Lamin A/C Mechanotransduction in Laminopathies




Abstract
1. Introduction
2. Lamins

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB Code | Segment | X-ray Resolution | a.a. | Chain |
|---|---|---|---|---|
| 1X8Y [94] | Coil 2B | 2.2 Å | 305–389 | Single chain (A) |
| 3V5B [95] | Coil 2B | 3 Å | 313–386 | Single chain (A) |
| 1IFR [54] | Globular domain | 1.4 Å | 436–552 | Single chain (A) |
| 2XV5 [96] | Coil 2B | 2.4 Å | 328–398 | Dimer (A + B) |
| 6SNZ [86] | Coil 1B | 2.6 Å | 65–222 | Tetramer (A + B + C + D) |
| 6JLB [23] | Head-coil 2 | 3.2 Å | 1–300 | Tetramer (A + B + C + D) |
| PDB Code | Segment | X-ray Resolution | a.a. | Chain |
|---|---|---|---|---|
| 3UMN [97] | Globular domain | 2 Å | 428–550 | Trimer (A + B + C) |
| 3TYY [97] | Coil 2B | 2.4 Å | 311–388 | Dimer (A + B) |
3. Lamin Binding Partners
4. Lamin A/C Roles in Cell Mechanotransduction
5. Lamin A/C Mechanotransduction in Laminopathies
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Boeri, L.; Albani, D.; Raimondi, M.T.; Jacchetti, E. Mechanical regulation of nucleocytoplasmic translocation in mesenchymal stem cells: Characterization and methods for investigation. Biophys. Rev. 2019, 11, 817–831. [Google Scholar] [CrossRef]
- Bennett, M.; Cantini, M.; Reboud, J.; Cooper, J.M.; Roca-Cusachs, P.; Salmeron-Sanchez, M. Molecular clutch drives cell response to surface viscosity. Proc. Natl. Acad. Sci. USA 2018, 115, 1192–1197. [Google Scholar] [CrossRef]
- Kassianidou, E.; Brand, C.A.; Schwarz, U.S.; Kumar, S. Geometry and network connectivity govern the mechanics of stress fibers. Proc. Natl. Acad. Sci. USA 2017, 114, 2622–2627. [Google Scholar] [CrossRef]
- Tsimbouri, P.; Gadegaard, N.; Burgess, K.; White, K.; Reynolds, P.; Herzyk, P.; Oreffo, R.; Dalby, M.J. Nanotopographical effects on mesenchymal stem cell morphology and phenotype. J. Cell Biochem. 2014, 115, 380–390. [Google Scholar] [CrossRef]
- Engler, A.J.; Griffin, M.A.; Sen, S.; Bonnetnann, C.G.; Sweeney, H.L.; Discher, D.E. Myotubes differentiate optimally on substrates with tissue-like stiffness: Pathological implications for soft or stiff microenvironments. J. Cell Biol. 2004, 166, 877–887. [Google Scholar] [CrossRef] [PubMed]
- Donnaloja, F.; Jacchetti, E.; Soncini, M.; Raimondi, M.T. Mechanosensing at the Nuclear Envelope by Nuclear Pore Complex Stretch Activation and Its Effect in Physiology and Pathology. Front. Physiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Crisp, M.; Liu, Q.; Roux, K.; Rattner, J.B.; Shanahan, C.; Burke, B.; Stahl, P.D.; Hodzic, D. Coupling of the nucleus and cytoplasm: Role of the LINC complex. J. Cell Biol. 2006, 172, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Lammerding, J.; Schulze, P.C.; Takahashi, T.; Kozlov, S.; Sullivan, T.; Kamm, R.D.; Stewart, C.L.; Lee, R.T. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J. Clin Investig. 2004, 113, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Turgay, Y.; Eibauer, M.; Goldman, A.E.; Shimi, T.; Khayat, M.; Ben-Harush, K.; Dubrovsky-Gaupp, A.; Sapra, K.T.; Goldman, R.D.; Medalia, O. The molecular architecture of lamins in somatic cells. Nature 2017, 543, 261–264. [Google Scholar] [CrossRef]
- Zwerger, M.; Roschitzki-Voser, H.; Zbinden, R.; Denais, C.; Herrmann, H.; Lammerding, J.; Grütter, M.G.; Medalia, O. Altering lamina assembly reveals lamina-dependent and -independent functions for A-type lamins. J. Cell Sci. 2015, 128, 3607–3620. [Google Scholar] [CrossRef]
- Broers, J.L.; Peeters, E.A.; Kuijpers, H.J.; Endert, J.; Bouten, C.V.; Oomens, C.W.; Baaijens, F.P.; Ramaekers, F.C. Decreased mechanical stiffness in LMNA-/- cells is caused by defective nucleo-cytoskeletal integrity: Implications for the development of laminopathies. Hum. Mol Genet. 2004, 13, 2567–2580. [Google Scholar] [CrossRef] [PubMed]
- Tajik, A.; Zhang, Y.; Wei, F.; Sun, J.; Jia, Q.; Zhou, W.; Singh, R.; Khanna, N.; Belmont, A.S.; Wang, N. Transcription upregulation via force-induced direct stretching of chromatin. Nat. Mater. 2016, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Ramdas, N.M.; Shivashankar, G.V. Cytoskeletal control of nuclear morphology and chromatin organization. J. Mol Biol. 2015, 427, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Davidson, P.M.; Lammerding, J. Broken nuclei—Lamins, nuclear mechanics, and disease. Trends Cell Biol. 2014, 24, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Hale, C.M.; Panorchan, P.; Khatau, S.B.; George, J.P.; Tseng, Y.; Stewart, C.L.; Hodzic, D.; Wirtz, D. Nuclear lamin A/C deficiency induces defects in cell mechanics, polarization, and migration. Biophys. J. 2007, 93, 2542–2552. [Google Scholar] [CrossRef] [PubMed]
- Lammerding, J.; Fong, L.G.; Ji, J.Y.; Reue, K.; Stewart, C.L.; Young, S.G.; Lee, R.T. Lamins A and C but not lamin B1 regulate nuclear mechanics. J. Biol. Chem. 2006, 281, 25768–25780. [Google Scholar] [CrossRef]
- Dahl, K.N.; Kahn, S.M.; Wilson, K.L.; Discher, D.E. The nuclear envelope lamina network has elasticity and a compressibility limit suggestive of a molecular shock absorber. J. Cell Sci. 2004, 117, 4779–4786. [Google Scholar] [CrossRef]
- Denais, C.M.; Gilbert, R.M.; Isermann, P.; McGregor, A.L.; te Lindert, M.; Weigelin, B.; Davidson, P.M.; Friedl, P.; Wolf, K.; Lammerding, J. Nuclear envelope rupture and repair during cancer cell migration. Science 2016, 352, 353–358. [Google Scholar] [CrossRef]
- Bonello-Palot, N.; Simoncini, S.; Robert, S.; Bourgeois, P.; Sabatier, F.; Levy, N.; Dignat-George, F.; Badens, C. Prelamin A accumulation in endothelial cells induces premature senescence and functional impairment. Atherosclerosis 2014, 237, 45–52. [Google Scholar] [CrossRef]
- Vargas, J.D.; Hatch, E.M.; Anderson, D.J.; Hetzer, M.W. Transient nuclear envelope rupturing during interphase in human cancer cells. Nucleus 2012, 3, 88–100. [Google Scholar] [CrossRef]
- Kubben, N.; Voncken, J.W.; Konings, G.; van Weeghel, M.; van den Hoogenhof, M.M.; Gijbels, M.; van Erk, A.; Schoonderwoerd, K.; van den Bosch, B.; Dahlmans, V.; et al. Post-natal myogenic and adipogenic developmental: Defects and metabolic impairment upon loss of A-type lamins. Nucleus 2011, 2, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Broers, J.L.; Kuijpers, H.J.; Ostlund, C.; Worman, H.J.; Endert, J.; Ramaekers, F.C. Both lamin A and lamin C mutations cause lamina instability as well as loss of internal nuclear lamin organization. Exp. Cell Res. 2005, 304, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Jo, I.; Kang, S.M.; Hong, S.; Kim, S.; Jeong, S.; Kim, Y.H.; Park, B.J.; Ha, N.C. Structural basis for lamin assembly at the molecular level. Nat. Commun. 2019, 10, 3757. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Chojnowski, A.; Boudier, T.; Lim, J.S.; Ahmed, S.; Ser, Z.; Stewart, C.; Burke, B. A-type lamins form distinct filamentous networks with differential nuclear pore complex associations. Curr. Biol. 2016, 26, 2651–2658. [Google Scholar] [CrossRef]
- Dittmer, T.A.; Misteli, T. The lamin protein family. Genome Biol. 2011, 12, 222. [Google Scholar] [CrossRef]
- Constantinescu, D.; Gray, H.L.; Sammak, P.J.; Schatten, G.P.; Csoka, A.B. Lamin A/C expression is a marker of mouse and human embryonic stem cell differentiation. Stem. Cells. 2006, 24, 177–185. [Google Scholar] [CrossRef]
- Machiels, B.M.; Zorenc, A.H.; Endert, J.M.; Kuijpers, H.J.; van Eys, G.J.; Ramaekers, F.C.; Broers, J.L. An alternative splicing product of the lamin A/C gene lacks exon 10. J. Biol. Chem. 1996, 271, 9249–9253. [Google Scholar] [CrossRef]
- Furukawa, K.; Inagaki, H.; Hotta, Y. Identification and cloning of an mRNA coding for a germ cell-specific A-type lamin in mice. Exp. Cell Res. 1994, 212, 426–430. [Google Scholar] [CrossRef]
- Furukawa, K.; Hotta, Y. cDNA cloning of a germ cell specific lamin B3 from mouse spermatocytes and analysis of its function by ectopic expression in somatic cells. EMBO J. 1993, 12, 97–106. [Google Scholar] [CrossRef]
- Klapper, M.; Exner, K.; Kempf, A.; Gehrig, C.; Stuurman, N.; Fisher, P.A.; Krohne, G. Assembly of A- and B-type lamins studied in vivo with the baculovirus system. J. Cell Sci. 1997, 110, 2519–2532. [Google Scholar]
- Heitlinger, E.; Peter, M.; Haner, M.; Lustig, A.; Aebi, U.; Nigg, E.A. Expression of chicken lamin B2in Escherichia coli: Characterization of its structure, assembly, and molecular interactions. J. Cell Biol. 1991, 113, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Shimi, T.; Kittisopikul, M.; Tran, J.; Goldman, A.E.; Adam, S.A.; Zheng, Y.; Jaqaman, K.; Goldman, R.D. Structural organization of nuclear lamins A, C, B1, and B2 revealed by super resolution microscopy. Mol. Biol. Cell. 2015, 26, 4075–4086. [Google Scholar] [CrossRef] [PubMed]
