Crucial Role of Lamin A/C in the Migration and Differentiation of MSCs in Bone


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Lamin A/C and Bone Formation
3. Lamin A/C and MSCs
3.1. MSCs’ Migration
3.1.1. Chemical Factors Involved in MSCs’ Migration
3.1.2. Mechanical Factors Involved in MSCs’ Migration
3.2. MSCs’ Differentiation
3.2.1. Response of MSCs to Biochemical Cues
3.2.2. Response of MSCs to Mechanical Cues
3.2.3. Response of MSCs to Other Mechanosensitive Pathways
3.3. MSCs’ Migration versus Differentiation
4. Lamin A/C Dysfunction in MSCs, Aging and Bone Disease
4.1. Lamin A/C Levels Alterations
4.2. Premature Aging Syndromes
4.2.1. Hutchinson-Gilford Progeria Syndrome (HGPS)
4.2.2. Mandibuloacral Dysplasia Type A (MADA)
4.2.3. Atypical Progeroid Syndromes (APS)
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Definition: | Abbreviation: |
| Two-dimensional | 2D |
| Three-dimensional | 3D |
| Atypical progeroid syndromes | APS |
| Bone marrow mesenchymal stem cells | BMSCs |
| Bone morphogenetic protein 2 | BMP-2 |
| Bone sialo-protein | BSP |
| Circulating osteoprogenitors | COPs |
| Cluster of differentiation | CD |
| Dentin matrix acidic phosphoprotein 1 | DMP-1 |
| Extracellular Matrix | ECM |
| Human leucocyte antigen-DR | HLA-DR |
| Hutchinson-Gildford progeria syndrome | HGPS |
| Interleukin | IL |
| Linker of nucleus and cytoskeletonLow density lipoprotein | LINCLDL |
| Mandibuloacral dysplasia type A | MADA |
| Inner nuclear membrane proteinMegakaryoblastic leukaemia 1 | MAN-1MKL1 |
| Mesenchymal stem cells | MSCs |
| Mouse embryonic fibroblasts | MEFs |
| Nuclear envelope spectrin-repeat-containing proteins | Nesprins |
| Osteocalcin | OCN |
| Osteoprotegerin | OPG |
| Osterix | OSX |
| Peroxisome proliferator-activated receptor | PPAR |
| Platelet-derived growth factors | PDGFs |
| Receptor activator of nuclear factor k-B ligand | RANKL |
| Retinoic acid | RA |
| Retinoic acid receptors | RAR |
| Run-t related transcription factor | RUNX2 |
| Serum response factor | SRF |
| Sry-related high-mobility group box 9 | SOX9 |
| Transcriptional coactivator with PDZ-binding motif | TAZ |
| Transforming growth factor-β1 | TGF-β1 |
| Tumor necrosis factor-α | TNF-α |
| Vascular endothelial growth factor A | VEGFA |
| Yes-associated protein | YAP |
References
- Burke, B.; Stewart, C.L. The nuclear lamins: Flexibility in function. Nat. Rev. Mol. Cell. Biol. 2013, 14, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Beck, L.A.; Hosick, T.J.; Sinensky, M. Isoprenylation is required for the processing of the lamin A precursor. J. Cell. Biol. 1990, 110, 1489–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtz, D.; Tanaka, R.A.; Hartwig, J.; McKeon, F. The CaaX motif of lamin A functions in conjunction with the nuclear localization signal to target assembly to the nuclear envelope. Cell 1989, 59, 969–977. [Google Scholar] [CrossRef]
- Fong, L.G.; Ng, J.K.; Lammerding, J.; Vickers, T.A.; Meta, M.; Cote, N.; Gavino, B.; Qiao, X.; Chang, S.Y.; Young, S.R.; et al. Prelamin A and lamin A appear to be dispensable in the nuclear lamina. J. Clin. Investig. 2006, 116, 743–752. [Google Scholar] [CrossRef]
- Shimi, T.; Pfleghaar, K.; Kojima, S.; Pack, C.G.; Solovei, I.; Goldman, A.E.; Adam, S.A.; Shumaker, D.K.; Kinjo, M.; Cremer, T.; et al. The A- and B-type nuclear lamin networks: Microdomains involved in chromatin organization and transcription. Genes Dev. 2008, 22, 3409–3421. [Google Scholar] [CrossRef] [Green Version]
- Xie, W.; Chojnowski, A.; Boudier, T.; Lim, J.S.; Ahmed, S.; Ser, Z.; Stewart, C.; Burke, B. A-type Lamins Form Distinct Filamentous Networks with Differential Nuclear Pore Complex Associations. Curr. Biol. 2016, 26, 2651–2658. [Google Scholar] [CrossRef] [Green Version]
- Shimi, T.; Kittisopikul, M.; Tran, J.; Goldman, A.E.; Adam, S.A.; Zheng, Y.; Jaqaman, K.; Goldman, R.D. Structural organization of nuclear lamins A, C, B1, and B2 revealed by superresolution microscopy. Mol. Biol. Cell 2015, 26, 4075–4086. [Google Scholar] [CrossRef]
- Turgay, Y.; Medalia, O. The structure of lamin filaments in somatic cells as revealed by cryo-electron tomography. Nucleus 2017, 8, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Nmezi, B.; Xu, J.; Fu, R.; Armiger, T.J.; Rodriguez-Bey, G.; Powell, J.S.; Ma, H.; Sullivan, M.; Tu, Y.; Chen, N.Y.; et al. Concentric organization of A- and B-type lamins predicts their distinct roles in the spatial organization and stability of the nuclear lamina. Proc. Natl. Acad. Sci. USA 2019, 116, 4307–4315. [Google Scholar] [CrossRef] [Green Version]
- Figueiras, E.; Silvestre, O.F.; Ihalainen, T.O.; Nieder, J.B. Phasor-assisted nanoscopy reveals differences in the spatial organization of major nuclear lamina proteins. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118530. [Google Scholar] [CrossRef]
- Dechat, T.; Pfleghaar, K.; Sengupta, K.; Shimi, T.; Shumaker, D.K.; Solimando, L.; Goldman, R.D. Nuclear lamins: Major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008, 22, 832–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dechat, T.; Adam, S.A.; Taimen, P.; Shimi, T.; Goldman, R.D. Nuclear lamins. Cold Spring Harb. Perspect. Biol. 2010, 2, a000547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martino, F.; Perestrelo, A.R.; Vinarsky, V.; Pagliari, S.; Forte, G. Cellular Mechanotransduction: From Tension to Function. Front. Physiol. 2018, 9, 824. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Jaalouk, D.E.; Lammerding, J. Novel insights into the disease etiology of laminopathies. Rare Dis. 2013, 1, e27002. [Google Scholar] [CrossRef] [PubMed]
- Guilluy, C.; Osborne, L.D.; Van Landeghem, L.; Sharek, L.; Superfine, R.; Garcia-Mata, R.; Burridge, K. Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nat. Cell. Biol. 2014, 16, 376–381. [Google Scholar] [CrossRef]
