 Expression and Clinical Parameters in Colorectal Cancer. PLoS ONE 2013, 8, e65821. [Google Scholar] [CrossRef]
- Zhao, L.; Li, C.; Liu, F.; Zhao, Y.; Liu, J.; Hua, Y.; Liu, J.; Huang, J.; Ge, C. A blockade of PD-L1 produced antitumor and antimetastatic effects in an orthotopic mouse pancreatic cancer model via the PI3K/Akt/mTOR signaling pathway. Onco. Targets. Ther. 2017, 10, 2115–2126. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Fan, J.; Chen, W.; Luan, J.; Mei, X.; Wang, S.; Li, Y.; Ye, L.; Li, S.; et al. Blocking CD47 efficiently potentiated therapeutic effects of anti-angiogenic therapy in non-small cell lung cancer. J. Immunother. Cancer 2019, 7, 346. [Google Scholar] [CrossRef]
- Huang, Y.; Yuan, J.; Righi, E.; Kamoun, W.S.; Ancukiewicz, M.; Nezivar, J.; Santosuosso, M.; Martin, J.D.; Martin, M.R.; Vianello, F.; et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 17561–17566. [Google Scholar] [CrossRef]
- Aghi, M.K.; Liu, T.C.; Rabkin, S.; Martuza, R.L. Hypoxia enhances the replication of oncolytic herpes simplex virus. Mol. Ther. 2009, 17, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Pin, R.H.; Reinblatt, M.; Fong, Y. Employing tumor hypoxia to enhance oncolytic viral therapy in breast cancer. Surgery 2004, 136, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.S. The impact of hypoxia on oncolytic virotherapy. Virus Adapt. Treat. 2011, 3, 71. [Google Scholar] [CrossRef]
- Hackstein, H.; Taner, T.; Logar, A.J.; Thomson, A.W. Rapamycin inhibits macropinocytosis and mannose receptor-mediated endocytosis by bone marrow-derived dendritic cells. Blood 2002, 100, 1084–1087. [Google Scholar] [CrossRef]
- Hackstein, H.; Taner, T.; Zahorchak, A.F.; Morelli, A.E.; Logar, A.J.; Gessner, A.; Thomson, A.W. Rapamycin inhibits IL-4-induced dendritic cell maturation in vitro and dendritic cell mobilization and function in vivo. Blood 2003, 101, 4457–4463. [Google Scholar] [CrossRef]
- Thomson, A.W.; Turnquist, H.R.; Raimondi, G. Immunoregulatory functions of mTOR inhibition. Nat. Rev. Immunol. 2009, 9, 324–337. [Google Scholar] [CrossRef]
- Chaoul, N.; Fayolle, C.; Desrues, B.; Oberkampf, M.; Tang, A.; Ladant, D.; Leclerc, C. Rapamycin impairs antitumor CD8p T-cell responses and vaccine-induced tumor eradication. Cancer Res. 2015, 75, 3279–3291. [Google Scholar] [CrossRef]
- Battaglia, M.; Stabilini, A.; Roncarolo, M.G. Rapamycin selectively expands CD4+CD25+FoxP3 + regulatory T cells. Blood 2005, 105, 4743–4748. [Google Scholar] [CrossRef]
- Battaglia, M.; Stabilini, A.; Migliavacca, B.; Horejs-Hoeck, J.; Kaupper, T.; Roncarolo, M.-G. Rapamycin Promotes Expansion of Functional CD4 + CD25 + FOXP3 + Regulatory T Cells of Both Healthy Subjects and Type 1 Diabetic Patients. J. Immunol. 2006, 177, 8338–8347. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Changes in Function and Secretome | References |
|---|---|---|
| DCs | ↓ Antigen uptake | [22,23] |
| ↓ CD1a, CD40, CD80, CD83, CD86, MHC class II | ||
| Macrophages | ↓ M1 phenotype | [27,28,29] |
| ↑ M2 phenotype | ||
| ↓ IL-2 | ||
| ↑ IL-4, IL-10, IL-13 | ||
| B cells | ↑ Development | [34,35] |
| ↑ IL-10 | ||
| T cells | ↓ Cytotoxic function | [41,44,48] |
| ↑ Anergy | ||
| ↑ Treg expansion | ||
| ↑ Memory function | ||
| ↑ Antibody production | ||
| ↑ PD-1 | ||
| NK cells | ↓ Cytolytic activity | [51,52] |
| ↑ VEGF | ||
| MDSCs | ↑ Differentiation and function | [55,56,59] |
| ↑ Recruitment to tumor site | ||
| ↑ Extracellular remodeling |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vito, A.; El-Sayes, N.; Mossman, K. Hypoxia-Driven Immune Escape in the Tumor Microenvironment. Cells 2020, 9, 992. https://doi.org/10.3390/cells9040992
Vito A, El-Sayes N, Mossman K. Hypoxia-Driven Immune Escape in the Tumor Microenvironment. Cells. 2020; 9(4):992. https://doi.org/10.3390/cells9040992
Chicago/Turabian StyleVito, Alyssa, Nader El-Sayes, and Karen Mossman. 2020. "Hypoxia-Driven Immune Escape in the Tumor Microenvironment" Cells 9, no. 4: 992. https://doi.org/10.3390/cells9040992
APA StyleVito, A., El-Sayes, N., & Mossman, K. (2020). Hypoxia-Driven Immune Escape in the Tumor Microenvironment. Cells, 9(4), 992. https://doi.org/10.3390/cells9040992