- Grossman, E.; Dahan, I.; Stick, R.; Goldberg, M.W.; Gruenbaum, Y.; Medalia, O. Filaments assembly of ectopically expressed caenorhabditis elegans lamin within xenopus oocytes. J. Struct Biol. 2012, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Kapinos, L.E.; Schumacher, J.; Mücke, N.; Machaidze, G.; Burkhard, P.; Aebi, U.; Strelkov, S.V.; Herrmann, H. Characterization of the head-to-tail overlap complexes formed by human lamin A, B1 and B2 “half-minilamin” dimers. J. Mol. Biol. 2010, 396, 719–731. [Google Scholar] [CrossRef]
- Goldberg, M.W.; Huttenlauch, I.; Hutchison, C.J.; Stick, R. Filaments made from A- and B-type lamins differ in structure and organization. J. Cell Sci. 2008, 121, 215–225. [Google Scholar] [CrossRef]
- Schirmer, E.C.; Gerace, L. The stability of the nuclear lamina polymer changes with the composition of lamin subtypes according to their individual binding strengths. J. Biol. Chem. 2004, 279, 42811–42817. [Google Scholar] [CrossRef]
- Schirmer, E.C.; Guan, T.; Gerace, L. Involvement of the lamin rod domain in heterotypic lamin interactions important for nuclear organization. J. Cell Biol. 2001, 153, 479–489. [Google Scholar] [CrossRef]
- Moir, R.D.; Yoon, M.; Khuon, S.; Goldman, R.D. Nuclear lamins A and B1: Different pathways of assembly during nuclear envelope formation in living cells. J. Cell Biol. 2000, 151, 1155–1168. [Google Scholar] [CrossRef]
- Lee, J.M.; Tu, Y.; Tatar, A.; Wu, D.; Nobumori, C.; Jung, H.J.; Yoshinaga, Y.; Coffinier, C.; de Jong, P.J.; Fong, L.G.; et al. Reciprocal knock-in mice to investigate the functional redundancy of lamin B1 and lamin B2. Mol. Biol. Cell. 2014, 25, 1666–1675. [Google Scholar] [CrossRef]
- Harborth, J.; Elbashir, S.M.; Bechert, K.; Tuschl, T.; Weber, K. Identification of essential genes in cultured mammalian cells using small interfering RNAs. J. Cell Sci. 2001, 114, 4557–4565. [Google Scholar]
- Coffinier, C.; Jung, H.J.; Nobumori, C.; Chang, S.; Tu, Y.; Barnes, R.H., 2nd; Yoshinaga, Y.; de Jong, P.J.; Vergnes, L.; Reue, K.; et al. Deficiencies in lamin b1 and lamin b2 cause neurodevelopmental defects and distinct nuclear shape abnormalities in neurons. Mol. Biol. Cell. 2011, 22, 4683–4693. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Sharov, A.A.; McDole, K.; Cheng, M.; Hao, H.; Fan, C.M.; Gaiano, N.; Ko, M.S.; Zheng, Y. Mouse B-type lamins are required for proper organogenesis but not by embryonic stem cells. Science 2011, 334, 1706–1710. [Google Scholar] [CrossRef] [PubMed]
- Vergnes, L.; Péterfy, M.; Bergo, M.O.; Young, S.G.; Reue, K. Lamin B1 is required for mouse development and nuclear integrity. Proc. Natl. Acad. Sci. USA 2004, 101, 10428–10433. [Google Scholar] [CrossRef] [PubMed]
- Hegele, R.A.; Cao, H.; Liu, D.M.; Costain, G.A.; Charlton-Menys, V.; Rodger, N.W.; Durrington, P.N. Sequencing of the reannotated LMNB2 gene reveals novel mutations in patients with acquired partial lipodystrophy. Am. J. Hum. Genet. 2006, 79, 383–389. [Google Scholar] [CrossRef]
- Padiath, Q.S.; Saigoh, K.; Schiffmann, R.; Asahara, H.; Yamada, T.; Koeppen, A.; Hogan, K.; Ptácek, L.J.; Fu, Y.H. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat. Genet. 2006, 38, 1114–1123. [Google Scholar] [CrossRef]
- Saito, N.; Araya, J.; Ito, S.; Tsubouchi, K.; Minagawa, S.; Hara, H.; Ito, A.; Nakano, T.; Hosaka, Y.; Ichikawa, A.; et al. Involvement of lamin B1 reduction in accelerated cellular senescence during chronic obstructive pulmonary disease pathogenesis. J. Immunol. 2019, 202, 1428–1440. [Google Scholar] [CrossRef]
- Freund, A.; Laberge, R.M.; Demaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell. 2012, 23, 2066–2075. [Google Scholar] [CrossRef]
- Shimi, T.; Butin-Israeli, V.; Adam, S.A.; Hamanaka, R.B.; Goldman, A.E.; Lucas, C.A.; Shumaker, D.K.; Kosak, S.T.; Chandel, N.S.; Goldman, R.D. The role of nuclear lamin b1 in cell proliferation and senescence. Genes. Dev. 2011, 25, 2579–2593. [Google Scholar] [CrossRef]
- Maresca, G.; Natoli, M.; Nardella, M.; Arisi, I.; Trisciuoglio, D.; Desideri, M.; Brandi, R.; D’Aguanno, S.; Nicotra, M.R.; D’Onofrio, M.; et al. LMNA knock-down affects differentiation and progression of human neuroblastoma cells. PLoS ONE 2012, 7, e45513. [Google Scholar] [CrossRef]
- Akter, R.; Rivas, D.; Geneau, G.; Drissi, H.; Duqu, G. Effect of Lamin A/C Knockdown on Osteoblast Differentiation and Function. J. Bone Min. Res. 2009, 24, 283–293. [Google Scholar] [CrossRef]
- Röber, R.A.; Weber, K.; Osborn, M. Differential timing of nuclear lamin A/C expression in the various organs of the mouse embryo and the young animal: A developmental study. Development 1989, 105, 365–378. [Google Scholar] [PubMed]
- Corrigan, D.P.; Kuszczak, D.; Rusinol, A.E.; Thewke, D.P.; Hrycyna, C.A.; Michaelis, S.; Sinensky, M.S. Prelamin A endoproteolytic processing in vitro by recombinant Zmpste24. Biochem J. 2005, 387, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Sinensky, M.; Fantle, K.; Trujillo, M.; McLain, T.; Kupfer, A.; Dalton, M. The processing pathway of prelamin A. J. Cell Sci. 1994, 107, 61–67. [Google Scholar] [PubMed]
- Dhe-Paganon, S.; Werner, E.D.; Chi, Y.I.; Shoelson, S.E. Structure of the globular tail of nuclear lamin. J. Biol. Chem. 2002, 277, 17381–17384. [Google Scholar] [CrossRef] [PubMed]
- Adam, S.A.; Butin-Israeli, V.; Cleland, M.M.; Shimi, T.; Goldman, R.D. Disruption of lamin B1 and lamin B2 processing and localization by farnesyltransferase inhibitors. Nucleus 2013, 4, 142–150. [Google Scholar] [CrossRef]
- Fong, L.G.; Ng, J.K.; Lammerding, J.; Vickers, T.A.; Meta, M.; Coté, N.; Gavino, B.; Qiao, X.; Chang, S.Y.; Young, S.R.; et al. Prelamin A and lamin A appear to be dispensable in the nuclear lamina. J. Clin. Investig. 2006, 116, 743–752. [Google Scholar] [CrossRef]
- Fisher, D.Z.; Chaudhary, N.; Blobel, G. cDNA sequencing of nuclear lamins A and C reveals primary and secondary structural homology to intermediate filament proteins. Proc. Natl. Acad. Sci. USA 1986, 83, 6450–6454. [Google Scholar] [CrossRef]
- Machowska, M.; Piekarowicz, K.; Rzepecki, R. Regulation of lamin properties and functions: Does phosphorylation do it all? Open Biol. 2015, 5, 150094. [Google Scholar] [CrossRef]
- Wu, D.; Flannery, A.R.; Cai, H.; Ko, E.; Cao, K. Nuclear localization signal deletion mutants of lamin A and progerin reveal insights into lamin A processing and emerin targeting. Nucleus 2014, 5, 66–74. [Google Scholar] [CrossRef][Green Version]
- Maske, C.P.; Hollinshead, M.S.; Higbee, N.C.; Bergo, M.O.; Young, S.G.; Vaux, D.J. A carboxyl-terminal interaction of lamin B1 is dependent on the CAAX endoprotease Rce1 and carboxymethylation. J. Cell Biol. 2003, 162, 1223–1232. [Google Scholar] [CrossRef]
- Kitten, G.T.; Nigg, E.A. The CaaX motif is required for isoprenylation, carboxyl methylation, and nuclear membrane association of lamin B2. J. Cell Biol. 1991, 113, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Dominici, S.; Fiori, V.; Magnani, M.; Capanni, C.; Camozzi, D.; D’Apice, M.R.; Le Dour, C.; Auclair, M.; Caron, M.; Novelli, G.; et al. Different prelamin A forms accumulate in human fibroblasts: A study in experimental models and progeria. Eur. J. Histochem. 2009, 53, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Dalton, M.B.; Fantle, K.S.; Bechtold, H.A.; DeMaio, L.; Evans, R.M.; Krystosek, A.; Sinensky, M. The farnesyl protein transferase inhibitor BZA-5B blocks farnesylation of nuclear lamins and p21ras but does not affect their function or localization. Cancer Res. 1995, 55, 3295–3304. [Google Scholar] [PubMed]
- Leung, G.K.; Schmidt, W.K.; Bergo, M.O.; Gavino, B.; Wong, D.H.; Tam, A.; Ashby, M.N.; Michaelis, S.; Young, S.G. Biochemical studies of Zmpste24-deficient mice. J. Biol. Chem. 2001, 276, 29051–29058. [Google Scholar] [CrossRef] [PubMed]
- Boyartchuk, V.L.; Ashby, M.N.; Rine, J. Modulation of Ras and a-factor function by carboxyl-terminal proteolysis. Science 1997, 275, 1796–1800. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.X.; Sayin, V.I.; Akula, M.K.; Liu, M.; Fong, L.G.; Young, S.G.; Bergo, M.O. Targeting isoprenylcysteine methylation ameliorates disease in a mouse model of progeria. Science 2013, 340, 1330–1333. [Google Scholar] [CrossRef]
- Casasola, A.; Scalzo, D.; Nandakumar, V.; Halow, J.; Recillas-Targa, F.; Groudine, M.; Rincón-Arano, H. Prelamin A processing, accumulation and distribution in normal cells and laminopathy disorders. Nuclues 2016, 7, 84–102. [Google Scholar] [CrossRef]
- Simon, D.N.; Wilson, K.L. Partners and post-translational modifications of nuclear lamins. Chromosoma 2013, 122, 13–31. [Google Scholar] [CrossRef]