- Osmanagic-Myers, S.; Dechat, T.; Foisner, R. Lamins at the crossroads of mechanosignaling. Genes Dev. 2015, 29, 225–237. [Google Scholar] [CrossRef] [Green Version]
- Osmanagic-Myers, S.; Foisner, R. The structural and gene expression hypotheses in laminopathic diseases-not so different after all. Mol. Biol. Cell 2019, 30, 1786–1790. [Google Scholar] [CrossRef]
- Swift, J.; Ivanovska, I.L.; Buxboim, A.; Harada, T.; Dingal, P.C.; Pinter, J.; Pajerowski, J.D.; Spinler, K.R.; Shin, J.W.; Tewari, M.; et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 2013, 341, 1240104. [Google Scholar] [CrossRef] [Green Version]
- Harada, T.; Swift, J.; Irianto, J.; Shin, J.W.; Spinler, K.R.; Athirasala, A.; Diegmiller, R.; Dingal, P.C.; Ivanovska, I.L.; Discher, D.E. Nuclear lamin stiffness is a barrier to 3D migration, but softness can limit survival. J. Cell Biol. 2014, 204, 669–682. [Google Scholar] [CrossRef] [Green Version]
- Lammerding, J. Mechanics of the nucleus. Compr. Physiol. 2011, 1, 783–807. [Google Scholar]
- Ozcivici, E.; Luu, Y.K.; Adler, B.; Qin, Y.X.; Rubin, J.; Judex, S.; Rubin, C.T. Mechanical signals as anabolic agents in bone. Nat. Rev. Rheumatol. 2010, 6, 50–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engler, A.J.; Sen, S.; Sweeney, H.L.; Discher, D.E. Matrix elasticity directs stem cell lineage specification. Cell 2006, 126, 677–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muncie, J.M.; Weaver, V.M. The Physical and Biochemical Properties of the Extracellular Matrix Regulate Cell Fate. Curr. Top. Dev. Biol. 2018, 130, 1–37. [Google Scholar] [PubMed]
- Medhat, D.; Rodriguez, C.I.; Infante, A. Immunomodulatory Effects of MSCs in Bone Healing. Int. J. Mol. Sci. 2019, 20, 5467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bermeo, S.; Vidal, C.; Zhou, H.; Duque, G. Lamin A/C Acts as an Essential Factor in Mesenchymal Stem Cell Differentiation Through the Regulation of the Dynamics of the Wnt/beta-Catenin Pathway. J. Cell. Biocehm. 2015, 116, 2344–2353. [Google Scholar] [CrossRef]
- Yang, L.; Tsang, K.Y.; Tang, H.C.; Chan, D.; Cheah, K.S. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc. Natl. Acad. Sci. USA 2014, 111, 12097–12102. [Google Scholar] [CrossRef] [Green Version]
- Su, P.; Tian, Y.; Yang, C.; Ma, X.; Wang, X.; Pei, J.; Qian, A. Mesenchymal Stem Cell Migration during Bone Formation and Bone Diseases Therapy. Int. J. Mol. Sci. 2018, 19, 2343. [Google Scholar] [CrossRef] [Green Version]
- Fakhry, M.; Hamade, E.; Badran, B.; Buchet, R.; Magne, D. Molecular mechanisms of mesenchymal stem cell differentiation towards osteoblasts. World J. Stem Cells 2013, 5, 136–148. [Google Scholar] [CrossRef]
- Berendsen, A.D.; Olsen, B.R. Bone development. Bone 2015, 80, 14–18. [Google Scholar] [CrossRef] [Green Version]
- Akter, R.; Rivas, D.; Geneau, G.; Drissi, H.; Duque, G. Effect of lamin A/C knockdown on osteoblast differentiation and function. J. Bone Miner. Res. 2009, 24, 283–293. [Google Scholar] [CrossRef]
- Rauner, M.; Sipos, W.; Goettsch, C.; Wutzl, A.; Foisner, R.; Pietschmann, P.; Hofbauer, L.C. Inhibition of lamin A/C attenuates osteoblast differentiation and enhances RANKL-dependent osteoclastogenesis. J. Bone Miner. Res. 2009, 24, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Tsukune, N.; Naito, M.; Kubota, T.; Ozawa, Y.; Nagao, M.; Ohashi, A.; Sato, S.; Takahashi, T. Lamin A overexpression promotes osteoblast differentiation and calcification in the MC3T3-E1 preosteoblastic cell line. Biophys. Res. Commun. 2017, 488, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.R.R.; Dahlke, M.H. Immunomodulation by Mesenchymal Stem Cells (MSCs): Mechanisms of Action of Living, Apoptotic, and Dead MSCs. Front. Immunol. 2019, 10, 1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keating, A. Mesenchymal stromal cells. Curr. Opin. Hematol. 2006, 13, 419–425. [Google Scholar] [CrossRef]
- Wagner, W.; Wein, F.; Seckinger, A.; Frankhauser, M.; Wirkner, U.; Krause, U.; Blake, J.; Schwager, C.; Eckstein, V.; Ansorge, W.; et al. Comparative characteristics of mesenchymal stem cells from human bone marrow, adipose tissue, and umbilical cord blood. Exp. Hematol. 2005, 33, 1402–1416. [Google Scholar] [CrossRef]
- Ullah, I.; Subbarao, R.B.; Rho, G.J. Human mesenchymal stem cells—current trends and future prospective. Biosci. Rep. 2015, 35, e00191. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Pittenger, M.F.; Discher, D.E.; Péault, B.M.; Phinney, D.G.; Hare, J.M.; Caplan, A.I. Mesenchymal stem cell perspective: Cell biology to clinical progress. NPJ Regen. Med. 2019, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Oh, E.J.; Lee, H.W.; Kalimuthu, S.; Kim, T.J.; Kim, H.M.; Baek, S.H.; Zhu, L.; Oh, J.M.; Son, S.H.; Chung, H.Y.; et al. In Vivo migration of mesenchymal stem cells to burn injury sites and their therapeutic effects in a living mouse model. J. Control Release 2018, 279, 79–88. [Google Scholar] [CrossRef]
- Kawai, T.; Katagiri, W.; Osugi, M.; Sugimura, Y.; Hibi, H.; Ueda, M. Secretomes from bone marrow-derived mesenchymal stromal cells enhance periodontal tissue regeneration. Cytotherapy 2015, 17, 369–381. [Google Scholar] [CrossRef]
- Nakamura, Y.; Ishikawa, H.; Kawai, K.; Tabata, Y.; Suzuki, S. Enhanced wound healing by topical administration of mesenchymal stem cells transfected with stromal cell-derived factor-1. Biomaterials 2013, 34, 9393–9400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnecchi, M.; Danieli, P.; Malpasso, G.; Ciuffreda, M.C. Paracrine Mechanisms of Mesenchymal Stem Cells in Tissue Repair. Methods Mol. Biol. 2016, 1416, 123–146. [Google Scholar]
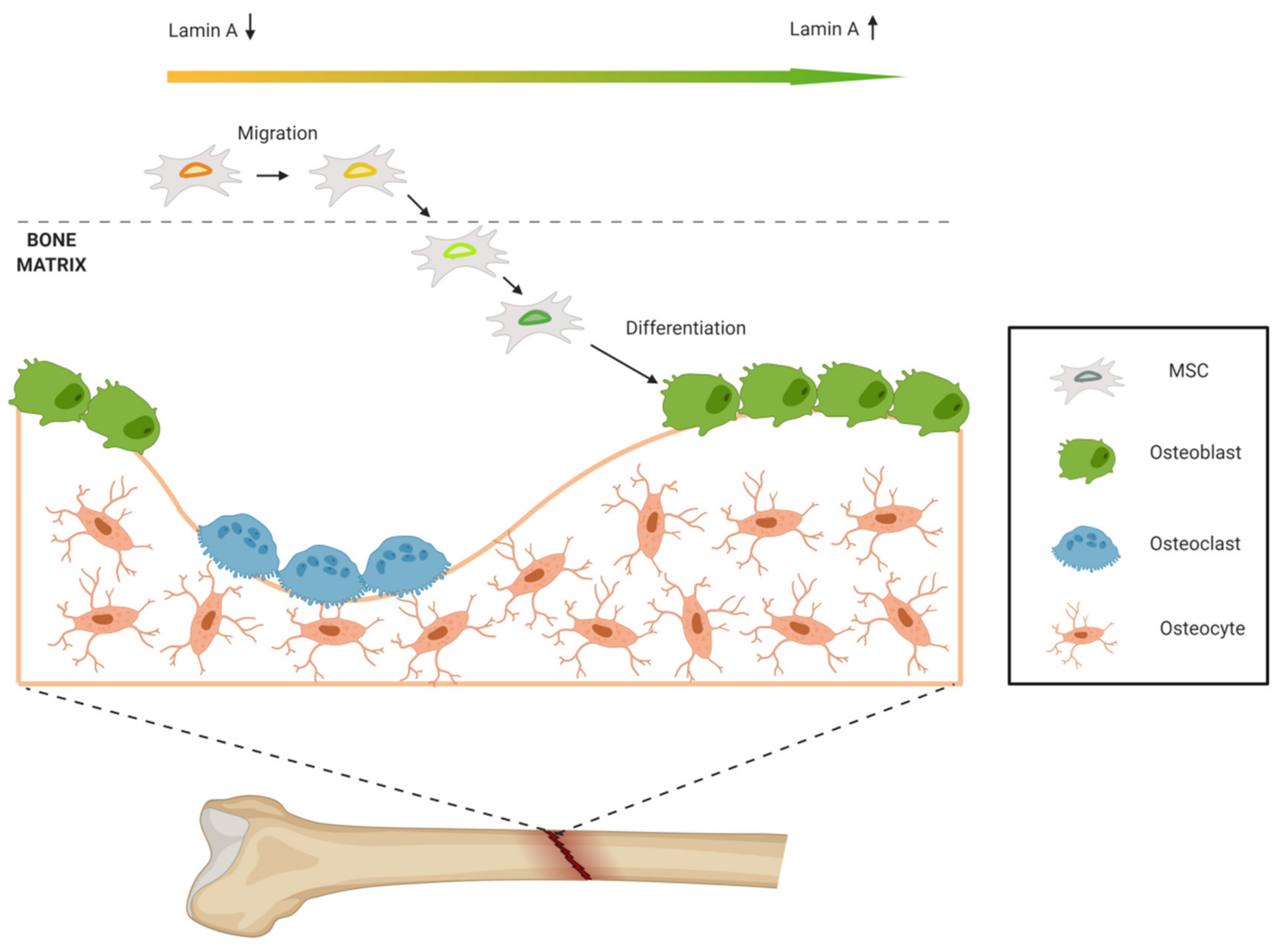
