1. Introduction
Being sessile organisms, plants cannot avoid being exposed to stressful conditions. They constantly fight with stressful factors such as high or low temperature, drought, soil salinity, and flooding. Plants have evolved a set of stress tolerance mechanisms, which are different processes involving physiological and biochemical changes that result in adaptive or morphological changes. The study of the mechanisms of plants adaptation to adverse conditions, as well as their response to stresses, is of great interest in the selection of stress resistant varieties [
1,
2].
The response of plants to stress factors is complex, both in terms of the physiological and molecular systems involved [
3,
4]. For example, a turgor pressure in plant cells decreases under drought stress, and pH is changed as well as cell size. The pressure drop is captured by receptor kinases and they activate the abscisic acid signaling which regulate a series of effector genes [
5]. This leads to a physiological reaction of the plant: stomata closure and osmoprotectant synthesis, which ensures the adaptation of the plant to stressful conditions. As a result of the heat stress action, the activation of a series of regulatory pathways occurs: kinase cascades, sumoylation, protein–protein interactions, Ca
2+, etc. They eventually activate heat shock transcription factors [
6,
7], which leads to the expression of chaperones and enzymes that provide acclimation, the primary adaptation to stress. Thus, the response to stress has several stages involving different genetic components. These components are common and include stress sensing, hormonal signal transduction [
8], specific transcription factors [
7], and protein kinase cascades [
9], leading to phenotypic and physiological changes in plant [
5,
10]. All these multilevel processes can be described using gene networks in which many genes are involved [
11,
12]. Analysis of the structure of stress gene networks reveals hub genes that may be important for the response to stress [
13,
14], thus greatly expediting the progress of discovery and functional characterization of the stress-tolerant genes/QTLs [
15].
The stress response gene networks also change in the process of evolution; therefore, analysis of the systems of interacting genes, taking into account evolutionary patterns and expression data, allows the identification of features of the organization of response systems to stress factors and their most significant components promising for gene prioritization [
16,
17,
18,
19]. One promising approach in this regard is phylostratigraphic analysis. It is of great interest in relation to identification of important stages of genome evolution, where appearance of new genes took place, and to identification of lineage specific genes [
20]. This analysis allows the determination of the time of occurrence of genes, assess their age and correlate these ages with the functional role of genes in an organism tissues [
21]. On the other hand, a close relationship between the age of genes and the level of their expression in the process of embryogenesis (‘hourglass-like pattern’) was shown for both animals [
22] and plants [
23]. Interestingly, an hourglass-like pattern of gene expression by age was also identified in the response of tobacco plants to biotic stress [
24]. One of the interesting tasks is to find functional features of genes that differ in age. In particular, several data suggest that genes associated with fundamental processes in cells usually are older than other genes. For instance, the study [
20] reported that human genes referring to such phylostrata as Cellular organisms and Eukaryota are generally associated with basal cellular functions (metabolic processes, transcription regulation), while the genes originating in the later stages of evolution are associated with the genes of the immune response and reproduction.
Phylostratigraphic analysis is a promising method for analyzing the evolution of gene networks. In addition to the structural features of networks, it allows one to identify important details of their evolution, in particular to locate functional modules in networks [
25]. This approach was used for large-scale analysis of the evolution of
A. thaliana gene co-expression networks [
26]. The authors showed that genes originating in the same evolutionary period tend to be connected, but extremely old and young genes tend to be disconnected.
As it was previously shown, the molecular mechanisms of response to stresses of different types are various and complex, both in the composition of genes involved in response to stress, and in the molecular mechanisms of this response. With this in mind, a more detailed study of phylostratigraphic indices for genes of stress response in plants and their relationship to the structure and functional role of gene networks is of great interest.
In this work, we used the Orthoscape application [
27] to carry out phylostratigraphic analysis of genes of plant stress, including the assessment of the distributions of these genes according to their evolutionary age as well as the reconstruction and structural analysis of gene networks by the example of the network of the heat stress response. Our results show that the substantial fraction of genes associated with various types of abiotic stress is of ancient origin and evolves under strong purifying selection. The interaction networks of genes associated with stress response have modular structure with regulatory component being one of the largest for five of seven stress types. We demonstrated a positive relationship between the number of interactions of gene in the stress gene network and its age. Moreover, genes of the same age tend to be connected in gene networks. We also demonstrated that old stress-related genes usually participate in the response for various types of stress and involved in numerous biological processes unrelated to stress. Our results demonstrate that stress response genes represent one of the most ancient and fundamental molecular systems in plants.