- Barrowman, J.; Hamblet, C.; Kane, M.S.; Michaelis, S. Requirements for efficient proteolytic cleavage of prelamin A by ZMPSTE24. PLoS ONE 2012, 7, e32120. [Google Scholar] [CrossRef]
- Pendás, A.M.; Zhou, Z.; Cadiñanos, J.; Freije, J.M.; Wang, J.; Hultenby, K.; Astudillo, A.; Wernerson, A.; Rodríguez, F.; Tryggvason, K.; et al. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat. Genet. 2002, 31, 94–99. [Google Scholar] [CrossRef]
- Kim, W.; Bennett, E.J.; Huttlin, E.L.; Guo, A.; Li, J.; Possemato, A.; Sowa, M.E.; Rad, R.; Rush, J.; Comb, M.J.; et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. 2011, 44, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef]
- Zhang, Y.Q.; Sarge, K.D. Sumoylation regulates lamin A function and is lost in lamin A mutants associated with familial cardiomyopathies. J. Cell Biol. 2008, 182, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Maeshima, K.; Yahata, K.; Sasaki, Y.; Nakatomi, R.; Tachibana, T.; Hashikawa, T.; Imamoto, F.; Imamoto, N. Cell-cycle-dependent dynamics of nuclear pores: Pore-free islands and lamins. J. Cell Sci. 2006, 119, 4442–4451. [Google Scholar] [CrossRef] [PubMed]
- Ottaviano, Y.; Gerace, L. Phosphorylation of the nuclear lamins during interphase and mitosis. J. Biol. Chem. 1985, 260, 624–632. [Google Scholar]
- Moiseeva, O.; Lopes-Paciencia, S.; Huot, G.; Lessard, F.; Ferbeyre, G. Permanent farnesylation of lamin A mutants linked to progeria impairs its phosphorylation at serine 22 during interphase. Aging 2016, 8, 366–381. [Google Scholar] [CrossRef]
- Chaffee, B.R.; Shang, F.; Chang, M.L.; Clement, T.M.; Eddy, E.M.; Wagner, B.D.; Nakahara, M.; Nagata, S.; Robinson, M.L.; Taylor, A. Nuclear removal during terminal lens fiber cell differentiation requires CDK1 activity: Appropriating mitosis-related nuclear disassembly. Development 2014, 141, 3388–3398. [Google Scholar] [CrossRef]
- Rzepecki, R.; Fisher, P.A. In vivo phosphorylation of Drosophila melanogaster nuclear lamins during both interphase and mitosis. Cell Mol. Biol. Lett. 2002, 7, 859–876. [Google Scholar]
- Schneider, U.; Mini, T.; Jenö, P.; Fisher, P.A.; Stuurman, N. Phosphorylation of the major Drosophila lamin in vivo: Site identification during both M-phase (meiosis) and interphase by electrospray ionization tandem mass spectrometry. Biochemistry 1999, 38, 4620–4632. [Google Scholar] [CrossRef]
- Buxboim, A.; Swift, J.; Irianto, J.; Spinler, K.R.; Dingal, P.C.D.P.; Athirasala, A.; Kao, Y.R.C.; Cho, S.; Harada, T.; Shin, J.W.; et al. Matrix elasticity regulates lamin-A,C phosphorylation and turnover with feedback to actomyosin. Curr. Biol. 2014, 24, 1909–1917. [Google Scholar] [CrossRef]
- Kochin, V.; Shimi, T.; Torvaldson, E.; Adam, S.A.; Goldman, A.; Pack, C.G.; Melo-Cardenas, J.; Imanishi, S.Y.; Goldman, R.D.; Eriksson, J.E. Interphase phosphorylation of lamin A. J. Cell Sci. 2014, 127, 2683–2696. [Google Scholar] [CrossRef] [PubMed]
- Swift, J.; Ivanovska, I.L.; Buxboim, A.; Harada, T.; Dingal, P.C.; Pinter, J.; Pajerowski, J.D.; Spinler, K.R.; Shin, J.W.; Tewari, M.; et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 2013, 341, 1240104. [Google Scholar] [CrossRef] [PubMed]
- Bronshtein, I.; Kepten, E.; Kanter, I.; Berezin, S.; Lindner, M.; Redwood, A.B.; Mai, S.; Gonzalo, S.; Foisner, R.; Shav-Tal, Y.; et al. Loss of lamin A function increases chromatin dynamics in the nuclear interior. Nat. Commun. 2015, 6, 8044. [Google Scholar] [CrossRef] [PubMed]
- Makarov, A.A.; Zou, J.; Houston, D.R.; Spanos, C.; Solovyova, A.S.; Cardenal-Peralta, C.; Rappsilber, J.; Schirmer, E.C. Lamin A molecular compression and sliding as mechanisms behind nucleoskeleton elasticity. Nat. Commun. 2019, 10, 3056. [Google Scholar] [CrossRef] [PubMed]
- Ben-Harush, K.; Wiesel, N.; Frenkiel-Krispin, D.; Moeller, D.; Soreq, E.; Aebi, U.; Herrmann, H.; Gruenbaum, Y.; Medalia, O. The supramolecular organization of the C. elegans nuclear lamin filament. J. Mol. Biol. 2009, 386, 1392–1402. [Google Scholar] [CrossRef] [PubMed]
- Lilina, A.V.; Chernyatina, A.A.; Guzenko, D.; Strelkov, S.V. Lateral A11 type tetramerization in lamins. J. Struct. Biol. 2020, 209, 107404. [Google Scholar] [CrossRef]
- Cenni, V.; Sabatelli, P.; Mattioli, E.; Marmiroli, S.; Capanni, C.; Ognibene, A.; Squarzoni, S.; Maraldi, N.M.; Bonne, G.; Columbaro, M.; et al. Lamin A N-terminal phosphorylation is associated with myoblast activation: Impairment in Emery-Dreifuss muscular dystrophy. J. Med. Genet. 2005, 42, 214–220. [Google Scholar] [CrossRef]
- Cho, S.; Vashisth, M.; Abbas, A.; Majkut, S.; Vogel, K.; Xia, Y.; Ivanovska, I.L.; Irianto, J.; Tewari, M.; Zhu, K.; et al. Mechanosensing by the lamina protects against nuclear rupture, DNA damage, and cell-cycle arrest. Dev. Cell. 2019, 49, 920–935.e5. [Google Scholar] [CrossRef]
- Harada, T.; Swift, J.; Irianto, J.; Shin, J.W.; Spinler, K.R.; Athirasala, A.; Diegmiller, R.; Dingal, P.C.; Ivanovska, I.L.; Discher, D.E. Nuclear lamin stiffness is a barrier to 3D migration, but softness can limit survival. J. Cell Biol. 2014, 204, 669–682. [Google Scholar] [CrossRef]
- Bermeo, S.; Vidal, C.; Zhou, H.; Duque, G. Lamin A/C acts as an essential factor in mesenchymal stem cell differentiation through the regulation of the dynamics of the Wnt/beta-catenin pathway. J. Cell Biochem. 2015, 116, 2344–2353. [Google Scholar] [CrossRef]
- Boguslavsky, R.L.; Stewart, C.L.; Worman, H.J. Nuclear lamin A inhibits adipocyte differentiation: Implications for Dunnigan-type familial partial lipodystrophy. Hum. Mol. Genet. 2006, 15, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Buxboim, A.; Irianto, J.; Swift, J.; Athirasala, A.; Shin, J.W.; Rehfeldt, F.; Discher, D.E. Coordinated increase of nuclear tension and lamin-A with matrix stiffness outcompetes lamin-B receptor that favors soft tissue phenotypes. Mol. Biol. Cell. 2017, 28, 3333–3348. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yeo, L.S.; Vidal, C.; McCorquodale, T.; Herrmann, M.; Fatkin, D.; Duque, G. Decreased bone formation and osteopenia in lamin A/C-deficient mice. PLoS ONE 2011, 6, e19313. [Google Scholar] [CrossRef] [PubMed]
- Strelkov, S.V.; Schumacher, J.; Burkhard, P.; Aebi, U.; Herrmann, H. Crystal structure of the human lamin A coil 2B dimer: Implications for the head-to-tail association of nuclear lamins. J. Mol. Biol. 2004, 343, 1067–1080. [Google Scholar] [CrossRef] [PubMed]
- Bollati, M.; Barbiroli, A.; Favalli, V.; Arbustini, E.; Charron, P.; Bolognesi, M. Structures of the lamin A/C R335W and E347K mutants: Implications for dilated cardiolaminopathies. Biochem. Biophys. Res. Commun. 2012, 418, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Kapinos, L.E.; Burkhard, P.; Herrmann, H.; Aebi, U.; Strelkov, S.V. Simultaneous formation of right- and left-handed anti-parallel coiled-coil interfaces by a coil2 fragment of human lamin A. J. Mol. Biol. 2011, 408, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Xu, C.; Bian, C.; Lam, R.; Wang, J.P.; Kania, J.; Min, J.; Zang, J. Crystal structures of the coil 2B fragment and the globular tail domain of human lamin B1. FEBS Lett. 2012, 586, 314–318. [Google Scholar] [CrossRef]
- Wilson, K.L.; Foisner, R. Lamin-binding Proteins. Cold Spring Harb Perspect Biol. 2010, 2, a000554. [Google Scholar] [CrossRef]
- Perovanovic, J.; Dell’Orso, S.; Gnochi, V.F.; Jaiswal, J.K.; Sartorelli, V.; Vigouroux, C.; Mamchaoui, K.; Mouly, V.; Bonne, G.; Hoffman, E.P. Laminopathies disrupt epigenomic developmental programs and cell fate. Sci. Transl. Med. 2016, 8. [Google Scholar] [CrossRef]
- Mattout, A.; Goldberg, M.; Tzur, Y.; Margalit, A.; Gruenbaum, Y. Specific and conserved sequences in D. melanogaster and C. elegans lamins and histone H2A mediate the attachment of lamins to chromosomes. J. Cell Sci. 2007, 120, 77–85. [Google Scholar] [CrossRef]
- Pekovic, V.; Harborth, J.; Broers, J.L.; Ramaekers, F.C.; van Engelen, B.; Lammens, M.; von Zglinicki, T.; Foisner, R.; Hutchison, C.; Markiewicz, E. Nucleoplasmic LAP2alpha-lamin A complexes are required to maintain a proliferative state in human fibroblasts. J. Cell Biol. 2007, 176, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Taniura, H.; Glass, C.; Gerace, L. A chromatin binding site in the tail domain of nuclear lamins that interacts with core histones. J. Cell Biol. 1995, 131, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Mogessie, B.; Schuh, M. Actin protects mammalian eggs against chromosome segregation errors. Science 2017, 357. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.N.; Zastrow, M.S.; Wilson, K.L. Direct actin binding to A- and B-type lamin tails and actin filament bundling by the lamin A tail. Nucleus 2010, 1, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Samson, C.; Petitalot, A.; Celli, F.; Herrada, I.; Ropars, V.; Le Du, M.H.; Nhiri, N.; Jacquet, E.; Arteni, A.A.; Buendia, B.; et al. Structural analysis of the ternary complex between lamin A/C, BAF and emerin identifies an interface disrupted in autosomal recessive progeroid diseases. Nucleic Acids Res. 2018, 46, 10460–10473. [Google Scholar] [CrossRef]