- Chen, L.; Jiang, F.; Qiao, Y.; Li, H.; Wei, Z.; Huang, T.; Lan, J.; Xia, Y.; Li, J. Nucleoskeletal stiffness regulates stem cell migration and differentiation through lamin A/C. J. Cell Physiol. 2018, 233, 5112–5118. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.; Liu, D.D.; Thakor, A.S. Mesenchymal Stromal Cell Homing: Mechanisms and Strategies for Improvement. iScience 2019, 15, 421–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loi, F.; Cordova, L.A.; Pajarinen, J.; Lin, T.H.; Yao, Z.; Goodman, S.B. Inflammation, fracture and bone repair. Bone 2016, 86, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.B.; Giannoudis, P.V.; Boxall, S.A.; McGonagle, D.; Jones, E. The systemic influence of platelet-derived growth factors on bone marrow mesenchymal stem cells in fracture patients. BMC Med. 2015, 13, 6. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Xia, X.; Yeh, J.; Kua, H.; Liu, H.; Mishina, Y.; Hao, A.; Li, B. PDGF-AA promotes osteogenic differentiation and migration of mesenchymal stem cell by down-regulating PDGFRalpha and derepressing BMP-Smad1/5/8 signaling. PLoS ONE 2014, 9, e113785. [Google Scholar]
- Kon, T.; Cho, T.J.; Aizawa, T.; Yamazaki, M.; Nooh, N.; Graves, D.; Gerstenfeld, L.C.; Einhorn, T.A. Expression of osteoprotegerin, receptor activator of NF-kappaB ligand (osteoprotegerin ligand) and related proinflammatory cytokines during fracture healing. J. Bone Miner. Res. 2001, 16, 1004–1014. [Google Scholar] [CrossRef]
- Cho, H.H.; Kyoung, K.M.; Seo, M.J.; Kim, Y.J.; Bae, Y.C.; Jung, J.S. Overexpression of CXCR4 increases migration and proliferation of human adipose tissue stromal cells. Stem Cells Dev. 2006, 15, 853–864. [Google Scholar] [CrossRef]
- Putra, A.; Ridwan, F.B.; Putridewi, A.I.; Kustiyah, A.R.; Wirastuti, K.; Sadyah, N.A.C.; Rosdiana, I.; Munir, D. The Role of TNF-alpha induced MSCs on Suppressive Inflammation by Increasing TGF-beta and IL-10. Open Access Maced. J. Med. Sci. 2018, 6, 1779–1783. [Google Scholar] [CrossRef] [Green Version]
- Claes, L.; Recknagel, S.; Ignatius, A. Fracture healing under healthy and inflammatory conditions. Nat. Rev. Rheumatol. 2012, 8, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wu, X.; Lei, W.; Pang, L.; Wan, C.; Shi, Z.; Zhao, L.; Nagy, T.R.; Peng, X.; Hu, J.; et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 2009, 15, 757–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Q.; Eberspaecher, H.; Lefebvre, V.; De Crombrugghe, B. Parallel expression of Sox9 and Col2a1 in cells undergoing chondrogenesis. Dev. Dyn. 1997, 209, 377–386. [Google Scholar] [CrossRef]
- Hattori, T.; Muller, C.; Gebhard, S.; Bauer, E.; Pausch, F.; Schlund, B.; Bosl, M.R.; Hess, A.; Surmann-Schmitt, C.; von der Mark, H.; et al. SOX9 is a major negative regulator of cartilage vascularization, bone marrow formation and endochondral ossification. Development 2010, 137, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Bear, J.E.; Haugh, J.M. Directed migration of mesenchymal cells: Where signaling and the cytoskeleton meet. Curr. Opin. Cell Biol. 2014, 30, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Friedl, P.; Wolf, K.; Lammerding, J. Nuclear mechanics during cell migration. Curr. Opin. Cell Biol. 2011, 23, 55–64. [Google Scholar] [CrossRef] [Green Version]
- Wintner, O.; Hirsch-Attas, N.; Schlossberg, M.; Brofman, F.; Friedman, R.; Kupervaser, M.; Kitsberg, D.; Buxboim, A. A Unified Linear Viscoelastic Model of the Cell Nucleus Defines the Mechanical Contributions of Lamins and Chromatin. Adv. Sci. 2020, 7, 1901222. [Google Scholar] [CrossRef] [Green Version]
- Davidson, P.M.; Denais, C.; Bakshi, M.C.; Lammerding, J. Nuclear deformability constitutes a rate-limiting step during cell migration in 3-D environments. Cell. Mol. Bioeng. 2014, 7, 293–306. [Google Scholar] [CrossRef] [Green Version]
- Denais, C.; Lammerding, J. Nuclear mechanics in cancer. Adv. Exp. Med. Biol. 2014, 773, 435–470. [Google Scholar]
- Lammerding, J.; Schulze, P.C.; Takahashi, T.; Kozlov, S.; Sullivan, T.; Kamm, R.D.; Stewart, C.L.; Lee, R.T. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J. Clin. Investig. 2004, 113, 370–378. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Hale, C.M.; Panorchan, P.; Khatau, S.B.; George, J.P.; Tseng, Y.; Stewart, C.L.; Hodzic, D.; Wirtz, D. Nuclear lamin A/C deficiency induces defects in cell mechanics, polarization, and migration. Biophys. J. 2007, 93, 2542–2552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lammerding, J.; Fong, L.G.; Ji, J.Y.; Reue, K.; Stewart, C.L.; Young, S.G.; Lee, R.T. Lamins A and C but not lamin B1 regulate nuclear mechanics. J. Biol. Chem. 2006, 281, 25768–25780. [Google Scholar] [CrossRef] [Green Version]
- Dorland, Y.L.; Cornelissen, A.S.; Kuijk, C.; Tol, S.; Hoogenboezem, M.; van Buul, J.D.; Nolte, M.A.; Voermans, C.; Huveneers, S. Nuclear shape, protrusive behaviour and in vivo retention of human bone marrow mesenchymal stromal cells is controlled by Lamin-A/C expression. Sci. Rep. 2019, 9, 14401. [Google Scholar] [CrossRef] [PubMed]
- Dahl, K.N.; Kalinowski, A. Nucleoskeleton mechanics at a glance. J. Cell. Sci. 2011, 124 Pt 5, 675–678. [Google Scholar] [CrossRef] [Green Version]
- Pajerowski, J.D.; Dahl, K.N.; Zhong, F.L.; Sammak, P.J.; Discher, D.E. Physical plasticity of the nucleus in stem cell differentiation. Proc. Natl. Acad. Sci. USA 2007, 104, 15619–15624. [Google Scholar] [CrossRef] [PubMed]
- Méjat, A.; Decostre, V.; Li, J.; Renou, L.; Kesari, A.; Hantaï, D.; Stewart, C.L.; Xiao, X.; Hoffman, E.; Bonne, G.; et al. Lamin A/C-mediated neuromuscular junction defects in Emery-Dreifuss muscular dystrophy. J. Cell Biol. 2009, 184, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Belaadi, N.; Aureille, J.; Guilluy, C. Under Pressure: Mechanical Stress Management in the Nucleus. Cells 2016, 5, 27. [Google Scholar] [CrossRef]
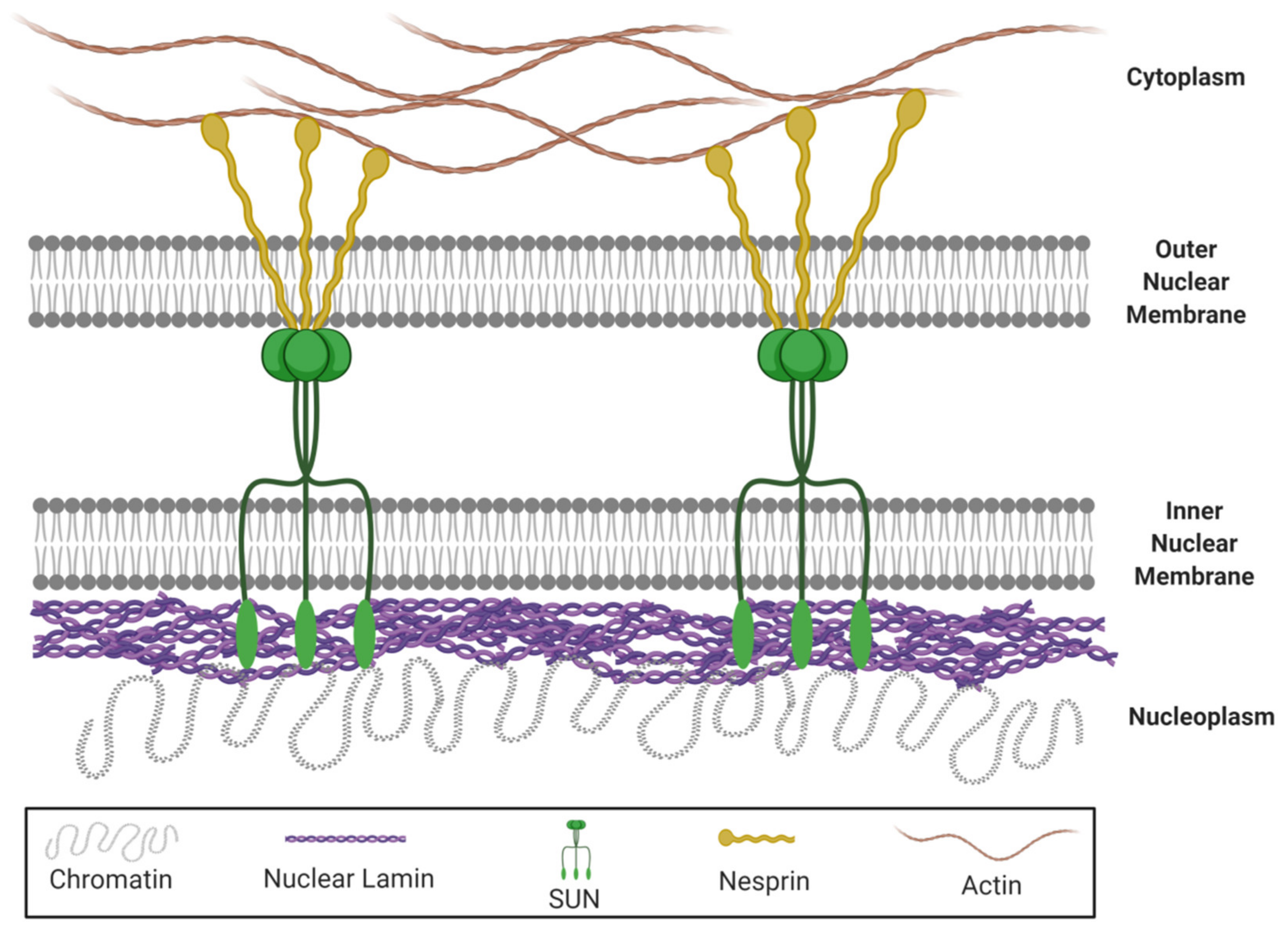
- Bouzid, T.; Kim, E.; Riehl, B.D.; Esfahani, A.M.; Rosenbohm, J.; Yang, R.; Duan, B.; Lim, J.Y. The LINC complex, mechanotransduction, and mesenchymal stem cell function and fate. J. Biol. Eng. 2019, 13, 68. [Google Scholar] [CrossRef]