4. Discussion
The study of the principles of evolution of the structure and function of gene networks is one of the most interesting and important tasks in biology [
71]. The results of such analysis not only have theoretical interest, but also predictive power [
72,
73].
Such evolutionary characteristics of genes as phylostratigraphic age [
21] and the index of divergence [
23] reflect the most important properties of genes: the order of their appearance in the genome in relation to other genes and the degree of selection pressure in the process of evolution. These metrics allow us to characterize the dynamics of changes in the function of gene sets, including gene networks. In particular, a comparison of the ages of genes and the annotation of their functions (GO) allows one to explore the relationship between the function of genes/organisms and the age of genes [
20,
74]. As a rule, this is done by searching for associations of genes of different ages with the GO terms [
26]. For example, Domazet-Loso and Tautz showed that genes related to human genetic diseases are significantly overrepresented among the genes that have emerged during the early evolution of the metazoan [
20]. Ruprecht and coauthors [
26] performed Mapman terms phylostrata enrichment analysis for three planta genomes, including
A. thaliana, and showed that significant occurrence of the terms in different phylostrata mostly happens only once, suggesting that new biological features emerged in (or during) distinct evolutionary periods, without a significant addition of new genetic material during later stages of evolution.
In this paper, we selected the genes associated with plant response to different types of stress and analyzed these gene sets, including reconstruction of interaction networks, evaluation of phylostratigraphic age and selection pressure. We compared the evolutionary properties of these sets of genes with those of the complete set of A. thaliana genes and found their significant differences.
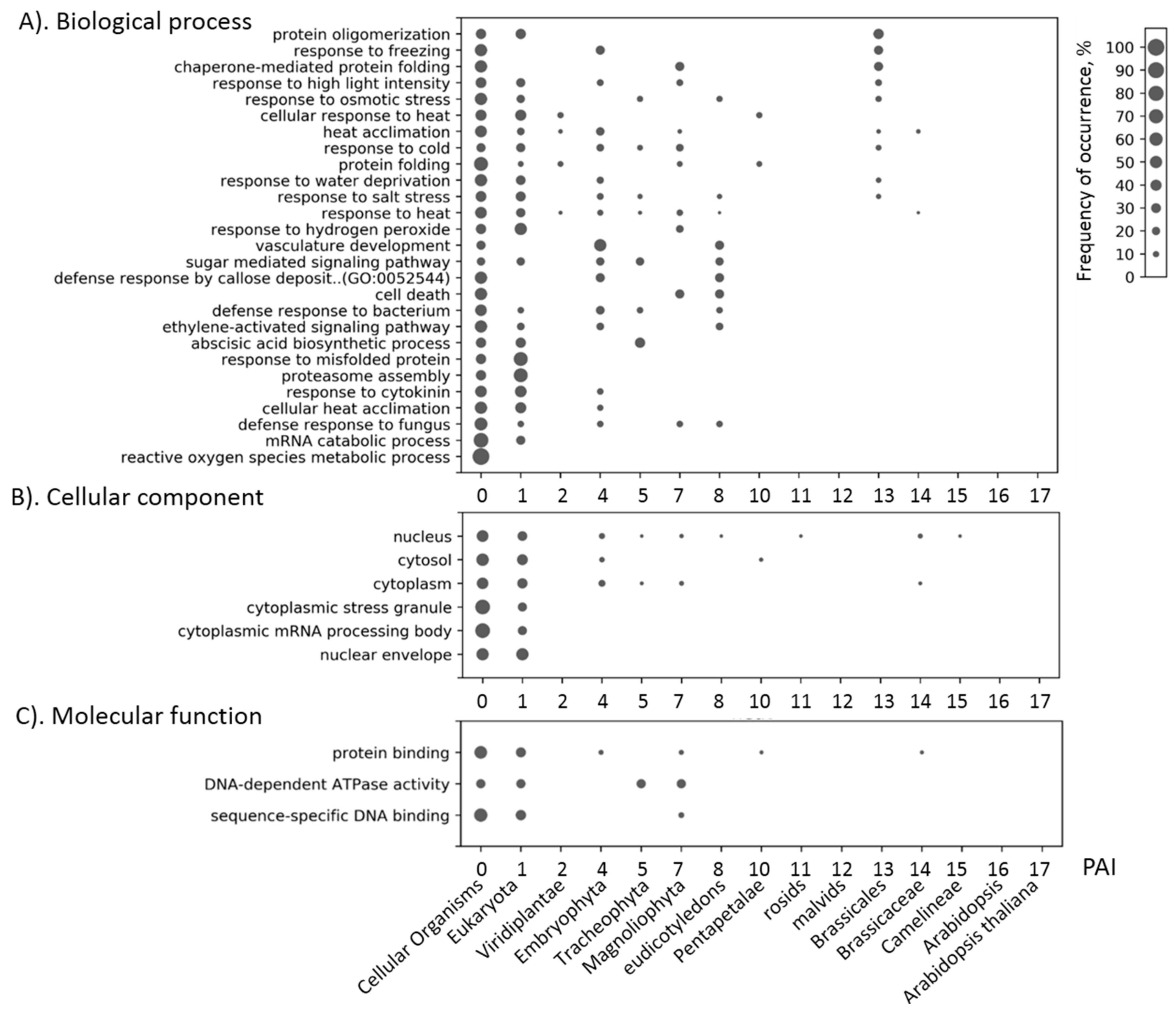
Our results show that the genes associated with the stress response generally contain a large number of ancient genes than would be expected from the distribution of such genes throughout the genome. In addition, stress genes are more conservative than would be expected from the conservatism of the whole set of genes. These data are in line with the concept that the more important genes in terms of function are older and are under strong pressure of stabilizing selection [
75,
76,
77]. Genes with high connectivity in the interaction networks have similar evolutionary properties [
78] and evolve under stabilizing selection [
79,
80,
81,
82,
83]. The response of plants to stress, of course, involves the basic functions of the cell that were formed at the earliest stages of evolution, such as, for example, the system of heat shock proteins forming a noticeable cluster in the heat stress response network (
Figure 4), or a cluster of genes of signaling pathways controlled by stress hormones (
Supplementary file 2,