- Holaska, J.M.; Kowalski, A.K.; Wilson, K.L. Emerin caps the pointed end of actin filaments: Evidence for an actin cortical network at the nuclear inner membrane. PLoS Biol. 2004, 2, E231. [Google Scholar] [CrossRef]
- Lee, K.K.; Haraguchi, T.; Lee, R.S.; Koujin, T.; Hiraoka, Y.; Wilson, K.L. Distinct functional domains in emerin bind lamin A and DNA-bridging protein BAF. J. Cell Sci. 2001, 114, 4567–4573. [Google Scholar]
- Sosa, B.A.; Demircioglu, F.E.; Chen, J.Z.; Ingram, J.; Ploegh, H.L.; Schwartz, T.U. How lamina-associated polypeptide 1 (LAP1) activates Torsin. Elife 2014, 3, e03239. [Google Scholar] [CrossRef]
- Gesson, K.; Rescheneder, P.; Skoruppa, M.P.; von Haeseler, A.; Dechat, T.; Foisner, R. A-type lamins bind both hetero- and euchromatin, the latter being regulated by lamina-associated polypeptide 2 alpha. Genome Res. 2016, 26, 462–473. [Google Scholar] [CrossRef]
- Dechat, T.; Korbei, B.; Vaughan, O.A.; Vlcek, S.; Hutchison, C.J.; Foisner, R. Lamina-associated polypeptide 2a binds intranuclear Atype lamins. J. Cell Sci. 2000, 19, 3473–3484. [Google Scholar]
- Gant, T.M.; Harris, C.A.; Wilson, K.L. Roles of LAP2 proteins in nuclear assembly and DNA replication: Truncated LAP2β proteins alter lamina assembly, envelope formation, nuclear size, and DNA replication efficiency in Xenopus laevis extracts. J. Cell Biol. 1999, 144, 1083–1096. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, M.A.; Davies, J.D.; Zhang, Q.; Emerson, L.J.; Hunt, J.; Shanahan, C.M.; Ellis, J.A. Distinct functional domains in nesprin-1alpha and nesprin-2beta bind directly to emerin and both interactions are disrupted in X-linked Emery-Dreifuss muscular dystrophy. Exp. Cell Res. 2007, 313, 2845–2857. [Google Scholar] [CrossRef] [PubMed]
- Mislow, J.M.; Holaska, J.M.; Kim, M.S.; Lee, K.K.; Segura-Totten, M.; Wilson, K.L.; McNally, E.M. Nesprin-1alpha self-associates and binds directly to emerin and lamin A in vitro. FEBS Lett. 2002, 525, 135–140. [Google Scholar] [CrossRef]
- Haque, F.; Mazzeo, D.; Patel, J.T.; Smallwood, D.T.; Ellis, J.A.; Shanahan, C.M.; Shackleton, S. Mammalian SUN protein interaction networks at the inner nuclear membrane and their role in laminopathy disease processes. J. Biol. Chem. 2010, 285, 3487–3498. [Google Scholar] [CrossRef] [PubMed]
- Haque, F.; Lloyd, D.J.; Smallwood, D.T.; Dent, C.L.; Shanahan, C.M.; Fry, A.M.; Trembath, R.C.; Shackleton, S. SUN1 interacts with nuclear lamin A and cytoplasmic nesprins to provide a physical connection between the nuclear lamina and the cytoskeleton. Mol. Cell Biol. 2006, 26, 3738–3751. [Google Scholar] [CrossRef]
- Lloyd, D.J.; Trembath, R.C.; Shackleton, S. A novel interaction between lamin A and SREBP1: Implications for partial lipodystrophy and other laminopathies. Hum. Mol. Genet. 2002, 11, 769–777. [Google Scholar] [CrossRef]
- Kim, J.B.; Spiegelman, B.M. ADD1/SREBP1 promotes adipocyte differentiation and gene expression linked to fatty acid metabolism. Genes Dev. 1996, 10, 1096–1107. [Google Scholar] [CrossRef]
- Ishimura, A.; Ng, J.K.; Taira, M.; Young, S.G.; Osada, S. Man1, an inner nuclear membrane protein, regulates vascular remodeling by modulating transforming growth factor beta signaling. Development 2006, 133, 3919–3928. [Google Scholar] [CrossRef]
- Mansharamani, M.; Wilson, K.L. Direct binding of nuclear membrane protein MAN1 to emerin in vitro and two modes of binding to barrier-to-autointegration factor. J. Biol. Chem. 2005, 280, 13863–13870. [Google Scholar] [CrossRef]
- Martelli, A.M.; Bortul, R.; Tabellini, G.; Faenza, I.; Cappellini, A.; Bareggi, R.; Manzoli, L.; Cocco, L. Molecular characterization of protein kinase C-alpha binding to lamin A. J. Cell Biochem. 2002, 86, 320–330. [Google Scholar] [CrossRef]
- Tang, K.; Finley, R.L.J.; Nie, D.; Honn, K.V. Identification of 12-lipoxygenase interaction with cellular proteins by yeast two-hybrid screening. Biochemistry 2000, 39, 3185–3191. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Maher, R.J.; Hannun, Y.A.; Porter, A.T.; Honn, K.V. 12(S)-HETE enhancement of prostate tumor cell invasion: Selective role of PKC alpha. J. Natl. Cancer Inst. 1994, 86, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- González, J.M.; Navarro-Puche, A.; Casar, B.; Crespo, P.; Andrés, V. Fast regulation of AP-1 activity through interaction of lamin A/C, ERK1/2, and c-Fos at the nuclear envelope. J. Cell Biol. 2008, 183, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Ivorra, C.; Kubicek, M.; González, J.M.; Sanz-González, S.M.; Alvarez-Barrientos, A.; O’Connor, J.E.; Burke, B.; Andrés, V. A mechanism of AP-1 suppression through interaction of c-Fos with lamin A/C. Genes Dev. 2006, 20, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Markiewicz, E.; Dechat, T.; Foisner, R.; Quinlan, R.A.; Hutchison, C.J. Lamin A/C binding protein LAP2alpha is required for nuclear anchorage of retinoblastoma protein. Mol. Biol. Cell. 2002, 13, 4401–4413. [Google Scholar] [CrossRef]
- Ozaki, T.; Saijo, M.; Murakami, K.; Enomoto, H.; Taya, Y.; Sakiyama, S. Complex formation between lamin A and the retinoblastoma gene product: Identification of the domain on lamin A required for its interaction. Oncogene 1994, 9, 2649–2653. [Google Scholar]
- Harper, M.; Tillit, J.; Kress, M.; Ernoult-Lange, M. Phosphorylation-dependent binding of human transcription factor MOK2 to lamin A/C. FEBS J. 2009, 276, 3137–3147. [Google Scholar] [CrossRef]
- Dreuillet, C.; Tillit, J.; Kress, M.; Ernoult-Lange, M. In vivo and in vitro interaction between human transcription factor MOK2 and nuclear lamin A/C. Nucleic Acids Res. 2002, 30, 4634–4642. [Google Scholar] [CrossRef]
- Adam, S.A.; Sengupta, K.; Goldman, R.D. Regulation of Nuclear Lamin Polymerization by Importin α. J. Biol. Chem. 2008, 283, 8462–8468. [Google Scholar] [CrossRef]
- Wang, X.; Xu, S.; Rivolta, C.; Li, L.Y.; Peng, G.H.; Swain, P.K.; Sung, C.H.; Swaroop, A.; Berson, E.L.; Dryja, T.P.; et al. Barrier to autointegration factor interacts with the cone-rod homeobox and represses its transactivation function. J. Biol. Chem. 2002, 277, 43288–43300. [Google Scholar] [CrossRef]
- Zheng, R.; Ghirlando, R.; Lee, M.S.; Mizuuchi, K.; Krause, M.; Craigie, R. Barrier-to-autointegration factor (BAF) bridges DNA in a discrete, higher-order nucleoprotein complex. Proc. Natl. Acad. Sci. USA 2000, 97, 8997–9002. [Google Scholar] [CrossRef]
- Zheng, X.; Hu, J.; Yue, S.; Kristiani, L.; Kim, M.; Sauria, M.; Taylor, J.; Kim, Y.; Zheng, Y. Lamins organize the global three-dimensional genome from the nuclear periphery. Mol. Cell. 2018, 71, 802–815.e7. [Google Scholar] [CrossRef] [PubMed]
- Guelen, L.; Pagie, L.; Brasset, E.; Meuleman, W.; Faza, M.B.; Talhout, W.; Eussen, B.H.; de Klein, A.; Wessels, L.; de Laat, W.; et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 2008, 453, 948–951. [Google Scholar] [CrossRef]
- Shumaker, D.K.; Solimando, L.; Sengupta, K.; Shimi, T.; Adam, S.A.; Grunwald, A.; Strelkov, S.V.; Aebi, U.; Cardoso, M.C.; Goldman, R.D. The highly conserved nuclear lamin Ig-fold binds to PCNA: Its role in DNA replication. J. Cell Biol. 2008, 181, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Bruston, F.; Delbarre, E.; Ostlund, C.; Worman, H.J.; Buendia, B.; Duband-Goulet, I. Loss of a DNA binding site within the tail of prelamin A contributes to altered heterochromatin anchorage by progerin. FEBS Lett. 2010, 584, 2999–3004. [Google Scholar] [CrossRef] [PubMed]
- Stierle, V.; Couprie, J.; Ostlund, C.; Krimm, I.; Zinn-Justin, S.; Hossenlopp, P.; Worman, H.J.; Courvalin, J.C.; Duband-Goulet, I. The carboxyl-terminal region common to lamins A and C contains a DNA binding domain. Biochemistry 2003, 42, 4819–4828. [Google Scholar] [CrossRef] [PubMed]
- Isermann, P.; Lammerding, J. Nuclear mechanics and mechanotransduction in health and disease. Curr. Biol. 2013, 23, R1113–R1121. [Google Scholar] [CrossRef]
- Guilluy, C.; Osborne, L.D.; Van Landeghem, L.; Sharek, L.; Superfine, R.; Garcia-Mata, R.; Burridge, K. Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nat. Cell Biol. 2014, 16, 376–381. [Google Scholar] [CrossRef]
- Mitsuhashi, H.; Hayashi, Y.K.; Matsuda, C.; Noguchi, S.; Wakatsuki, S.; Araki, T.; Nishino, I. Specific phosphorylation of Ser458 of A-type lamins in LMNA-associated myopathy patients. J. Cell Sci. 2010, 123, 3893–3900. [Google Scholar] [CrossRef]