- Haque, F.; Lloyd, D.J.; Smallwood, D.T.; Dent, C.L.; Shanahan, C.M.; Fry, A.M.; Trembath, R.C.; Shackleton, S. SUN1 interacts with nuclear lamin A and cytoplasmic nesprins to provide a physical connection between the nuclear lamina and the cytoskeleton. Mol. Cell. Biol. 2006, 26, 3738–3751. [Google Scholar] [CrossRef] [Green Version]
- Philip, J.T.; Dahl, K.N. Nuclear mechanotransduction: Response of the lamina to extracellular stress with implications in aging. J. Biomech. 2008, 41, 3164–3170. [Google Scholar] [CrossRef]
- Fu, X.; Liu, G.; Halim, A.; Ju, Y.; Luo, Q.; Song, A.G. Mesenchymal Stem Cell Migration and Tissue Repair. Cells 2019, 8, 784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.; Huang, X.; Zhou, Y.; Jin, R.; Li, Q. Mechanical Stretching Promotes Skin Tissue Regeneration via Enhancing Mesenchymal Stem Cell Homing and Transdifferentiation. Stem Cells Transl. Med. 2016, 5, 960–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.B.; Wang, J.; Chiang, C.A.; Sheng, L.L.; Li, Q.F. Mechanical stretch upregulates SDF-1α in skin tissue and induces migration of circulating bone marrow-derived stem cells into the expanded skin. Stem Cells 2013, 31, 2703–2713. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Luo, Q.; Chen, Z.; Sun, J.; Xu, B.; Ju, Y.; Song, G. Cyclic mechanical stretching promotes migration but inhibits invasion of rat bone marrow stromal cells. Stem Cell Res. 2015, 14, 155–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lammerding, J.; Lee, R.T. The nuclear membrane and mechanotransduction: Impaired nuclear mechanics and mechanotransduction in lamin A/C deficient cells. Novartis Found Symp. 2005, 264, 264–273. [Google Scholar]
- Vincent, L.G.; Choi, Y.S.; Alonso-Latorre, B.; del Álamo, J.C.; Engler, A.J. Mesenchymal stem cell durotaxis depends on substrate stiffness gradient strength. Biotechnol. J. 2013, 8, 472–484. [Google Scholar] [CrossRef] [Green Version]
- Raab, M.; Swift, J.; Dingal, P.C.; Shah, P.; Shin, J.W.; Discher, D.E. Crawling from soft to stiff matrix polarizes the cytoskeleton and phosphoregulates myosin-II heavy chain. J. Cell. Biol. 2012, 199, 669–683. [Google Scholar] [CrossRef] [Green Version]
- Isermann, P.; Lammerding, J. Nuclear mechanics and mechanotransduction in health and disease. Curr. Biol. 2013, 23, R1113–R1121. [Google Scholar] [CrossRef] [Green Version]
- Raab, M.; Discher, D.E. Matrix rigidity regulates microtubule network polarization in migration. Cytoskeleton 2017, 74, 114–124. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Luo, Q.; Sun, J.; Song, G. Nucleus and nucleus-cytoskeleton connections in 3D cell migration. Exp. Cell. Res. 2016, 348, 56–65. [Google Scholar] [CrossRef]
- Stephens, A.D.; Banigan, E.J.; Adam, S.A.; Goldman, R.D.; Marko, J.F. Chromatin and lamin A determine two different mechanical response regimes of the cell nucleus. Mol. Biol. Cell 2017, 28, 1984–1996. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.; Blanquer, S.B.; Haimi, S.P.; Korus, G.; Dunlop, J.W.; Duda, G.N.; Grijpma, D.W.; Petersen, A. Surface Curvature Differentially Regulates Stem Cell Migration and Differentiation via Altered Attachment Morphology and Nuclear Deformation. Adv. Sci. 2017, 4, 1600347. [Google Scholar] [CrossRef]
- Mao, X.; Chen, Z.; Luo, Q.; Zhang, B.; Song, G. Simulated microgravity inhibits the migration of mesenchymal stem cells by remodeling actin cytoskeleton and increasing cell stiffness. Cytotechnology 2016, 68, 2235–2243. [Google Scholar] [CrossRef] [PubMed]
- Koaykul, C.; Kim, M.H.; Kawahara, Y.; Yuge, L.; Kino-Oka, M. Alterations in Nuclear Lamina and the Cytoskeleton of Bone Marrow-Derived Human Mesenchymal Stem Cells Cultured Under Simulated Microgravity Conditions. Stem Cells Dev. 2019, 28, 1167–1176. [Google Scholar] [CrossRef] [PubMed]
- Malashicheva, A.; Bogdanova, M.; Zabirnyk, A.; Smolina, N.; Ignatieva, E.; Freilikhman, O.; Fedorov, A.; Dmitrieva, R.; Sjoberg, G.; Sejersen, T.; et al. Various lamin A/C mutations alter expression profile of mesenchymal stem cells in mutation specific manner. Mol. Genet. Metab. 2015, 115, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Almalki, S.G.; Agrawal, D.K. Key transcription factors in the differentiation of mesenchymal stem cells. Differentiation 2016, 92, 41–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.A.; Choi, H.K.; Kim, T.M.; Leem, S.H.; Oh, I.H. Regulation of mesenchymal stromal cells through fine tuning of canonical Wnt signaling. Stem Cell Res. 2015, 14, 356–368. [Google Scholar] [CrossRef] [Green Version]
- Liang, W.C.; Fu, W.M.; Wang, Y.B.; Sun, Y.X.; Xu, L.L.; Wong, C.W.; Chan, K.M.; Li, G.; Waye, M.M.; Zhang, J.F. H19 activates Wnt signaling and promotes osteoblast differentiation by functioning as a competing endogenous RNA. Sci. Rep. 2016, 6, 20121. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, X.; Bikle, D.D. Osteogenic Differentiation of Periosteal Cells during Fracture Healing. J. Cell Physiol. 2017, 232, 913–921. [Google Scholar] [CrossRef] [Green Version]
- Case, N.; Rubin, J. Beta-catenin--a supporting role in the skeleton. J. Cell Biochem. 2010, 110, 545–553. [Google Scholar] [CrossRef] [Green Version]
- Houschyar, K.S.; Tapking, C.; Borrelli, M.R.; Popp, D.; Duscher, D.; Maan, Z.N.; Chelliah, M.P.; Li, J.; Harati, K.; Wallner, C.; et al. Wnt Pathway in Bone Repair and Regeneration—What Do We Know So Far. Front. Cell Dev. Biol. 2018, 6, 170. [Google Scholar] [CrossRef]