Figures S4, S6, S8 and S9), where their fraction is significant. In response to stress factors of biotic and abiotic nature in animals and plants, many parallels can be found at the level of physiology [
84], and especially at a cellular level [
85,
86,
87]. It is known that stress response genes are homologous in plants and animals. For instance, stress associated proteins (SAPs) in plants contain A20/AN1 zinc finger domain homologous to proteins from the genomes of diverse organisms including protists, fungi, animals, and plants [
88]. We found these genes among the genes associated with the response to osmotic and water types of stress (SAP5/AT3G12630, A20/AN1-like zinc finger family protein,
Supplementary file 3).
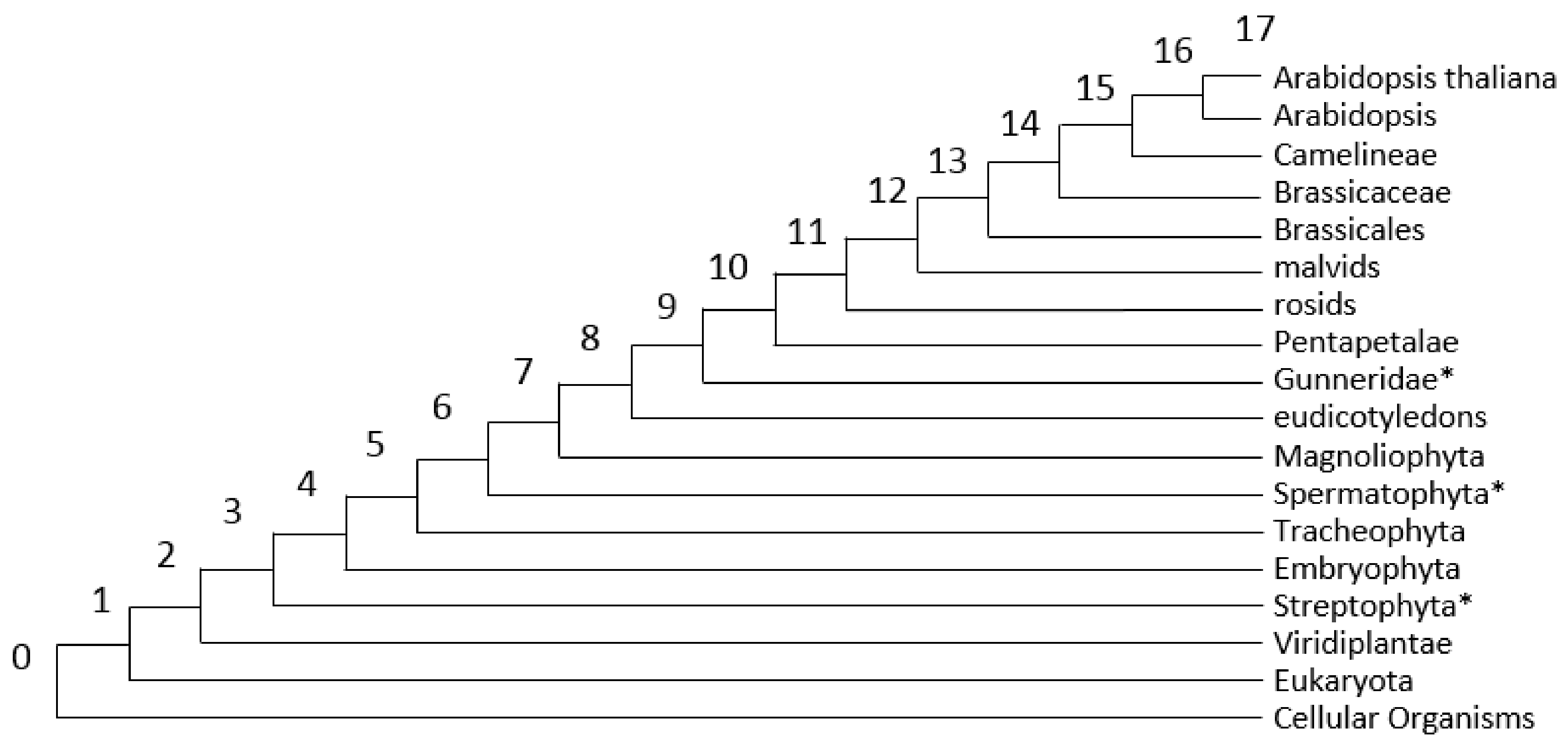
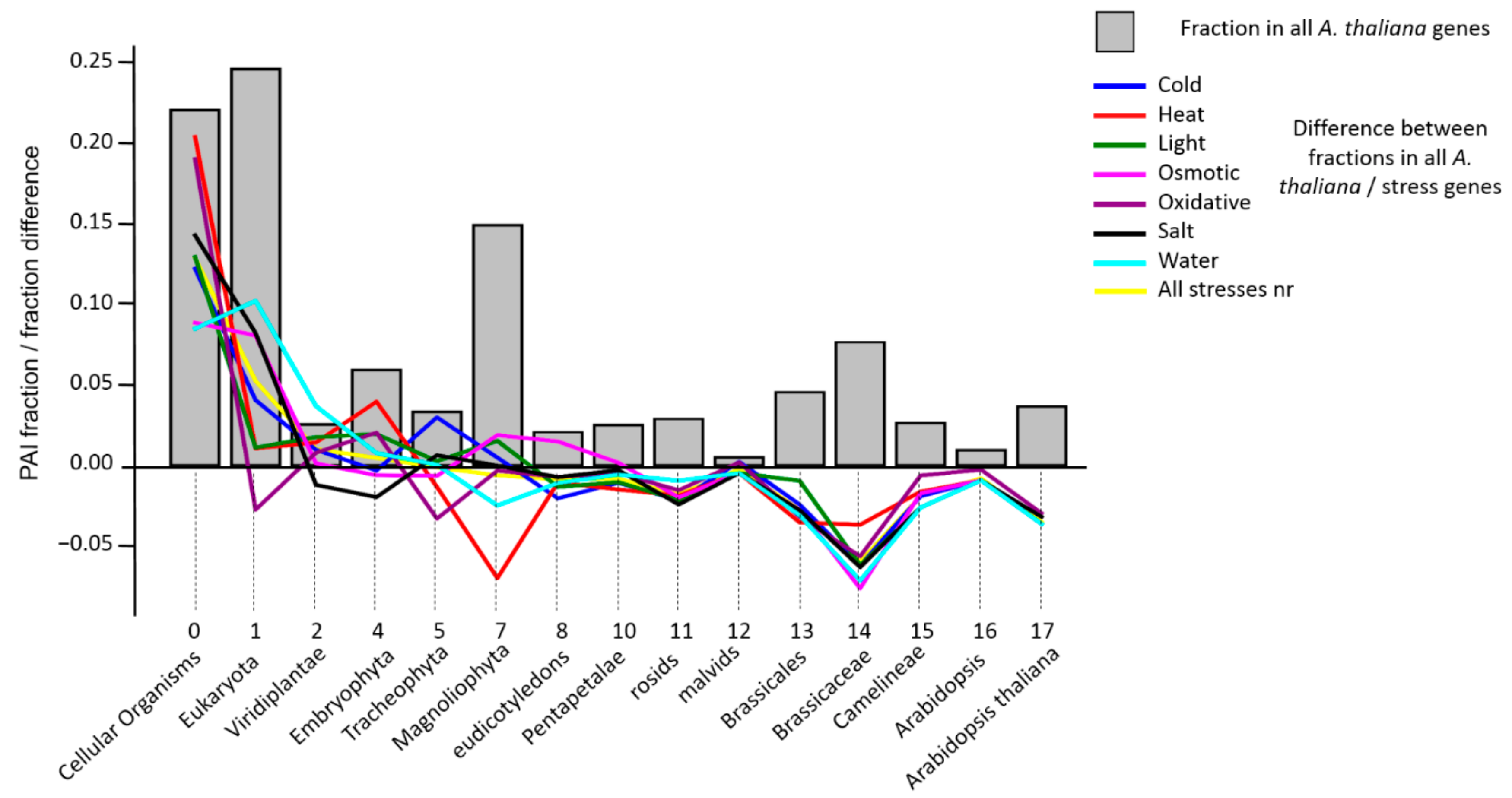
Our analysis involves 15 phylostrata of organisms represented in KEGG database [
33]. The distribution of genes across these phylostrata reveals an interesting feature: the presence of three peaks in the distribution (
Figure 2). The most ancient peak (PAI < 2), contains about a half of all genes. The other peaks correspond to PAI = 7 (Magnoliophyta) and to PAI = 14 (Brassicacea). The position of these peaks does not change from the change in the value of the identity threshold when determining orthologs (see
Supplementary file 2,
Figure S2), although the ratio of gene fractions varies. We can assume that these two peaks correspond to the events of the whole genome duplication in
A. thaliana lineage, an α-duplication that preceded the formation of the Brassecaceae clade [
89,
90,
91,
92] and more ancient γ-duplication, which corresponds to the angiosperms ancestor [
93]. It is known that whole genome duplications are substantial events in organisms’ evolution, leading to the emergence of many novel genes [
94,
95]. The presence of two younger peaks in the PAI distribution for
A. thaliana probably reflects this feature of evolution. It is interesting to note that for γ-duplication, the difference between the fractions of genes of this phylostratum in stress-related genes and in the whole genome of
A. thaliana is close to zero or even positive (except for the genes of heat and water stress). This means that during the diversification of duplicated genes at this stage of evolution, new stress response genes have emerged. For α-duplication (Brassicaceae phylostratum) on the contrary, this difference is negative, and large in absolute value for all stress genes. We can speculate that at this stage, after the duplication, there was a sharp loss of duplicated stress genes (relative to other types of genes). Apparently, by that time, in general, the systems of response to various stresses in plants were mostly formed and they did not have a need for evolutionary innovations (compared to the increase in innovations in other gene systems). It should be noted; however, that Orthoscape in its current version cannot account for the possible bias in the phylostratigraphic assignment due to these duplications because we did not consider genome synteny when defining orthologous groups. The influence of such bias can be resolved in a future research.