- Krimm, I.; Ostlund, C.; Gilquin, B.; Couprie, J.; Hossenlopp, P.; Mornon, J.P.; Bonne, G.; Courvalin, J.C.; Worman, H.J.; Zinn-Justin, S. The Ig-like structure of the C-terminal domain of lamin A/C, mutated in muscular dystrophies, cardiomyopathy, and partial lipodystrophy. Structure 2002, 10, 811–823. [Google Scholar] [CrossRef]
- Booth-Gauthier, E.A.; Alcoser, T.A.; Yang, G.; Dahl, K.N. Force-induced changes in subnuclear movement and rheology. Biophys. J. 2012, 103, 2423–2431. [Google Scholar] [CrossRef] [PubMed]
- Philip, J.T.; Dahl, K.N. Nuclear mechanotransduction: Response of the lamina to extracellular stress with implications in aging. J. Biomech. 2008, 41, 3164–3170. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, A.T.; Ziaei, S.; Ehret, C.; Duchemin, H.; Mamchaoui, K.; Bigot, A.; Mayer, M.; Quijano-Roy, S.; Desguerre, I.; Lainé, J.; et al. Cellular microenvironments reveal defective mechanosensing responses and elevated YAP signaling in LMNA-mutated muscle precursors. J. Cell Sci. 2014, 127, 2873–2884. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Miralles, F.; Posern, G.; Zaromytidou, A.I.; Treisman, R. Actin Dynamics Control SRF Activity by Regulation of Its Coactivator MAL. Cell 2003, 113, 329–342. [Google Scholar] [CrossRef]
- Le, H.Q.; Ghatak, S.; Yeung, C.Y.; Tellkamp, F.; Günschmann, C.; Dieterich, C.; Yeroslaviz, A.; Habermann, B.; Pombo, A.; Niessen, C.M.; et al. Mechanical regulation of transcription controls Polycomb-mediated gene silencing during lineage commitment. Nat. Cell Biol. 2016, 18, 864–875. [Google Scholar] [CrossRef]
- Liu, J.; Rolef Ben-Shahar, T.; Riemer, D.; Treinin, M.; Spann, P.; Weber, K.; Fire, A.; Gruenbaum, Y. Essential roles for Caenorhabditis elegans lamin gene in nuclear organization, cell cycle progression, and spatial organization of nuclear pore complexes. Mol. Biol. Cell. 2000, 11, 3937–3947. [Google Scholar] [CrossRef]
- Aaronson, R.P.; Blobel, G. Isolation of nuclear pore complexes in association with a lamina. Proc. Natl. Acad. Sci. USA 1975, 72, 1007–1011. [Google Scholar] [CrossRef]
- García-González, A.; Jacchetti, E.; Marotta, R.; Tunesi, M.; Rodríguez Matas, J.F.; Raimondi, M.T. The effect of cell morphology on the permeability of the nuclear envelope to diffusive factors. Front. Physiol. 2018, 9, 925. [Google Scholar] [CrossRef]
- Elosegui-Artola, A.; Andreu, I.; Beedle, A.E.M.; Lezamiz, A.; Uroz, M.; Kosmalska, A.J.; Oria, R.; Kechagia, J.Z.; Rico-Lastres, P.; Le Roux, A.L.; et al. Force Triggers YAP Nuclear Entry by Regulating Transport across Nuclear Pores. Cell 2017, 171, 1397–1410. [Google Scholar] [CrossRef]
- Mimura, Y.; Takemoto, S.; Tachibana, T.; Ogawa, Y.; Nishimura, M.; Yokota, H.; Imamoto, N. A statistical image analysis framework for pore-free islands derived from heterogeneity distribution of nuclear pore complexes. Sci Rep. 2017, 7, 16315. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, K.; Ishida, K.; Tsunoyama, T.A.; Toda, S.; Osoda, S.; Horigome, T.; Fisher, P.A.; Sugiyama, S. A-type and B-type lamins initiate layer assembly at distinct areas of the nuclear envelope in living cells. Exp. Cell Res. 2009, 315, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Nava, M.M.; Fedele, R.; Raimondi, M.T. Computational prediction of strain-dependent diffusion of transcription factors through the cell nucleus. Biomech. Model. Mechanobiol. 2016, 15, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, H.; Zhu, M.; Cao, C.; Xu, Z.; Tsatskis, Y.; Lau, K.; Kuok, C.; Filleter, T.; McNeill, H.; et al. Mechanical stability of the cell nucleus—Roles played by the cytoskeleton in nuclear deformation and strain recovery. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Stephens, A.D.; Banigan, E.J.; Adam, S.A.; Goldman, R.D.; Marko, J.F. Chromatin and lamin A determine two different mechanical response regimes of the cell nucleus. Mol. Biol. Cell. 2017, 28, 1984–1996. [Google Scholar] [CrossRef] [PubMed]
- Stephens, A.D.; Liu, P.Z.; Kandula, V.; Chen, H.; Almassalha, L.M.; Herman, C.; Backman, V.; O’Halloran, T.; Adam, S.A.; Goldman, R.D.; et al. Physicochemical mechanotransduction alters nuclear shape and mechanics via heterochromatin formation. Mol. Biol. Cell. 2019, 30, 2320–2330. [Google Scholar] [CrossRef] [PubMed]
- Heo, S.J.; Driscoll, T.P.; Thorpe, S.D.; Nerurkar, N.L.; Baker, B.M.; Yang, M.T.; Chen, C.S.; Lee, D.A.; Mauck, R.L. Differentiation alters stem cell nuclear architecture, mechanics, and mechano-sensitivity. Elife 2016, 30, 5. [Google Scholar] [CrossRef]
- Engler, A.J.; Sen, S.; Sweeney, H.L.; Discher, D.E. Matrix elasticity directs stem cell lineage specification. Cell 2006, 126, 677–689. [Google Scholar] [CrossRef]
- Raimondi, M.T.; Nava, M.M.; Eaton, S.M.; Bernasconi, A.; Vishnubhatla, K.C.; Cerullo, G.; Osellame, R. Optimization of femtosecond laser polymerized structural niches to control mesenchymal stromal cell fate in culture. Micromachines 2014, 5, 341–358. [Google Scholar] [CrossRef]
- Ranade, D.; Pradhan, R.; Jayakrishnan, M.; Hegde, S.; Sengupta, K. Lamin A/C and Emerin depletion impacts chromatin organization and dynamics in the interphase nucleus. BMC Mol. Cell Biol. 2019, 20, 11. [Google Scholar] [CrossRef]
- Peric-Hupkes, D.; Meuleman, W.; Pagie, L.; Bruggeman, S.W.; Solovei, I.; Brugman, W.; Gräf, S.; Flicek, P.; Kerkhoven, R.M.; van Lohuizen, M.; et al. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol. Cell. 2010, 38, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Finlan, L.E.; Sproul, D.; Thomson, I.; Boyle, S.; Kerr, E.; Perry, P.; Ylstra, B.; Chubb, J.R.; Bickmore, W.A. Recruitment to the nuclear periphery can alter expression of genes in human cells. PLoS Genet. 2008, 4, e1000039. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.L.; Zullo, J.M.; Bertolino, E.; Singh, H. Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature 2008, 452, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Leemans, C.; van der Zwalm, M.C.H.; Brueckner, L.; Comoglio, F.; van Schaik, T.; Pagie, L.; van Arensbergen, J.; van Steensel, B. Promoter-Intrinsic and Local Chromatin Features Determine Gene Repression in LADs. Cell 2019, 177, 852–864. [Google Scholar] [CrossRef]
- Zullo, J.M.; Demarco, I.A.; Piqué-Regi, R.; Gaffney, D.J.; Epstein, C.B.; Spooner, C.J.; Luperchio, T.R.; Bernstein, B.E.; Pritchard, J.K.; Reddy, K.L.; et al. DNA sequence-dependent compartmentalization and silencing of chromatin at the nuclear lamina. Cell 2012, 149, 1474–1487. [Google Scholar] [CrossRef]
- Kind, J.; Pagie, L.; Ortabozkoyun, H.; Boyle, S.; de Vries, S.S.; Janssen, H.; Amendola, M.; Nolen, L.D.; Bickmore, W.A.; van Steensel, B. Single-cell dynamics of genome-nuclear lamina interactions. Cell 2013, 153, 178–192. [Google Scholar] [CrossRef]
- Lund, E.; Oldenburg, A.R.; Delbarre, E.; Freberg, C.T.; Duband-Goulet, I.; Eskeland, R.; Buendia, B.; Collas, P. Lamin A/C-promoter interactions specify chromatin state-dependent transcription outcomes. Genome Res. 2013, 23, 1580–1589. [Google Scholar] [CrossRef]
- Meuleman, W.; Peric-Hupkes, D.; Kind, J.; Beaudry, J.B.; Pagie, L.; Kellis, M.; Reinders, M.; Wessels, L.; van Steensel, B. Constitutive nuclear lamina-genome interactions are highly conserved and associated with A/T-rich sequence. Genome Res. 2013, 23, 270–280. [Google Scholar] [CrossRef]
- Ricci, M.A.; Manzo, C.; García-Parajo, M.F.; Lakadamyali, M.; Cosma, M.P. Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo. Cell 2015, 160, 1145–1158. [Google Scholar] [CrossRef]
- Eckersley-Maslin, M.A.; Bergmann, J.H.; Lazar, Z.; Spector, D.L. Lamin A/C is expressed in pluripotent mouse embryonic stem cells. Nucleus 2013, 4, 53–60. [Google Scholar] [CrossRef]
- Kang., S.M.; Yoon, M.H.; Park, B.J. Laminopathies; Mutations on single gene and various human genetic diseases. BMB Rep. 2018, 51, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Quijano-Roy, S.; Mbieleu, B.; Bönnemann, C.G.; Jeannet, P.Y.; Colomer, J.; Clarke, N.F.; Cuisset, J.M.; Roper, H.; De Meirleir, L.; D’Amico, A.; et al. De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann. Neurol. 2008, 64, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Rudnik-Schöneborn, S.; Botzenhart, E.; Eggermann, T.; Senderek, J.; Schoser, B.G.; Schröder, R.; Wehnert, M.; Wirth, B.; Zerres, K. Mutations of the LMNA gene can mimic autosomal dominant proximal spinal muscular atrophy. Neurogenetics 2007, 8, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Ki, C.S.; Hong, J.S.; Jeong, G.Y.; Ahn, K.J.; Choi, K.M.; Kim, D.K.; Kim, J.W. Identification of lamin A/C (LMNA) gene mutations in Korean patients with autosomal dominant Emery-Dreifuss muscular dystrophy and limb-girdle muscular dystrophy 1B. J. Hum. Genet. 2002, 47, 225–228. [Google Scholar] [CrossRef][Green Version]