- Zanotti, S.; Canalis, E. Notch Signaling and the Skeleton. Endocr. Rev. 2016, 37, 223–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, E.R.; Sandberg, R.; Lendahl, U. Notch signaling: Simplicity in design, versatility in function. Development 2011, 138, 3593–3612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henrique, D.; Schweisguth, F. Mechanisms of Notch signaling: A simple logic deployed in time and space. Development 2019, 146, dev172148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Wei, Y.; Lian, J.; Yang, L.; Zhang, X.; Xie, J.; Liu, Q.; Luo, J.; He, B.; Tang, M. Notch signaling pathway promotes osteogenic differentiation of mesenchymal stem cells by enhancing BMP9/Smad signaling. Int. J. Mol. Med. 2017, 40, 378–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenova, D.; Bogdanova, M.; Kostina, A.; Golovkin, A.; Kostareva, A.; Malashicheva, A. Dose-dependent mechanism of Notch action in promoting osteogenic differentiation of mesenchymal stem cells. Cell Tissue Res. 2020, 379, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Song, B.Q.; Chi, Y.; Li, X.; Du, W.J.; Han, Z.B.; Tian, J.J.; Li, J.J.; Chen, F.; Wu, H.H.; Han, L.X.; et al. Inhibition of Notch Signaling Promotes the Adipogenic Differentiation of Mesenchymal Stem Cells Through Autophagy Activation and PTEN-PI3K/AKT/mTOR Pathway. Cell. Physiol. Biochem. 2015, 36, 1991–2002. [Google Scholar] [CrossRef] [PubMed]
- Bogdanova, M.A.; Gudkova, A.; Zabirnik, A.S.; Ignat’eva, E.V.; Dmitrieva, R.I.; Smolina, N.A.; Kostareva, A.A.; Malashicheva, A.B. Nuclear lamins regulate osteogenic differentiation of mesenchymal stem cells. Tsitologiia 2014, 56, 260–267. [Google Scholar] [CrossRef]
- Sakaki, M.; Koike, H.; Takahashi, N.; Sasagawa, N.; Tomioka, S.; Arahata, K.; Ishiura, S. Interaction between emerin and nuclear lamins. J. Biochem. 2001, 129, 321–327. [Google Scholar] [CrossRef]
- Lee, B.; Lee, T.H.; Shim, J. Emerin suppresses Notch signaling by restricting the Notch intracellular domain to the nuclear membrane. Biochim. Biophys. Acta. Mol. Cell Res. 2017, 1864, 303–313. [Google Scholar] [CrossRef]
- Perepelina, K.; Dmitrieva, R.; Ignatieva, E.; Borodkina, A.; Kostareva, A.; Malashicheva, A. Lamin A/C mutation associated with lipodystrophy influences adipogenic differentiation of stem cells through interaction with Notch signaling. Biochem. Cell Biol. 2018, 96, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Capanni, C.; Cenni, V.; Mattioli, E.; Sabatelli, P.; Ognibene, A.; Columbaro, M.; Parnaik, V.K.; Wehnert, M.; Maraldi, N.M.; Squarzoni, S.; et al. Failure of lamin A/C to functionally assemble in R482L mutated familial partial lipodystrophy fibroblasts: Altered intermolecular interaction with emerin and implications for gene transcription. Exp. Cell. Res. 2003, 291, 122–134. [Google Scholar] [CrossRef]
- Perepelina, K.; Klauzen, P.; Kostareva, A.; Malashicheva, A. Tissue-Specific Influence of Lamin A Mutations on Notch Signaling and Osteogenic Phenotype of Primary Human Mesenchymal Cells. Cells 2019, 8, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, A.; Shehadeh, L.A.; Yu, H.; Webster, K.A. Age-related molecular genetic changes of murine bone marrow mesenchymal stem cells. BMC Genom. 2010, 11, 229. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Kh Haider, H.; Ahmed, R.P.; Idris, N.M.; Salim, A.; Ashraf, M. Transcriptional profiling of young and old mesenchymal stem cells in response to oxygen deprivation and reparability of the infarcted myocardium. J. Mol. Cell. Cardiol. 2008, 44, 582–596. [Google Scholar] [CrossRef] [Green Version]
- Costa, N.; Paramanathan, S.; Mac Donald, D.; Wierzbicki, A.S.; Hampson, G. Factors regulating circulating vascular endothelial growth factor (VEGF): Association with bone mineral density (BMD) in post-menopausal osteoporosis. Cytokine 2009, 46, 376–381. [Google Scholar] [CrossRef]
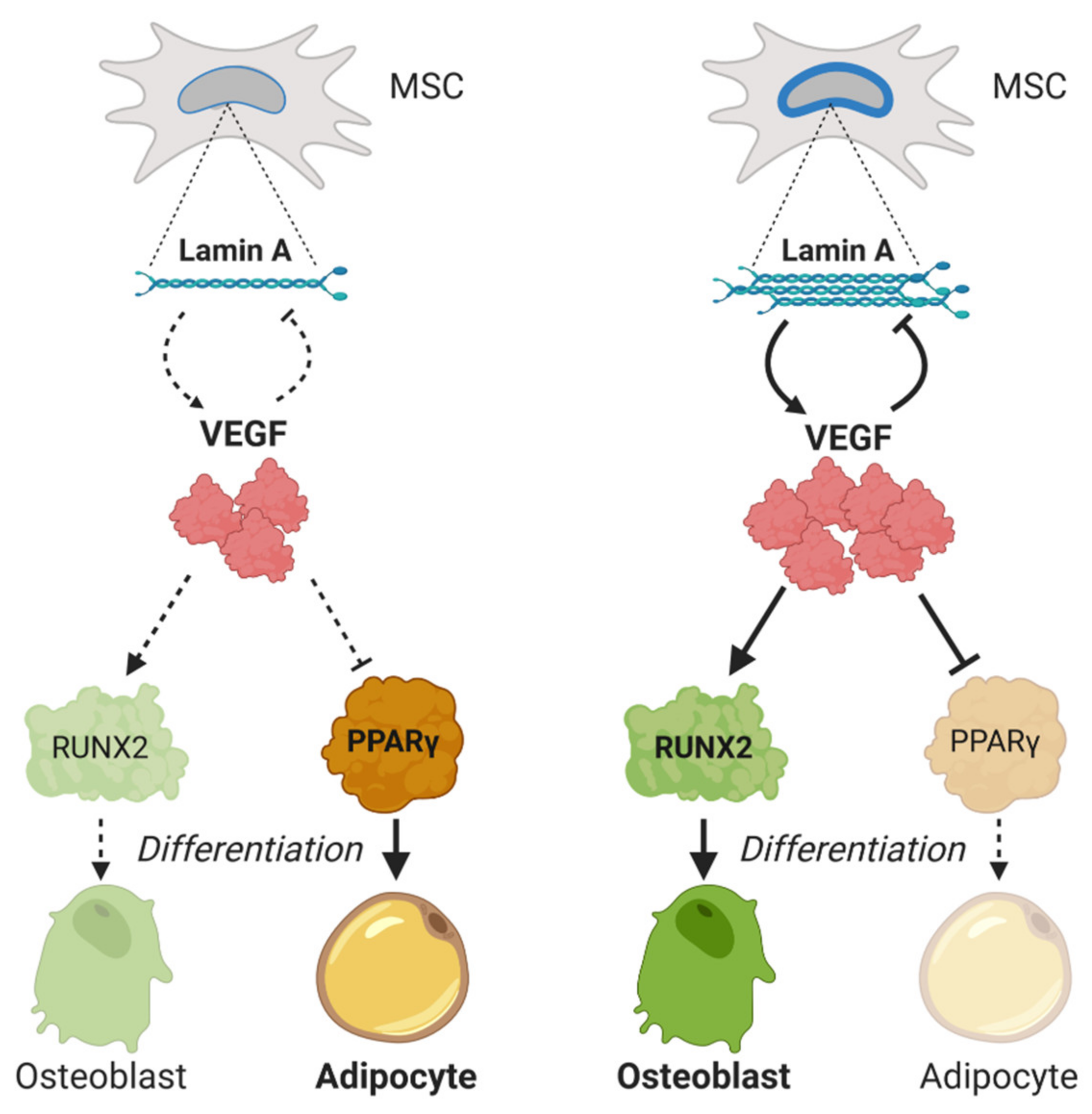
- Berendsen, A.D.; Olsen, B.R. Regulation of adipogenesis and osteogenesis in mesenchymal stem cells by vascular endothelial growth factor A. J. Inter. Med. 2015, 277, 674–680. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Berendsen, A.D.; Jia, S.; Lotinun, S.; Baron, R.; Ferrara, N.; Olsen, B.R. Intracellular VEGF regulates the balance between osteoblast and adipocyte differentiation. J. Clin. Investig. 2012, 122, 3101–3113. [Google Scholar] [CrossRef] [Green Version]
- Komori, T. Regulation of osteoblast differentiation by Runx2. Adv. Exp. Med. Biol. 2010, 658, 43–49. [Google Scholar]
- Xu, J.; Li, Z.; Hou, Y.; Fang, W. Potential mechanisms underlying the Runx2 induced osteogenesis of bone marrow mesenchymal stem cells. Am. J. Transl. Res. 2015, 7, 2527–2535. [Google Scholar]
- Komori, T. Regulation of Proliferation, Differentiation and Functions of Osteoblasts by Runx2. Int. J. Mol. Sci. 2019, 20, 1694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostlund, C.; Sullivan, T.; Stewart, C.L.; Worman, H.J. Dependence of diffusional mobility of integral inner nuclear membrane proteins on A-type lamins. Biochemistry 2006, 45, 1374–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Yeo, L.S.; Vidal, C.; McCorquodale, T.; Herrmann, M.; Fatkin, D.; Duque, G. Decreased bone formation and osteopenia in lamin a/c-deficient mice. PLoS ONE 2011, 6, e19313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heessen, S.; Fornerod, M. The inner nuclear envelope as a transcription factor resting place. EMBO Rep. 2007, 8, 914–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capanni, C.; Mattioli, E.; Columbaro, M.; Lucarelli, E.; Parnaik, V.K.; Novelli, G.; Wehnert, M.; Cenni, V.; Maraldi, N.M.; Squarzoni, S.; et al. Altered pre-lamin A processing is a common mechanism leading to lipodystrophy. Hum. Mol. Genet. 2005, 14, 1489–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz de Eguino, G.; Infante, A.; Schlangen, K.; Aransay, A.M.; Fullaondo, A.; Soriano, M.; Garcia-Verdugo, J.M.; Martin, A.G.; Rodriguez, C.I. Sp1 transcription factor interaction with accumulated prelamin a impairs adipose lineage differentiation in human mesenchymal stem cells: Essential role of sp1 in the integrity of lipid vesicles. Stem Cells Transl. Med. 2012, 1, 309–321. [Google Scholar] [CrossRef]