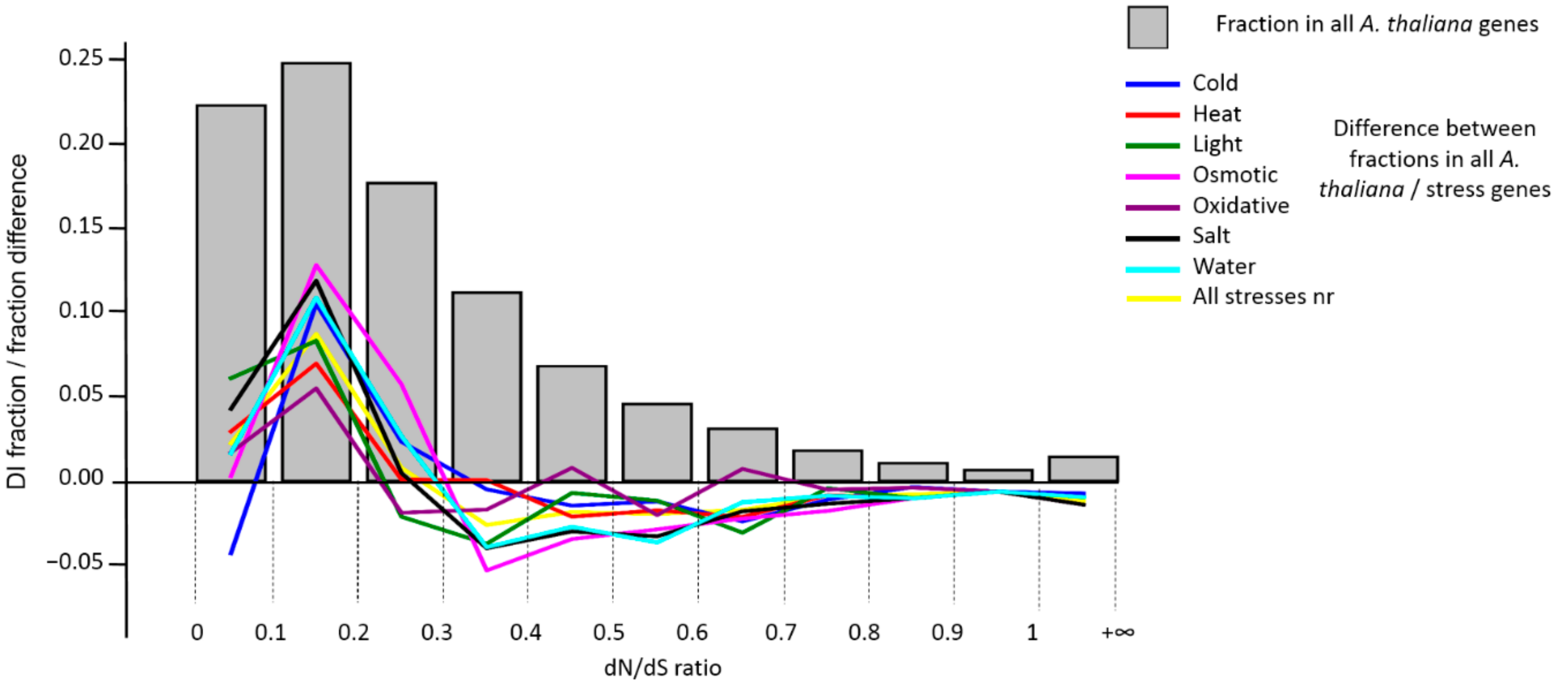
Stress genes are also under a strong pressure of purifying selection, which indicates their absolute importance for the organism (although few of them are absolutely conservative). Among them, very rarely (only two cases) are genes subject to positive Darwinian selection (DI > 1), which is very small compared to the proportion of such genes in the entire genome. This is generally consistent with data from Lei et al., according to which
A. thaliana genes of DI > 1 are enriched in lipid localization, transport and binding, and the endomembrane system (i.e., stress unrelated terms) [
37]. However, it should be noted that the use of the genome pair
A. thaliana vs.
A. lyrata for DI estimates (i.e., Ka/Ks) might provide insight into genes that evolved under different selection regimes only in the most recent past. Of course, in this case, it is hard to expect that any genes performing the basic functions of the organism can be affected by the positive selection. On the other hand, the results show that most stress genes at this stage of evolution are the subject to stabilizing selection, which is quite consistent with the hypothesis of their functional importance and the performance of their basic functions.