- Fatkin, D.; MacRae, C.; Sasaki, T.; Wolff, M.R.; Porcu, M.; Frenneaux, M.; Atherton, J.; Vidaillet, H.J., Jr.; Spudich, S.; De Girolami, U.; et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med. 1999, 341, 1715–1724. [Google Scholar] [CrossRef]
- Decaudain, A.; Vantyghem, M.C.; Guerci, B.; Hécart, A.C.; Auclair, M.; Reznik, Y.; Narbonne, H.; Ducluzeau, P.H.; Donadille, B.; Lebbé, C.; et al. New metabolic phenotypes in laminopathies: LMNA mutations in patients with severe metabolic syndrome. J. Clin. Endocrinol. Metab. 2007, 92, 4835–4844. [Google Scholar] [CrossRef]
- Speckman, R.A.; Garg, A.; Du, F.; Bennett, L.; Veile, R.; Arioglu, E.; Taylor, S.I.; Lovett, M.; Bowcock, A.M. Mutational and haplotype analyses of families with familial partial lipodystrophy (Dunnigan variety) reveal recurrent missense mutations in the globular C-terminal domain of lamin A/C. Am. J. Hum. Genet. 2000, 66, 1192–1198. [Google Scholar] [CrossRef]
- Chaouch, M.; Allal, Y.; De Sandre-Giovannoli, A.; Vallat, J.M.; Amer-el-Khedoud, A.; Kassouri, N.; Chaouch, A.; Sindou, P.; Hammadouche, T.; Tazir, M.; et al. The phenotypic manifestations of autosomal recessive axonal Charcot-Marie-Tooth due to a mutation in lamin A/C gene. Neuromuscul. Disord. 2003, 13, 60–67. [Google Scholar] [CrossRef]
- Osmanagic-Myers, S.; Kiss, A.; Manakanatas, C.; Hamza, O.; Sedlmayer, F.; Szabo, P.L.; Fischer, I.; Fichtinger, P.; Podesser, B.K.; Eriksson, M.; et al. Endothelial progerin expression causes cardiovascular pathology through an impaired mechanoresponse. J. Clin. Investig. 2019, 129, 531–545. [Google Scholar] [CrossRef]
- Motegi, S.; Yokoyama, Y.; Uchiyama, A.; Ogino, S.; Takeuchi, Y.; Yamada, K.; Hattori, T.; Hashizume, H.; Ishikawa, Y.; Goto, M.; et al. First Japanese case of atypical progeroid syndrome/atypical Werner Syndrome with heterozygous LMNA mutation. J. Dermatol. 2014, 41, 1047–1052. [Google Scholar] [CrossRef]
- Al-Haggar, M.; Madej-Pilarczyk, A.; Kozlowski, L.; Bujnicki, J.M.; Yahia, S.; Abdel-Hadi, D.; Shams, A.; Ahmad, N.; Hamed, S.; Puzianowska-Kuznicka, M. A novel homozygous p.Arg527Leu LMNA mutation in two unrelated Egyptian families causes overlapping mandibuloacral dysplasia and progeria syndrome. Eur. J. Hum. Genet. 2012, 20, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Dialynas, G.; Flannery, K.M.; Zirbel, L.N.; Nagy, P.L.; Mathews, K.D.; Moore, S.A.; Wallrath, L.L. LMNA variants cause cytoplasmic distribution of nuclear pore proteins in drosophila and human muscle. Hum. Mol. Genet. 2012, 21, 1544–1556. [Google Scholar] [CrossRef] [PubMed]
- Earle, A.J.; Kirby, T.J.; Fedorchak, G.R.; Isermann, P.; Patel, J.; Iruvanti, S.; Moore, S.A.; Bonne, G.; Wallrath, L.L.; Lammerding, J. Mutant lamins cause nuclear envelope rupture and DNA damage in skeletal muscle cells. Nat. Mater. 2020, 19, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Szeverenyi, I.; Cassidy, A.J.; Chung, C.W.; Lee, B.T.; Common, J.E.; Ogg, S.C.; Chen, H.; Sim, S.Y.; Goh, W.L.; Ng, K.W.; et al. The human intermediate filament database: Comprehensive information on a gene family involved in many human diseases. Hum. Mutat. 2008, 29, 351–360. [Google Scholar] [CrossRef]
- Bonne, G.; Mercuri, E.; Muchir, A.; Urtizberea, A.; Bécane, H.M.; Recan, D.; Merlini, L.; Wehnert, M.; Boor, R.; Reuner, U.; et al. Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann. Neurol. 2000, 48, 170–180. [Google Scholar] [CrossRef]
- Tesson, F.; Saj, M.; Uvaize, M.M.; Nicolas, H.; Płoski, R.; Bilińska, Z. Lamin A/C mutations in dilated cardiomyopathy. Cardiol. J. 2014, 21, 331–342. [Google Scholar] [CrossRef]
- Chen, W.; Huo, J.; Ma, A.; Bai, L.; Liu, P. A novel mutation of the LMNA gene in a family with dilated cardiomyopathy, conduction system disease, and sudden cardiac death of young females. Mol. Cell Biochem. 2013, 382, 307–311. [Google Scholar] [CrossRef]
- Jakobs, P.M.; Hanson, E.L.; Crispell, K.A.; Toy, W.; Keegan, H.; Schilling, K.; Icenogle, T.B.; Litt, M.; Hershberger, R.E. Novel lamin A/C mutations in two families with dilated cardiomyopathy and conduction system disease. J. Card Fail. 2001, 7, 249–256. [Google Scholar] [CrossRef]
- Verstraeten, V.L.; Caputo, S.; van Steensel, M.A.; Duband-Goulet, I.; Zinn-Justin, S.; Kamps, M.; Kuijpers, H.J.; Ostlund, C.; Worman, H.J.; Briedé, J.J.; et al. The R439C mutation in LMNA causes lamin oligomerization and susceptibility to oxidative stress. J. Cell Mol. Med. 2009, 13, 959–971. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat. Med. 2005, 11, 440–445. [Google Scholar] [CrossRef]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Lee, L.; Kudlow, B.A.; Dos Santos, H.G.; Sletvold, O.; Shafeghati, Y.; Botha, E.G.; Garg, A.; Hanson, N.B.; Martin, G.M.; et al. LMNA mutations in atypical Werner’s syndrome. Lancet 2003, 362, 440–445. [Google Scholar] [CrossRef]
- Navarro, C.L.; De Sandre-Giovannoli, A.; Bernard, R.; Boccaccio, I.; Boyer, A.; Geneviève, D.; Hadj-Rabia, S.; Gaudy-Marqueste, C.; Smitt, H.S.; Vabres, P.; et al. Lamin A and ZMPSTE24 (FACE-1) defects cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy. Hum. Mol. Genet. 2004, 13, 2493–2503. [Google Scholar] [CrossRef] [PubMed]
- Ozer, L.; Unsal, E.; Aktuna, S.; Baltaci, V.; Celikkol, P.; Akyigit, F.; Sen, A.; Ayvaz, O.; Balci, S. Mandibuloacral dysplasia and LMNA A529V mutation in Turkish patients with severe skeletal changes and absent breast development. Clin. Dysmorphol. 2016, 25, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Yassaee, V.R.; Khojaste, A.; Hashemi-Gorji, F.; Ravesh, Z.; Toosi, P. A novel homozygous LMNA mutation (P.MET540ILE) causes mandibuloacral dysplasia type A. Gene 2016, 577, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Zaragoza, M.V.; Hakim, S.A.; Hoang, V.; Elliott, A.M. Heart-hand syndrome IV: A second family with LMNA-related cardiomyopathy and brachydactyly. Clin. Genet. 2017, 91, 499–500. [Google Scholar] [CrossRef]
- Sullivan, T.; Escalante-Alcalde, D.; Bhatt, H.; Anver, M.; Bhat, N.; Nagashima, K.; Stewart, C.L.; Burke, B. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. Suj. Cell Biol. 1999, 147, 913–920. [Google Scholar] [CrossRef]
- Meaburn, K.J.; Cabuy, E.; Bonne, G.; Levy, N.; Morris, G.E.; Novelli, G.; Kill, I.R.; Bridger, J.M. Primary laminopathy fibroblasts display altered genome organization and apoptosis. Aging Cell. 2007, 6, 139–153. [Google Scholar] [CrossRef]
- Muchir, A.; Pavlidis, P.; Decostre, V.; Herron, A.J.; Arimura, T.; Bonne, G.; Worman, H.J. Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J. Clin. Investig. 2007, 117, 1282–1293. [Google Scholar] [CrossRef]
- Bakay, M.; Wang, Z.; Melcon, G.; Schiltz, L.; Xuan, J.; Zhao, P.; Sartorelli, V.; Seo, J.; Pegoraro, E.; Angelini, C.; et al. Nuclear envelope dystrophies show a transcriptional fingerprint suggesting disruption of Rb-MyoD pathways in muscle regeneration. Brain 2006, 129, 996–1013. [Google Scholar] [CrossRef]
- Dahl, K.N.; Scaffidi, P.; Islam, M.F.; Yodh, A.G.; Wilson, K.L.; Misteli, T. Distinct structural and mechanical properties of the nuclear lamina in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 10271–10276. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Jaalouk, D.E.; Vartiainen, M.K.; Lammerding, J. Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature 2013, 497, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Favreau, C.; Higuet, D.; Courvalin, J.C.; Buendia, B. Expression of a mutant lamin A that causes Emery-Dreifuss muscular dystrophy inhibits in vitro differentiation of C2C12 myoblasts. Mol. Cell Biol. 2004, 24, 1481–1492. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.H.; Luu, J.; Heizer, P.; Tu, Y.; Weston, T.A.; Chen, N.; Lim, C.; Li, R.L.; Lin, P.Y.; Dunn, J.C.Y.; et al. Disrupting the LINC complex in smooth muscle cells reduces aortic disease in a mouse model of Hutchinson-Gilford progeria syndrome. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Dechat, T.; Shimi, T.; Adam, S.A.; Rusinol, A.E.; Andres, D.A.; Spielmann, H.P.; Sinensky, M.S.; Goldman, R.D. Alterations in mitosis and cell cycle progression caused by a mutant lamin A known to accelerate human aging. Proc. Natl.Acad. Sci. USA 2007, 104, 4955–4960. [Google Scholar] [CrossRef]
- McClintock, D.; Ratner, D.; Lokuge, M.; Owens, D.M.; Gordon, L.B.; Collins, F.S.; Djabali, K. The mutant form of lamin A that causes Hutchinson-Gilford progeria is a biomarker of cellular aging in human skin. PLoS ONE 2007, 2, e1269. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T. Lamin A-dependent nuclear defects in human aging. Science 2006, 312, 1059–1063. [Google Scholar] [CrossRef]