- Infante, A.; Gago, A.; de Eguino, G.R.; Calvo-Fernandez, T.; Gomez-Vallejo, V.; Llop, J.; Schlangen, K.; Fullaondo, A.; Aransay, A.M.; Martin, A.; et al. Prelamin A accumulation and stress conditions induce impaired Oct-1 activity and autophagy in prematurely aged human mesenchymal stem cell. Aging 2014, 6, 264–280. [Google Scholar] [CrossRef] [Green Version]
- Infante, A.; Rodriguez, C.I. Pathologically Relevant Prelamin A Interactions with Transcription Factors. Methods Enzymol. 2016, 569, 485–501. [Google Scholar]
- Casciaro, F.; Beretti, F.; Zavatti, M.; McCubrey, J.A.; Ratti, S.; Marmiroli, S.; Follo, M.Y.; Maraldi, T. Nuclear Nox4 interaction with prelamin A is associated with nuclear redox control of stem cell aging. Aging 2018, 10, 2911–2934. [Google Scholar] [CrossRef]
- Zhuang, H.; Zhang, X.; Zhu, C.; Tang, X.; Yu, F.; Shang, G.W.; Cai, X. Molecular Mechanisms of PPAR-gamma Governing MSC Osteogenic and Adipogenic Differentiation. Curr. Stem Cell Res. Ther. 2016, 11, 255–264. [Google Scholar] [CrossRef]
- Moerman, E.J.; Teng, K.; Lipschitz, D.A.; Lecka-Czernik, B. Aging activates adipogenic and suppresses osteogenic programs in mesenchymal marrow stroma/stem cells: The role of PPAR-gamma2 transcription factor and TGF-beta/BMP signaling pathways. Aging Cell 2004, 3, 379–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, S.M.; Guo, Y.; Wang, Q.; Xu, Y.; Wang, M.; Chen, H.N.; Shen, W.M. Over-expression of PPAR-gamma2 gene enhances the adipogenic differentiation of hemangioma-derived mesenchymal stem cells in vitro and in vivo. Oncotarget 2017, 8, 115817–115828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boguslavsky, R.L.; Stewart, C.L.; Worman, H.J. Nuclear lamin A inhibits adipocyte differentiation: Implications for Dunnigan-type familial partial lipodystrophy. Hum. Mol. Genet. 2006, 15, 653–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
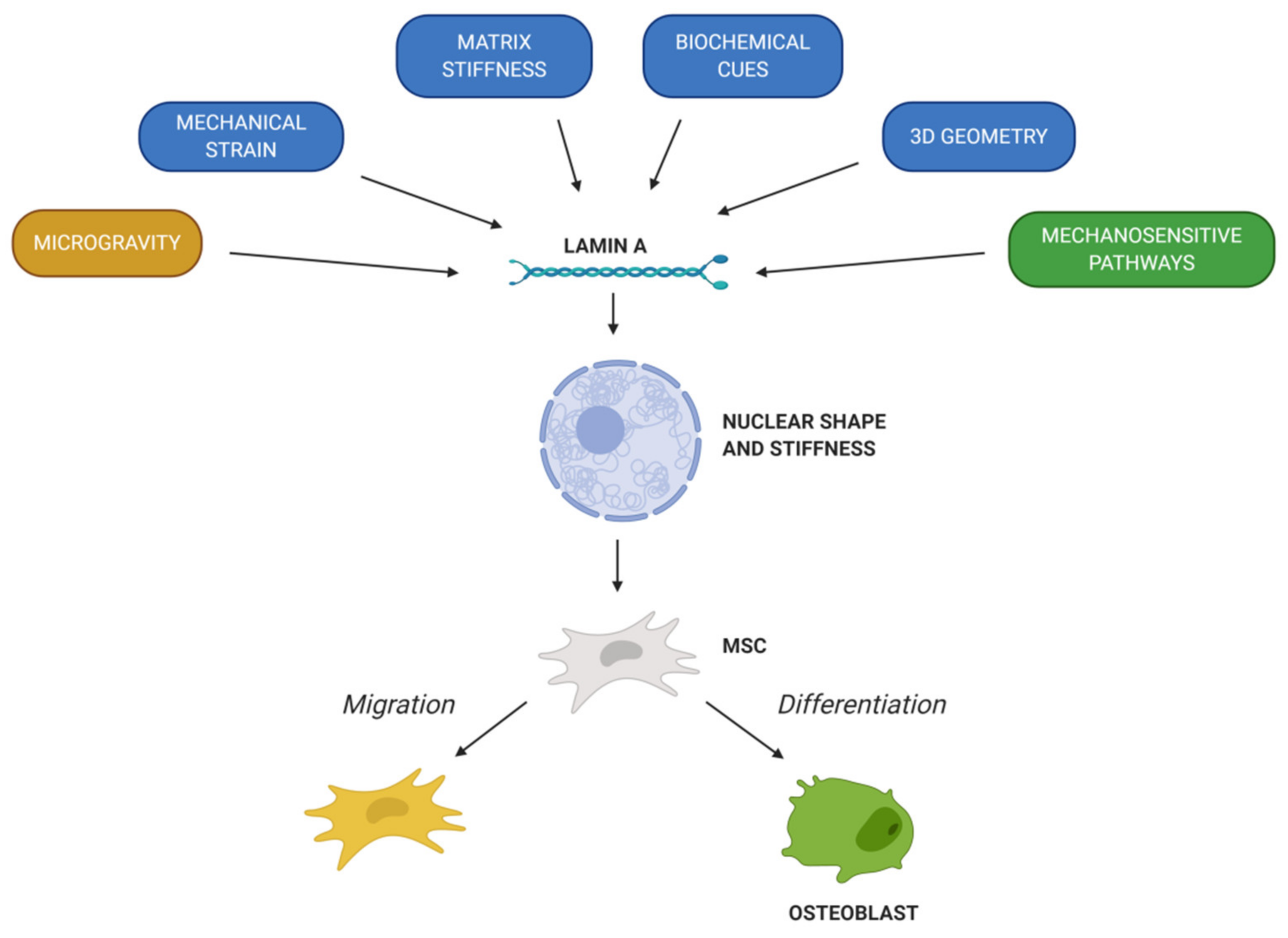
- Wang, Y.K.; Chen, C.S. Cell adhesion and mechanical stimulation in the regulation of mesenchymal stem cell differentiation. J. Cell. Mol. Med. 2013, 17, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Zhang, Y.; Jing, D.; Shen, Y.; Tang, G.; Huang, S.; Zhao, Z. Mechanobiology of mesenchymal stem cells: Perspective into mechanical induction of MSC fate. Acta Biomater. 2015, 20, 1–9. [Google Scholar] [CrossRef]
- Uzer, G.; Rubin, C.T.; Rubin, J. Cell Mechanosensitivity is Enabled by the LINC Nuclear Complex. Curr. Mol. Biol. Rep. 2016, 2, 36–47. [Google Scholar] [CrossRef] [Green Version]
- McBeath, R.; Pirone, D.M.; Nelson, C.M.; Bhadriraju, K.; Chen, C.S. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev. Cell 2004, 6, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Bacil, E.D.; Mazzardo Junior, O.; Rech, C.R.; Legnani, R.F.; de Campos, W. Physical activity and biological maturation: A systematic review. Rev. Paul. Pediatr. 2015, 33, 114–121. [Google Scholar] [CrossRef] [Green Version]
- Ho, C.Y.; Lammerding, J. Lamins at a glance. J. Cell Sci. 2012, 125 Pt 9, 2087–2093. [Google Scholar] [CrossRef] [Green Version]
- Constantinescu, D.; Gray, H.L.; Sammak, P.J.; Schatten, G.P.; Csoka, A.B. Lamin A/C expression is a marker of mouse and human embryonic stem cell differentiation. Stem Cells 2006, 24, 177–185. [Google Scholar] [CrossRef]
- Fedorchak, G.R.; Kaminski, A.; Lammerding, J. Cellular mechanosensing: Getting to the nucleus of it all. Prog. Biophys. Mol. Biol. 2014, 115, 76–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, L.; Cho, S.; Discher, D.E. Mechanosensing of matrix by stem cells: From matrix heterogeneity, contractility, and the nucleus in pore-migration to cardiogenesis and muscle stem cells in vivo. Semin. Cell Dev. Biol. 2017, 71, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Chi, G.; Li, P.; Lv, S.; Xu, J.; Xu, Z.; Xia, Y.; Tan, Y.; Xu, J.; Li, L.; et al. Effects of Matrix Stiffness on the Morphology, Adhesion, Proliferation and Osteogenic Differentiation of Mesenchymal Stem Cells. Int. J. Med. Sci. 2018, 15, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Miroshnikova, Y.A.; Nava, M.M.; Wickstrom, S.A. Emerging roles of mechanical forces in chromatin regulation. J. Cell Sci. 2017, 130, 2243–2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, R.P.; Finan, J.D.; Guilak, F.; Lee, D.A. Mechanical regulation of nuclear structure and function. Annu. Rev. Biomed. Eng. 2012, 14, 431–455. [Google Scholar] [CrossRef] [Green Version]
- Rubin, J.; Styner, M.; Uzer, G. Physical Signals May Affect Mesenchymal Stem Cell Differentiation via Epigenetic Controls. Exerc. Sport Sci. Rev. 2018, 46, 42–47. [Google Scholar] [CrossRef]
- Cho, S.; Vashisth, M.; Abbas, A.; Majkut, S.; Vogel, K.; Xia, Y.; Ivanovska, I.L.; Irianto, J.; Tewari, M.; Zhu, K.; et al. Mechanosensing by the Lamina Protects against Nuclear Rupture, DNA Damage, and Cell-Cycle Arrest. Dev. Cell 2019, 49, 920–935. [Google Scholar] [CrossRef]
- Sen, B.; Xie, Z.; Case, N.; Ma, M.; Rubin, C.; Rubin, J. Mechanical strain inhibits adipogenesis in mesenchymal stem cells by stimulating a durable beta-catenin signal. Endocrinology 2008, 149, 6065–6075. [Google Scholar] [CrossRef] [Green Version]
- Styner, M.; Meyer, M.B.; Galior, K.; Case, N.; Xie, Z.; Sen, B.; Thompson, W.R.; Pike, J.W.; Rubin, J. Mechanical strain downregulates C/EBPbeta in MSC and decreases endoplasmic reticulum stress. PLoS ONE 2012, 7, e51613. [Google Scholar] [CrossRef]
- Li, R.; Liang, L.; Dou, Y.; Huang, Z.; Mo, H.; Wang, Y.; Yu, B. Mechanical strain regulates osteogenic and adipogenic differentiation of bone marrow mesenchymal stem cells. BioMed Res. Int. 2015, 2015, 873251. [Google Scholar] [CrossRef]
- Swift, J.; Discher, D.E. The nuclear lamina is mechano-responsive to ECM elasticity in mature tissue. J. Cell Sci. 2014, 127 Pt 14, 3005–3015. [Google Scholar] [CrossRef] [Green Version]