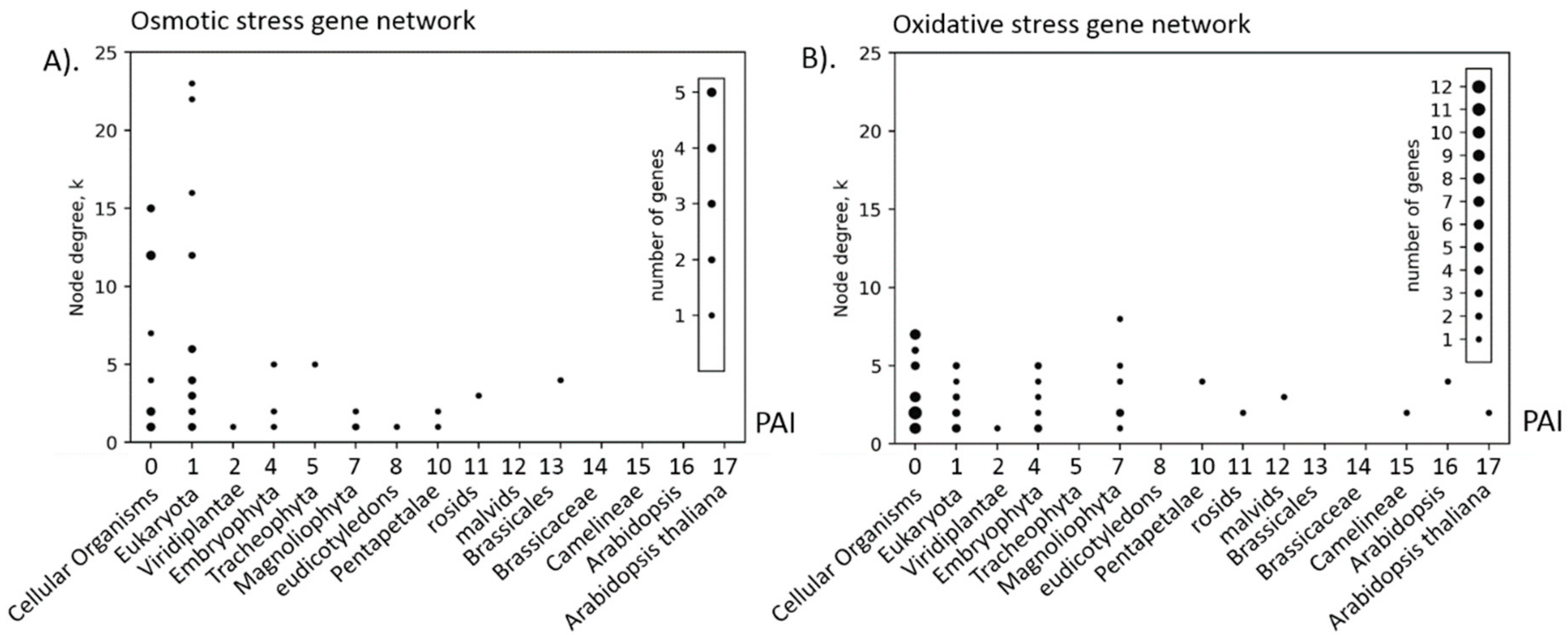
Analysis of the relationship between the structural characteristics of the reconstructed networks showed that for such networks as heat, osmotic and salt stress, the higher is the order of gene interactions in the network, the greater is the age of the gene. However, for other networks, we found no significant correlations. Apparently, these results are affected by the topology of the networks, which turned out to be different. For example, the oxidative stress network does not have a clearly defined large regulatory cluster; the light stress response network does not contain nodes with a large number of connections. It was also shown that a significant part of the interacting pairs of genes have the same age (except for light stress genes). This suggests that interactions in the network are preferable for genes of similar age (or that clusters of genes are generally homogeneous in age of genes). This trend is also indicated by the positive assortativity coefficients of the age of genes. Similar results were obtained in the study of Ruprecht [
26], where it was shown that genes from the same evolutionary period tend to be connected, whereas old and young genes tend to be disconnected. This trend is not, however, a general rule, as confirmed by the structural analysis of the light stress network.