- Wang, Y.; Lichter-Konecki, U.; Anyane-Yeboa, K.; Shaw, J.E.; Lu, J.T.; Östlund, C.; Shin, J.Y.; Clark, L.N.; Gundersen, G.G.; Nagy, P.L.; et al. A mutation abolishing the ZMPSTE24 cleavage site in prelamin A causes a progeroid disorder. J. Cell Sci. 2016, 129, 1975–1980. [Google Scholar] [CrossRef]
- Song, M.; San, H.; Anderson, S.A.; Cannon, R.O., 3rd; Orlic, D. Shear stress-induced mechanotransduction protein deregulation and vasculopathy in a mouse model of progeria. Stem Cell Res. 2014, 5, 41. [Google Scholar] [CrossRef]
- Apte, K.; Stick, R.; Radmacher, M. Mechanics in human fibroblasts and progeria: Lamin A mutation E145K results in stiffening of nuclei. J. Mol. Recognit. 2017, 30. [Google Scholar] [CrossRef]
- Goldman, R.D.; Shumaker, D.K.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Gordon, L.B.; Gruenbaum, Y.; Khuon, S.; Mendez, M.; Varga, R.; et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 8963–8968. [Google Scholar] [CrossRef] [PubMed]
- Verstraeten, V.L.; Ji, J.Y.; Cummings, K.S.; Lee, R.T.; Lammerding, J. Increased mechanosensitivity and nuclear stiffness in Hutchinson-Gilford progeria cells: Effects of farnesyltransferase inhibitors. Aging Cell. 2008, 7, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Bikkul, M.U.; Clements, C.S.; Godwin, L.S.; Goldberg, M.W.; Kill, I.R.; Bridger, J.M. Farnesyltransferase inhibitor and rapamycin correct aberrant genome organisation and decrease DNA damage respectively, in Hutchinson-Gilford progeria syndrome fibroblasts. Biogerontology 2018, 19, 579–602. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J.; Wang, W.P.; Chen, Y.C.; Wang, J.Y.; Lin, W.H.; Tai, L.A.; Liou, G.G.; Yang, C.S.; Chi, Y.H. Dysregulated interactions between lamin A and SUN1 induce abnormalities in the nuclear envelope and endoplasmic reticulum in progeric laminopathies. J. Cell Sci. 2014, 127, 1792–1804. [Google Scholar] [CrossRef] [PubMed]
- Baarlink, C.; Plessner, M.; Sherrard, A.; Morita, K.; Misu, S.; Virant, D.; Kleinschnitz, E.M.; Harniman, R.; Alibhai, D.; Baumeister, S.; et al. A transient pool of nuclear F-actin at mitotic exit controls chromatin organization. Nat. Cell Biol. 2017, 19, 1389–1399. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Chen, S.; Jackson, D.A. Inheriting nuclear organization: Can nuclear lamins impart spatial memory during post-mitotic nuclear assembly? Chromosome Res. 2010, 18, 525–541. [Google Scholar] [CrossRef]
- Busch, A.; Kiel, T.; Heupel, W.M.; Wehnert, M.; Hübner, S. Nuclear protein import is reduced in cells expressing nuclear envelopathy-causing lamin A mutants. Exp. Cell Res. 2009, 315, 2373–2385. [Google Scholar] [CrossRef]
- Pujol, G.; Söderqvist, H.; Radu, A. Age-associated reduction of nuclear protein import in human fibroblasts. Biochem Biophys. Res. Commun. 2002, 294, 354–358. [Google Scholar] [CrossRef]
- McCord, R.P.; Nazario-Toole, A.; Zhang, H.; Chines, P.S.; Zhan, Y.; Erdos, M.R.; Collins, F.S.; Dekker, J.; Cao, K. Correlated alterations in genome organization, histone methylation, and DNA-lamin A/C interactions in Hutchinson-Gilford progeria syndrome. Genome Res. 2013, 23, 260–269. [Google Scholar] [CrossRef]
- Cao, K.; Capell, B.C.; Erdos, M.R.; Djabali, K.; Collins, F.S. A lamin A protein isoform overexpressed in Hutchinson-Gilford progeria syndrome interferes with mitosis in progeria and normal cells. Proc Natl. Acad. Sci. USA 2007, 104, 4949–4954. [Google Scholar] [CrossRef]
- Shumaker, D.K.; Dechat, T.; Kohlmaier, A.; Adam, S.A.; Bozovsky, M.R.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Khuon, S.; Collins, F.S.; et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc. Natl. Acad. Sci. USA 2006, 103, 8703–8708. [Google Scholar] [CrossRef] [PubMed]
- Naetar, N.; Korbei, B.; Kozlov, S.; Kerenyi, M.A.; Dorner, D.; Kral, R.; Gotic, I.; Fuchs, P.; Cohen, T.V.; Bittner, R.; et al. Loss of nucleoplasmic LAP2alpha-lamin A complexes causes erythroid and epidermal progenitor hyperproliferation. Nat. Cell Biol. 2008, 10, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo, S.; García-Cao, M.; Fraga, M.F.; Schotta, G.; Peters, A.H.; Cotter, S.E.; Eguía, R.; Dean, D.C.; Esteller, M.; Jenuwein, T.; et al. Role of the RB1 family in stabilizing histone methylation at constitutive heterochromatin. Nat. Cell Biol. 2005, 7, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Le Dour, C.; Macquart, C.; Sera, F.; Homma, S.; Bonne, G.; Morrow, J.P.; Worman, H.J.; Muchir, A. Decreased WNT/β-catenin signalling contributes to the pathogenesis of dilated cardiomyopathy caused by mutations in the lamin A/C gene. Hum. Mol. Genet. 2017, 26, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, L.; Roux, K.J.; Wong, E.S.; Mounkes, L.C.; Mutalif, R.; Navasankari, R.; Rai, B.; Cool, S.; Jeong, J.W.; Wang, H.; et al. Functional coupling between the extracellular matrix and nuclear lamina by Wnt signaling in progeria. Dev. Cell. 2010, 19, 413–425. [Google Scholar] [CrossRef]
- Andrés, V.; González, J.M. Role of A-type lamins in signaling, transcription, and chromatin organization. J. Cell Biol. 2009, 187, 945–957. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T. Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat. Cell Biol. 2008, 10, 452–459. [Google Scholar] [CrossRef]
- Zuela, N.; Zwerger, M.; Levin, T.; Medalia, O.; Gruenbaum, Y. Impaired mechanical response of an EDMD mutation leads to motility phenotypes that are repaired by loss of prenylation. J. Cell Sci. 2016, 129, 1781–1791. [Google Scholar] [CrossRef]
- Dreifuss, F.E.; Hogan, G.R. Survival in X-chromosomal muscular dystrophy. Neurology 1961, 11, 734–737. [Google Scholar] [CrossRef]
- Hausmanowa-Petrusewicz, I.; Madej-Pilarczyk, A.; Marchel, M.; Opolski, G. Emery-Dreifuss dystrophy: A 4-year follow-up on a laminopathy of special interest. Neurol. Neurochir. Polska 2009, 43, 415–420. [Google Scholar]
- Bernasconi, P.; Carboni, N.; Ricci, G.; Siciliano, G.; Politano, L.; Maggi, L.; Mongini, T.; Vercelli, L.; Rodolico, C.; Biagini, E.; et al. Elevated TGF β2 serum levels in Emery-Dreifuss muscular dystrophy: Implications for myocyte and tenocyte differentiation and fibrogenic processes. Nuclues 2018, 9, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Van Berlo, J.H.; Voncken, J.W.; Kubben, N.; Broers, J.L.; Duisters, R.; van Leeuwen, R.E.; Crijns, H.J.; Ramaekers, F.C.; Hutchison, C.J.; Pinto, Y.M. A-type lamins are essential for TGF-beta1 induced PP2A to dephosphorylate transcription factors. Hum. Mol. Genet. 2005, 14, 2839–2849. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Das, J.K.; Maganti, L.; Bhattacharyya, M.; Bhattacharyya, D.; Mukherjee, S.; Sengupta, K. Skeletal Muscle Dystrophy mutant of lamin A alters the structure and dynamics of the Ig fold domain. Sci. Rep. 2018, 8, 13793. [Google Scholar] [CrossRef] [PubMed]
- Mio, M.; Sugiki, T.; Matsuda, C.; Mitsuhashi, H.; Kojima, C.; Chan, S.Y.; Hayashi, Y.K.; Mio, K. Structural instability of lamin A tail domain modulates its assembly and higher order function in Emery-Dreifuss muscular dystrophy. Biochem. Biophys. Res. Commun. 2019, 512, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Gangemi, F.; Degano, M. Disease-associated mutations in the coil 2B domain of human lamin A/C affect structural properties that mediate dimerization and intermediate filament formation. J. Struct. Biol. 2013, 181, 17–28. [Google Scholar] [CrossRef]
- Mattout, A.; Pike, B.L.; Towbin, B.D.; Bank, E.M.; Gonzalez-Sandoval, A.; Stadler, M.B.; Meister, P.; Gruenbaum, Y.; Gasser, S.M. An EDMD mutation in C. elegans lamin blocks muscle-specific gene relocation and compromises muscle integrity. Curr. Biol. 2011, 21, 1603–1614. [Google Scholar] [CrossRef]
- Owens, D.J.; Fischer, M.; Jabre, S.; Moog, S.; Mamchaoui, K.; Butler-Browne, G.; Coirault, C. Lamin mutations cause increased YAP nuclear entry in muscle stem cells. Cells 2020, 27, 816. [Google Scholar] [CrossRef]
- Zuela, N.; Dorfman, J.; Gruenbaum, Y. Global transcriptional changes caused by an EDMD mutation correlate to tissue specific disease phenotypes in C. elegans. Nucleus 2017, 8, 60–69. [Google Scholar] [CrossRef]
- Sabatelli, P.; Lattanzi, G.; Ognibene, A.; Columbaro, M.; Capanni, C.; Merlini, L.; Maraldi, N.M.; Squarzoni, S. Nuclear alterations in autosomal-dominant Emery-Dreifuss muscular dystrophy. Muscle Nerve 2001, 24, 826–829. [Google Scholar] [CrossRef]
- Cheedipudi, S.M.; Matkovich, S.J.; Coarfa, C.; Hu, X.; Robertson, M.J.; Sweet, M.; Taylor, M.; Mestroni, L.; Cleveland, J.; Willerson, J.T.; et al. Genomic reorganization of lamin-associated domains in cardiac myocytes is associated with differential gene expression and DNA methylation in human dilated cardiomyopathy. Circ Res. 2019, 124, 1198–1213. [Google Scholar] [CrossRef]