- Naito, M.; Omoteyama, K.; Mikami, Y.; Takagi, M.; Takahashi, T. Suppression of lamin A/C by short hairpin RNAs promotes adipocyte lineage commitment in mesenchymal progenitor cell line, ROB-C26. Histochem. Cell Biol. 2012, 137, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Buxboim, A.; Irianto, J.; Swift, J.; Athirasala, A.; Shin, J.W.; Rehfeldt, F.; Discher, D.E. Coordinated increase of nuclear tension and lamin-A with matrix stiffness outcompetes lamin-B receptor that favors soft tissue phenotypes. Mol. Biol. Cell 2017, 28, 3333–3348. [Google Scholar] [CrossRef] [PubMed]
- Sastry, S.K.; Burridge, K. Focal adhesions: A nexus for intracellular signaling and cytoskeletal dynamics. Exp. Cell Res. 2000, 261, 25–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Yang, Y.; Keyimu, R.; Hao, J.; Zhao, Z.; Ye, R. The role of lamin A/C in mesenchymal stem cell differentiation. J. Physiol. Biochem. 2019, 75, 11–18. [Google Scholar] [CrossRef]
- Gudas, L.J.; Wagner, J.A. Retinoids regulate stem cell differentiation. J. Cell. Physiol. 2011, 226, 322–330. [Google Scholar] [CrossRef] [Green Version]
- Roa, L.A.; Bloemen, M.; Carels, C.E.L.; Wagener, F.; Von den Hoff, J.W. Retinoic acid disrupts osteogenesis in pre-osteoblasts by down-regulating WNT signaling. Int. J. Biochem. Cell Biol. 2019, 116, 105597. [Google Scholar] [CrossRef]
- Ivanovska, I.L.; Swift, J.; Spinler, K.; Dingal, D.; Cho, S.; Discher, D.E. Cross-linked matrix rigidity and soluble retinoids synergize in nuclear lamina regulation of stem cell differentiation. Mol. Biol. Cell 2017, 28, 2010–2022. [Google Scholar] [CrossRef]
- Dupont, S. Role of YAP/TAZ in cell-matrix adhesion-mediated signalling and mechanotransduction. Exp. Cell Res. 2016, 343, 42–53. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Lorthongpanich, C.; Thumanu, K.; Tangkiettrakul, K.; Jiamvoraphong, N.; Laowtammathron, C.; Damkham, N.; Yaowalak, U.P.; Issaragrisil, S. YAP as a key regulator of adipo-osteogenic differentiation in human MSCs. Stem Cell Res. Ther. 2019, 10, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Driscoll, T.P.; Cosgrove, B.D.; Heo, S.J.; Shurden, Z.E.; Mauck, R.L. Cytoskeletal to Nuclear Strain Transfer Regulates YAP Signaling in Mesenchymal Stem Cells. Biophys. J. 2015, 108, 2783–2793. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, A.T.; Ziaei, S.; Ehret, C.; Duchemin, H.; Mamchaoui, K.; Bigot, A.; Mayer, M.; Quijano-Roy, S.; Desguerre, I.; Laine, J.; et al. Cellular microenvironments reveal defective mechanosensing responses and elevated YAP signaling in LMNA-mutated muscle precursors. J. Cell Sci. 2014, 127 Pt 13, 2873–2884. [Google Scholar] [CrossRef] [Green Version]
- Baarlink, C.; Wang, H.; Grosse, R. Nuclear actin network assembly by formins regulates the SRF coactivator MAL. Science 2013, 340, 864–867. [Google Scholar] [CrossRef] [PubMed]
- Nobusue, H.; Onishi, N.; Shimizu, T.; Sugihara, E.; Oki, Y.; Sumikawa, Y.; Chiyoda, T.; Akashi, K.; Saya, H.; Kano, K. Regulation of MKL1 via actin cytoskeleton dynamics drives adipocyte differentiation. Nat. Commun. 2014, 5, 3368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenwald, M.; Efthymiou, V.; Opitz, L.; Wolfrum, C. SRF and MKL1 Independently Inhibit Brown Adipogenesis. PLoS ONE 2017, 12, e0170643. [Google Scholar] [CrossRef] [Green Version]
- Simon, D.N.; Zastrow, M.S.; Wilson, K.L. Direct actin binding to A- and B-type lamin tails and actin filament bundling by the lamin A tail. Nucleus 2010, 1, 264–272. [Google Scholar] [CrossRef]
- Ho, C.Y.; Jaalouk, D.E.; Vartiainen, M.K.; Lammerding, J. Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature 2013, 497, 507–511. [Google Scholar] [CrossRef] [Green Version]
- Buxboim, A.; Swift, J.; Irianto, J.; Spinler, K.R.; Dingal, P.C.; Athirasala, A.; Kao, Y.R.; Cho, S.; Harada, T.; Shin, J.W.; et al. Matrix elasticity regulates lamin-A,C phosphorylation and turnover with feedback to actomyosin. Curr. Biol. 2014, 24, 1909–1917. [Google Scholar] [CrossRef]
- Tong, J.; Li, W.; Vidal, C.; Yeo, L.S.; Fatkin, D.; Duque, G. Lamin A/C deficiency is associated with fat infiltration of muscle and bone. Mech. Ageing Dev. 2011, 132, 552–559. [Google Scholar] [CrossRef]
- Duque, G.; Rivas, D. Age-related changes in lamin A/C expression in the osteoarticular system: Laminopathies as a potential new aging mechanism. Mech. Ageing Dev. 2006, 127, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Infante, A.; Rodriguez, C.I. Osteogenesis and aging: Lessons from mesenchymal stem cells. Stem Cell Res. Ther. 2018, 9, 244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunawardene, P.; Bermeo, S.; Vidal, C.; Al-Saedi, A.; Chung, P.; Boersma, D.; Phu, S.; Pokorski, I.; Suriyaarachchi, P.; Demontiero, O.; et al. Association Between Circulating Osteogenic Progenitor Cells and Disability and Frailty in Older Persons: The Nepean Osteoporosis and Frailty Study. J. Gerontol. Ser. A Biomed. Sci. Med Sci. 2016, 71, 1124–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feehan, J.; Nurgali, K.; Apostolopoulos, V.; Al Saedi, A.; Duque, G. Circulating osteogenic precursor cells: Building bone from blood. EBioMedicine 2019, 39, 603–611. [Google Scholar] [CrossRef] [Green Version]
- Al Saedi, A.; Gunawardene, P.; Bermeo, S.; Vogrin, S.; Boersma, D.; Phu, S.; Singh, L.; Suriyaarachchi, P.; Duque, G. Lamin A expression in circulating osteoprogenitors as a potential biomarker for frailty: The Nepean Osteoporosis and Frailty (NOF) Study. Exp. Gerontol. 2018, 102, 69–75. [Google Scholar] [CrossRef]
- Golpanian, S.; DiFede, D.L.; Khan, A.; Schulman, I.H.; Landin, A.M.; Tompkins, B.A.; Heldman, A.W.; Miki, R.; Goldstein, B.J.; Mushtaq, M.; et al. Allogeneic Human Mesenchymal Stem Cell Infusions for Aging Frailty. J. Gerontol. Ser. A 2017, 72, 1505–1512. [Google Scholar] [CrossRef]
- Tompkins, B.A.; DiFede, D.L.; Khan, A.; Landin, A.M.; Schulman, I.H.; Pujol, M.V.; Heldman, A.W.; Miki, R.; Goldschmidt-Clermont, P.J.; Goldstein, B.J.; et al. Allogeneic Mesenchymal Stem Cells Ameliorate Aging Frailty: A Phase II Randomized, Double-Blind, Placebo-Controlled Clinical Trial. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2017, 72, 1513–1522. [Google Scholar] [CrossRef]