Our results show polyfunctionality of the stress-associated genes in agreement with the current knowledge [
96]. A lot of GO terms, in addition to the stress terms we used, was found among the annotations of our gene lists. This can be explained by the fact that response of plants to stress of any nature affects a large number of molecular processes [
3]. For example, the heat stress leads to the triggering of such processes in plant cells as change in membrane fluidity, increase of the reactive oxygen species (ROS), change in the transport of Ca
+ ions and restructuring of the cytoskeleton, the denaturation of proteins and RNA, changing the structure of chromatin and the expression of miRNAs [
97]. The heat stress activates heat shock proteins, sumoylation systems, chromatin remodeling, dehydration control [
7]. The drought stress activates specific signaling pathways and transcription factors, detoxification enzymes, enzymes of the biosynthesis of osmolytes, the system of transporters and water channels, response to protein denaturation [
98]. In response to the salt stress, genes of photosynthesis and carbon production, cell wall components, water channels, ion transport, ROS protection system, a detoxification system, signaling pathways and specific transcription factors are involved [
99]. It should be noted that the system of response to the osmotic and the oxidative stress themselves are involved in responses to other types of abiotic stress [
100]. Thus, the systems of response to abiotic stresses in plants are closely interconnected. Our analysis of annotations of the stress genes in
A. thaliana indeed has shown that the involvement of some genes in several stress responses is one of the features of stress genes (
Figure S1,
Supplementary file 2).
The presence of common and unique genes can be explained also by the multilevel structure of molecular systems of response to stress [
3,
101]: as a rule, these systems include stress sensors, signal transmission systems (including hormonal response), triggering transcription of stress response genes, molecular response to the occurrence of stress conditions to minimize its consequences. Systems of the first and second level, as well as partly the regulation of genes, are mainly specific for each type of abiotic stress. At the same time, the molecular response to cell stress for different types of stress has many common features: control of reactive oxygen species (ROS), change of ion transport, cell detoxification, control of protein denaturation. In our work, we demonstrated the existence of large regulatory cluster of genes common to cold, osmotic, heat, salt and water-related types of stress, which includes various regulatory, hormone-related and signal transduction genes.
Another possible reason for the generality of genes for different types of stress is that in nature, stress factors often act together, and in the course of evolution plants develop shared responses which are common to individual stresses and stress combinations [
102].
Several ancient genes of stress response were involved in biological processes that occurred at much later stages of evolution compared to the time of occurrence of these genes. The explanation for this may lie in the fact that during the evolution of plants the old genes were intensively involved for the formation of new functions, so they are involved in a new functional context [
103].
Interestingly, our analysis shows that ancient stress genes have many properties in common with so-called multifunctional genes [
104]: they are also highly conservative, involved in several biological processes, tend to form a large number of connections in the structure of gene networks and are involved in the performance of important functions in the life of organisms.
It should be noted that the analysis strongly depends on the results obtained at the stage of the formation of stress gene sets, as well as the reconstruction of gene networks, since it is completely based on these data. We had this in mind and chose rather strict criteria for the selection of genes by the GO terms and for the reconstruction of gene networks by the STRING method. Of course, these data may be incomplete and contain errors. However, a comparison of the composition of stress genes (
Figure S1) demonstrates the commonality of several genes for different types of stress especially noticeable for stresses for which their hormonal control (ABA, ethylene and jasmonates) is known: salt, cold, osmotic, water stresses and to a lesser extent heat one. The structure of gene networks for these stresses also demonstrates the presence of a large cluster represented by genes associated with the perception and transmission of hormone signals (
Figure 4,
Figures S4, S6, S8 and S9,
Supplementary file 2). This is in good agreement with the known role of hormones in regulating the response to abiotic stress [
57,
58,
59]. Gene networks built on the basis of co-expression are definitely more common, if we talk about choosing a strict threshold to establish intergenic associations and can include a large number of genes, in comparison with networks built via STRING. However, the level of gene expression in response to stress strongly depends on the time elapsed after stress exposure, with many side processes that are secondary to the stress response itself, which complicates the interpretation of expression data. Therefore, we decided to use data based not only on co-expression, but also on broader information (GO annotation, which is-based, among other things, on expert data; STRING networks, which include information on co-expression along with protein-protein interactions and other additional information) in the selection of stress genes and reconstruction of their networks.



 and
and 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}