- Emerson, L.J.; Holt, M.R.; Wheeler, M.A.; Wehnert, M.; Parsons, M.; Ellis, J.A. Defects in cell spreading and ERK1/2 activation in fibroblasts with lamin A/C mutations. Biochim. Biophys. Acta 2009, 1792, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Markiewicz, E.; Ledran, M.; Hutchison, C.J. Remodelling of the nuclear lamina and nucleoskeleton is required for skeletal muscle differentiation in vitro. J. Cell Sci. 2005, 118, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Hale, C.M.; Shrestha, A.L.; Khatau, S.B.; Stewart-Hutchinson, P.J.; Hernandez, L.; Stewart, C.L.; Hodzic, D.; Wirtz, D. Dysfunctional connections between the nucleus and the actin and microtubule networks in laminopathic models. Biophys. J. 2008, 95, 5462–5475. [Google Scholar] [CrossRef] [PubMed]
- Essawy, N.; Samson, C.; Petitalot, A.; Moog, S.; Bigot, A.; Herrada, I.; Marcelot, A.; Arteni, A.-A.; Coirault, C.; Zinn-Justin, S. An Emerin LEM-Domain Mutation Impairs Cell Response to Mechanical Stress. Cells 2019, 8, 570. [Google Scholar] [CrossRef]








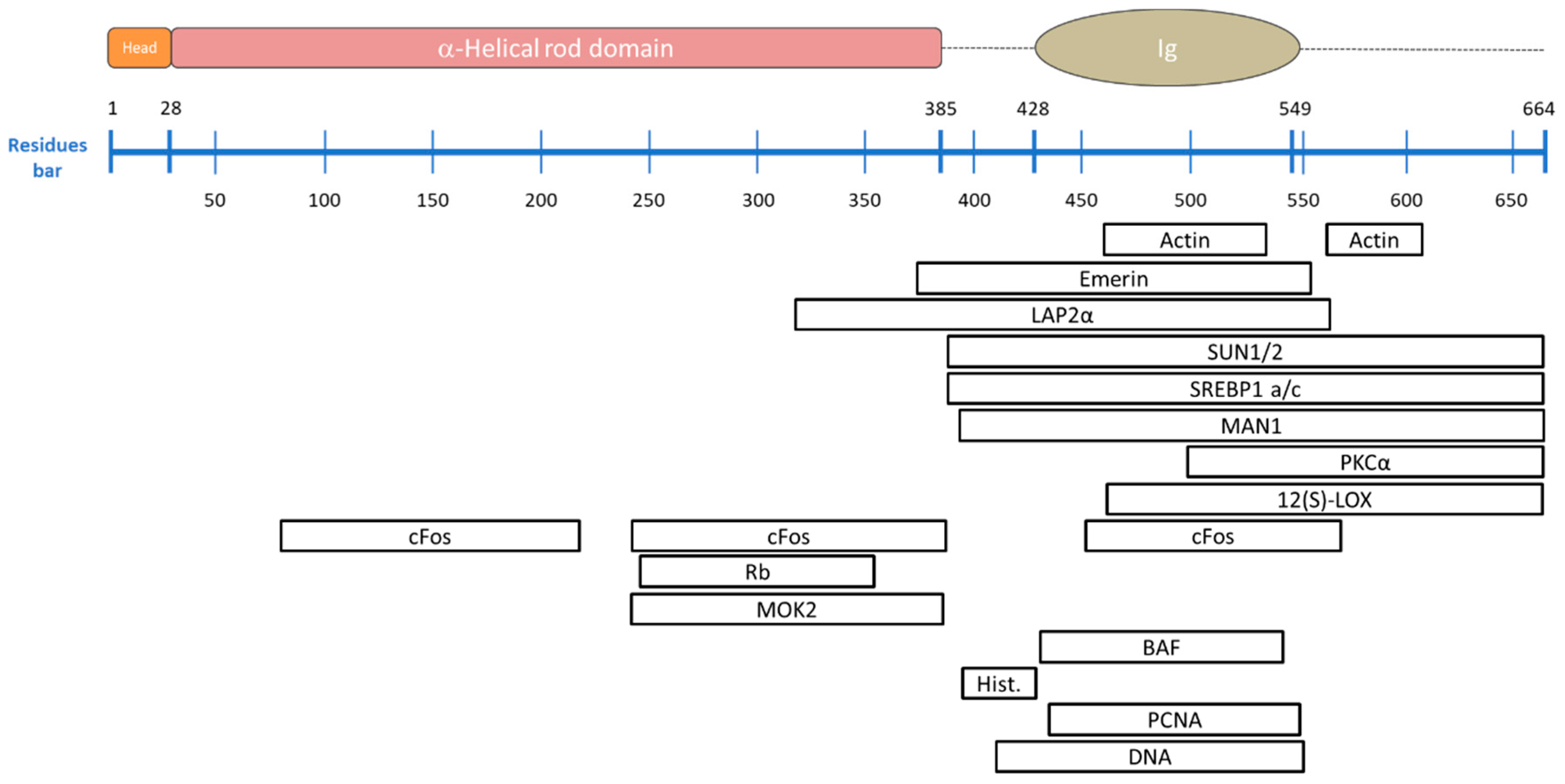
| Binding Partner | Description | A-Type Lamin Binding Region | Reference |
|---|---|---|---|
| Lamin B | Despite their distinct assembly pathways, “the stage is set” for the assembly of A-type lamins. | ND * | [36] |
| Nuclear Actin | Essential for the integrity of the nuclear envelope, it mediates chromatin movement during transcription and mitosis. Failure of this binding impairs the role of nuclear actin, as happens in Hutchinson-Gilford Progeria Syndrome (see details in Section 5). | 461–536 and 564–608 | [103,104] |
| Emerin | Protein of the LEM-domain family; it is relatively immobile in the INM and it anchors the lamina. It also binds directly to the barrier-to-autointegration factor (BAF), retaining chromatin close to the nuclear envelope during cell interphase and acts on gene expression inhibition. Loss of emerin causes Emery-Dreifuss muscular dystrophy (see details in Section 5). | 384–566 | [105,106,107] |
| LAP1 | Integral membrane protein that binds both A- and B-type lamins. Its role has not been characterized yet, but it is involved in Primary dystonia, a central nervous system laminopathy caused by a mutation in torsin A. | ND * | [108] |
| LAP2α | The most-studied architectural partner for A-type lamins. It is located inside the nucleus and is necessary to maintain lamin A/C in a soluble and low-assembly state. Its binding to transcriptional regulators suggests its influence in gene regulation either directly, or indirectly through the lamins. Mutations in LAP2α which disrupt the binding to the A lamin are known to cause dilated cardiomyopathy. | 319–566 | [101,109,110,111] |
| Nesprin 1α | Nuclear membrane protein that directly binds A-type lamins and emerin and anchors them at the nuclear envelope. Human fibroblasts lacking A-type lamins present mis-localized nesprin 1α and emerin (which are located at the endoplasmic reticulum level) inducing an impaired nuclear geometry and peripheral chromatin loss as occurs in Emery-Dreifuss muscular dystrophy. | ND * | [112,113] |
| SUN1/2 | Essential during cell mitosis. Its important role has been recently suggested in anchoring and opening the nuclear pore complex, and therefore, regulating the nuclear influx of transcription factors. | 389–664 | [6,7,114,115] |
| SREBP1 a/c | Known to activate genes required for cholesterol biosynthesis and adipocyte differentiation. They bind the Ig-fold domain of A-type lamins. Deregulation of this binding is involved in lipodystrophies. | 389–664 | [116,117] |
| MAN1 | LEM-domain protein; it binds BAF directly, but also DNA. It is involved in TGF-β-signaling, important for bone development. | 394–664 | [118,119] |
| PKCα | Serine/threonine kinase, activated by many signal pathways and involved in lamin phosphorylation. Once activated, it translocates to the nucleus and binds to the A-type lamin tail to trigger post translational modifications. | 500–664 | [120] |
| 12(S)-LOX | Lamin binding enzyme 12(S)-lipoxygenase converts arachidonic acid (AA) to 12(S)-hydroxy eicosatetraenoic acid [12(S)-HETE] and is involved in the lipid signaling pathway. It also activates PKCα mediating prostate tumor cell metastasis. | 463–664 | [121,122] |
| cFos | Early response transcription factor sequestrated at the nuclear envelope by A-type lamins. During MAP kinase signaling, this binding is released and c-Fos can facilitate cell proliferation. | 81–219, 243–388 and 453–571 | [123,124] |
| Rb | Transcriptional regulator that has a central role in cell-cycle control and in apoptosis mechanisms. It directly binds to A-type lamins and to LAP2α. It appears that Rb tumor suppressor activity depends on its attachment to both proteins. | 247–355 | [125,126] |
| MOK2 | DNA-binding transcriptional repressor that modulates gene expression activated by the cone-rod homeobox protein (Crx), by competing binding to the same binding sites. It also seems to influence RNA processing. | 243–387 | [127,128] |
| IMPORTIN α | Nuclear import receptor. It is supposed to prevent lamins from assembling in the nucleoplasm. | ND * | [129] |
| BAF | Non-specific double-stranded DNA-binding protein. It can bridge DNA and interacts with histones. It also binds several transcription activators including Crx, with an analogous function to MOK2. Alterations in BAF expression lead to impaired chromatin structure, nuclear envelope defects and altered gene expression. | 432–544 | [105,107,130,131] |
| LAD | Lamina-associated domains containing lowly transcribed genes. They are dynamic structures involved in chromosomes organization, gene repression, and cell differentiation. LAD disruptions have been correlated to diseases such as Hutchinson Gilford progeria syndrome (see details in Section 4 and Section 5). | ND * | [109,132,133] |
| Core histones | Their interaction with A-type lamins affects chromatin localization and gene expression. | 396–430 | [100,102] |
| PCNA | Necessary to activate the DNA replication machinery, it binds to the Ig-fold domain. | 436–552 | [134] |
| DNA | The lamin-DNA interaction occurs directly, but non-specifically, by contacting the minor groove. The DNA-binding region is identical in both lamin A and lamin C. Some lamin A mutations drastically reduce the DNA affinity, leading to gene regulation problems. | 411–553 | [135,136] |
| Group | Pathology | OMIM Code | Gene Involved | Reference |
|---|---|---|---|---|
| 1 | Emery-Dreifuss muscular dystrophy, autosomal dominant (EDMD2) | 181350 | LMNA | [171,174,183,184,185] |
| 1 | Emery-Dreifuss muscular dystrophy, autosomal recessive (EDMD3) | 616516 | LMNA | [174,184,185] |
| 1 | Limb-girdle muscular dystrophy, type 1B (LGMD1B) | 159001 | LMNA | [171,174] |
| 1 | Congenital muscular dystrophy (CMD) | 613205 | LMNA | [172] |
| 1 | Autosomal dominant spinal muscular atrophy (AD-SMA) | 182980 | LMNA | [173] |
| 1 | Dilated cardiomyopathy 1A (CMD1A) | 115200 | LMNA | [175,184,186] |
| 1 | Dilated cardiomyopathy with conduction system defects (DCM-CD) | n/a | LMNA | [187,188] |
| 2 | Dunnigan-type familial partial lipodystrophy (FPLD2) | 151660 | LMNA | [171,177,184,189] |
| 2 | Metabolic syndrome (MS) | n/a | LMNA | [176] |
| 2 | Barraquer-Simons syndrome (acquired partial lipodystrophy -APL) | 608709 | LMNB2 | [44] |
| 3 | Charcot-Marie-Tooth disease, type 2B1 (CMT2B1) | 605588 | LMNA | [178] |
| 3 | Autosomal dominant leukodystrophy (ADLD) | 169500 | LMNB1: present an extra copy of the gene | [45] |
| 4 | Hutchinson-Gilford progeria syndrome (HGPS) | 176670 | LMNA: LaminA-Δ50 permanently farnesylated | [171,184,190,191,192] |
| 4 | Atypical Werner syndrome (WRN) | 277700 | LMNA | [171,180,187] |
| 4 | Restrictive dermopathy (RD) | 275210 | LMNA | [193] |
| 4 | Mandibuloacral dysplasia with type A lipodystrophy (MADA) | 248370 | LMNA | [171,181,184,194,195] |
| 4 | Heart-hand syndrome, Slovenian type (HHS) | 610140 | LMNA | [196] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donnaloja, F.; Carnevali, F.; Jacchetti, E.; Raimondi, M.T. Lamin A/C Mechanotransduction in Laminopathies. Cells 2020, 9, 1306. https://doi.org/10.3390/cells9051306
Donnaloja F, Carnevali F, Jacchetti E, Raimondi MT. Lamin A/C Mechanotransduction in Laminopathies. Cells. 2020; 9(5):1306. https://doi.org/10.3390/cells9051306
Chicago/Turabian StyleDonnaloja, Francesca, Federica Carnevali, Emanuela Jacchetti, and Manuela Teresa Raimondi. 2020. "Lamin A/C Mechanotransduction in Laminopathies" Cells 9, no. 5: 1306. https://doi.org/10.3390/cells9051306
APA StyleDonnaloja, F., Carnevali, F., Jacchetti, E., & Raimondi, M. T. (2020). Lamin A/C Mechanotransduction in Laminopathies. Cells, 9(5), 1306. https://doi.org/10.3390/cells9051306