- Ho, R.; Hegele, R.A. Complex effects of laminopathy mutations on nuclear structure and function. Clin. Genet. 2019, 95, 199–209. [Google Scholar] [CrossRef]
- Capell, B.C.; Collins, F.S. Human laminopathies: Nuclei gone genetically awry. Nat. Rev. Genet. 2006, 7, 940–952. [Google Scholar] [CrossRef]
- Merideth, M.A.; Gordon, L.B.; Clauss, S.; Sachdev, V.; Smith, A.C.; Perry, M.B.; Brewer, C.C.; Zalewski, C.; Kim, H.J.; Solomon, B.; et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N. Engl. J. Med. 2008, 358, 592–604. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Sandre-Giovannoli, A.; Bernard, R.; Cau, P.; Navarro, C.; Amiel, J.; Boccaccio, I.; Lyonnet, S.; Stewart, C.L.; Munnich, A.; Le Merrer, M.; et al. Lamin a truncation in Hutchinson-Gilford progeria. Science 2003, 300, 2055. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.M.; Gordon, L.B.; Snyder, B.D.; Nazarian, A.; Quinn, N.; Huh, S.; Giobbie-Hurder, A.; Neuberg, D.; Cleveland, R.; Kleinman, M.; et al. Hutchinson-Gilford progeria is a skeletal dysplasia. J. Bone Min. Res. 2011, 26, 1670–1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Lian, Q.; Zhu, G.; Zhou, F.; Sui, L.; Tan, C.; Mutalif, R.A.; Navasankari, R.; Zhang, Y.; Tse, H.F.; et al. A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell 2011, 8, 31–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scaffidi, P.; Misteli, T. Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat. Cell Biol. 2008, 10, 452–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blondel, S.; Egesipe, A.L.; Picardi, P.; Jaskowiak, A.L.; Notarnicola, M.; Ragot, J.; Tournois, J.; Le Corf, A.; Brinon, B.; Poydenot, P.; et al. Drug screening on Hutchinson Gilford progeria pluripotent stem cells reveals aminopyrimidines as new modulators of farnesylation. Cell Death Dis. 2016, 7, e2105. [Google Scholar] [CrossRef] [Green Version]
- Lo Cicero, A.; Jaskowiak, A.L.; Egesipe, A.L.; Tournois, J.; Brinon, B.; Pitrez, P.R.; Ferreira, L.; de Sandre-Giovannoli, A.; Levy, N.; Nissan, X. A High Throughput Phenotypic Screening reveals compounds that counteract premature osteogenic differentiation of HGPS iPS-derived mesenchymal stem cells. Sci. Rep. 2016, 6, 34798. [Google Scholar] [CrossRef]
- Bergo, M.O.; Gavino, B.; Ross, J.; Schmidt, W.K.; Hong, C.; Kendall, L.V.; Mohr, A.; Meta, M.; Genant, H.; Jiang, Y.; et al. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc. Natl. Acad. Sci. USA 2002, 99, 13049–13054. [Google Scholar] [CrossRef] [Green Version]
- Infante, A.; Rodriguez, C.I. Secretome analysis of in vitro aged human mesenchymal stem cells reveals IGFBP7 as a putative factor for promoting osteogenesis. Sci. Rep. 2018, 8, 4632. [Google Scholar] [CrossRef] [Green Version]
- Cenni, V.; D’Apice, M.R.; Garagnani, P.; Columbaro, M.; Novelli, G.; Franceschi, C.; Lattanzi, G. Mandibuloacral dysplasia: A premature ageing disease with aspects of physiological ageing. Ageing Res. Rev. 2018, 42, 1–13. [Google Scholar] [CrossRef]
- Novelli, G.; Muchir, A.; Sangiuolo, F.; Helbling-Leclerc, A.; D’Apice, M.R.; Massart, C.; Capon, F.; Sbraccia, P.; Federici, M.; Lauro, R.; et al. Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am. J. Hum. Genet. 2002, 71, 426–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avnet, S.; Pallotta, R.; Perut, F.; Baldini, N.; Pittis, M.G.; Saponari, A.; Lucarelli, E.; Dozza, B.; Greggi, T.; Maraldi, N.M.; et al. Osteoblasts from a mandibuloacral dysplasia patient induce human blood precursors to differentiate into active osteoclasts. Biochim. Biophys. Acta 2011, 1812, 711–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renard, D.; Fourcade, G.; Milhaud, D.; Bessis, D.; Esteves-Vieira, V.; Boyer, A.; Roll, P.; Bourgeois, P.; Levy, N.; De Sandre-Giovannoli, A. Novel LMNA mutation in atypical Werner syndrome presenting with ischemic disease. Stroke 2009, 40, e11–e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motegi, S.; Yokoyama, Y.; Uchiyama, A.; Ogino, S.; Takeuchi, Y.; Yamada, K.; Hattori, T.; Hashizume, H.; Ishikawa, Y.; Goto, M.; et al. First Japanese case of atypical progeroid syndrome/atypical Werner syndrome with heterozygous LMNA mutation. J. Dermatol. 2014, 41, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Jiajue, R.; Feng, K.; Wang, R.; Xia, W. Recurrent Femoral Fractures in a Boy with an Atypical Progeroid Syndrome: A Case Report. Calcif. Tissue Int. 2020, 106, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Subramanyam, L.; Agarwal, A.K.; Simha, V.; Levine, B.; D’Apice, M.R.; Novelli, G.; Crow, Y. Atypical progeroid syndrome due to heterozygous missense LMNA mutations. J. Clin. Endocrinol. Metab. 2009, 94, 4971–4983. [Google Scholar] [CrossRef]
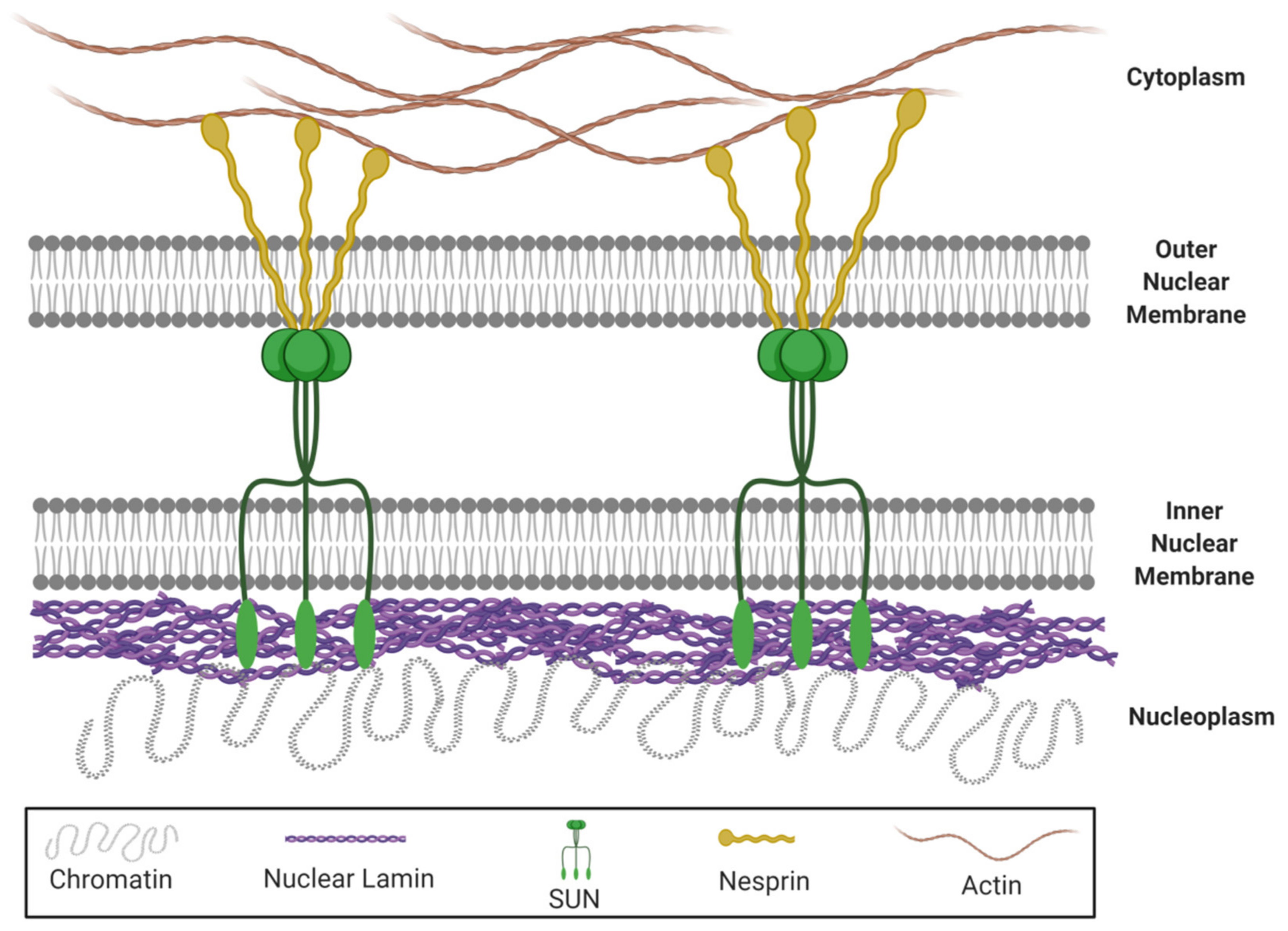


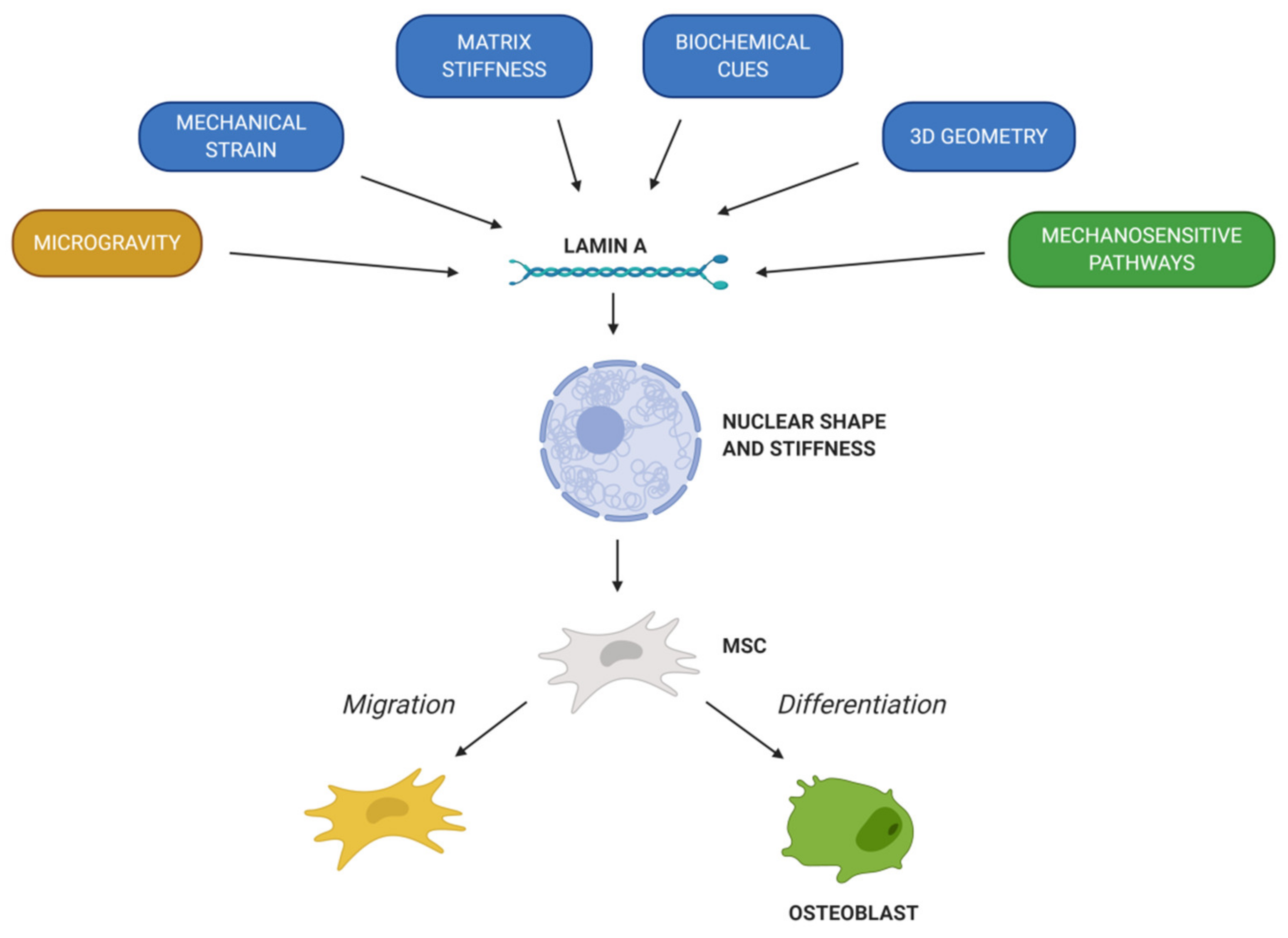
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alcorta-Sevillano, N.; Macías, I.; Rodríguez, C.I.; Infante, A. Crucial Role of Lamin A/C in the Migration and Differentiation of MSCs in Bone. Cells 2020, 9, 1330. https://doi.org/10.3390/cells9061330
Alcorta-Sevillano N, Macías I, Rodríguez CI, Infante A. Crucial Role of Lamin A/C in the Migration and Differentiation of MSCs in Bone. Cells. 2020; 9(6):1330. https://doi.org/10.3390/cells9061330
Chicago/Turabian StyleAlcorta-Sevillano, Natividad, Iratxe Macías, Clara I. Rodríguez, and Arantza Infante. 2020. "Crucial Role of Lamin A/C in the Migration and Differentiation of MSCs in Bone" Cells 9, no. 6: 1330. https://doi.org/10.3390/cells9061330
APA StyleAlcorta-Sevillano, N., Macías, I., Rodríguez, C. I., & Infante, A. (2020). Crucial Role of Lamin A/C in the Migration and Differentiation of MSCs in Bone. Cells, 9(6), 1330. https://doi.org/10.3390/cells9061330