
Insulin-like Growth Factor 1 Signaling in Mammalian Hearing

 , and
, and
Abstract
:1. The Insulin-Like Growth Factor System (IGF System)
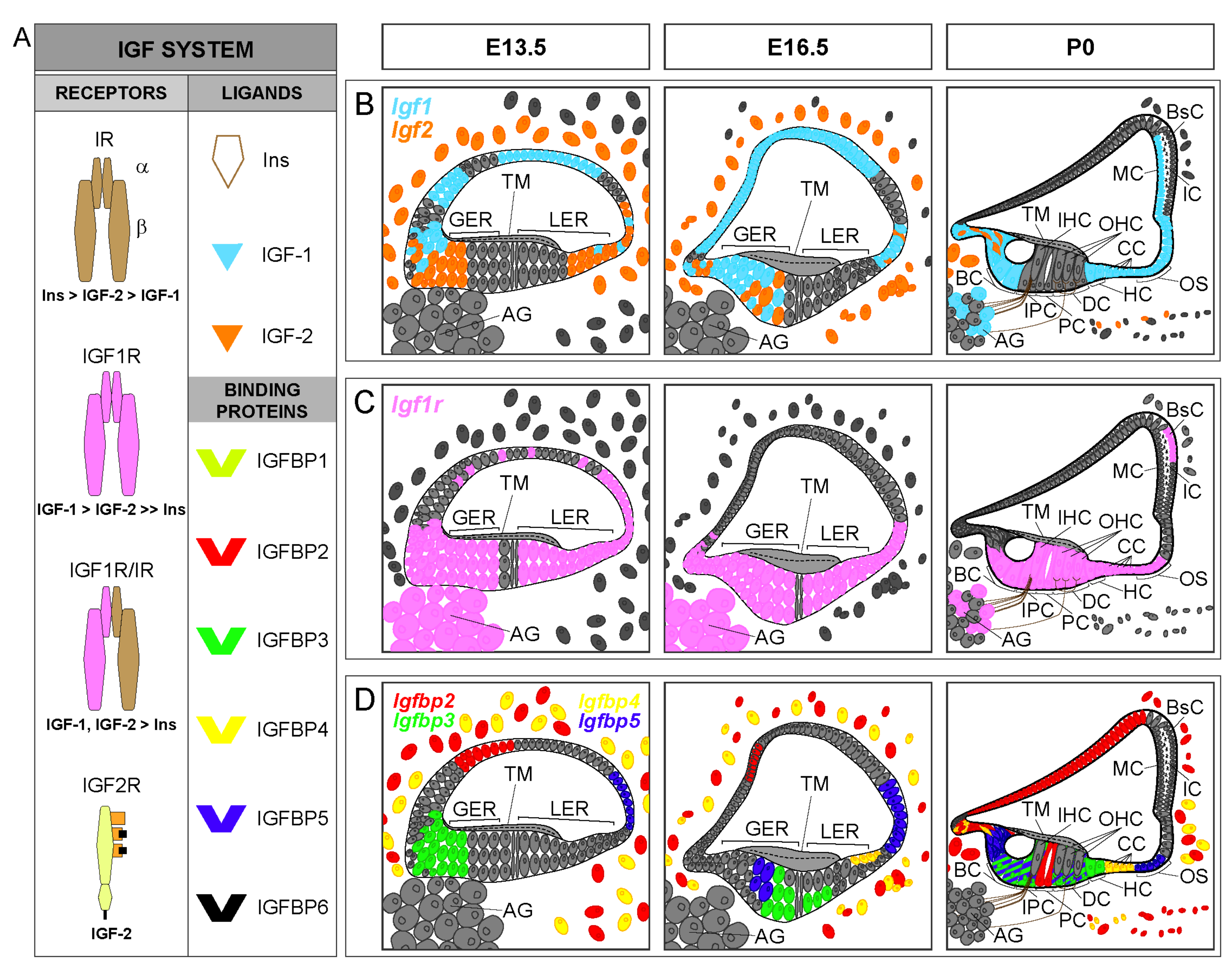
2. IGF-1 Signaling Network

2.1. RAF/MEK/ERK Cascade
2.2. Stress Kinase (p38 and JNK) Cascades
2.3. PI3K-AKT Pathway
3. Animal Models of Hearing Loss Associated with Human Mutations in the IGF1 System
3.1. GH System
3.2. IGF System
3.3. IGF-1 Signaling
4. GH-IGF-1 Axis Human Mutations Associated with Hearing Impairment
4.1. IGF System and Signaling Mutations

{kind=link}
{kind=link}
| Woods et al., 1996 [78] | Walenkamp et al., 2005 [96] | Netchine et al., 2009 [98] | Keselman et al., 2019 [97] | Batey et al., 2014 [99] | Fuqua et al., 2012 [100] | |
|---|---|---|---|---|---|---|
| Mutation type | Deletion Homozygous | Missense mutation Homozygous | Missense mutation Homozygous | Missense mutation Homozygous | Deletion Heterozygous | Splicing mutation Heterozygous |
| Mutational analysis | 181 bp ex. 4–5 | c.274G ˃ A, p.V92M (1) | c.251G > A, p.R84Q (2) | c.332T > C, p.Y108H | 262 kb, entire IGF-1 gene | Splicing excision ex 4 c.402+1G > C, p.N74Rfs*8 |
| Coordinates (GRCh38) | Not mapped | chr12: 102419637 | chr12: 102419660 | chr12: 102419589 | Not mapped | chr12: 102419508 |
| Age & clinical data | ♂ 15.8 yr Pre- and postnatal growth failure Microcephaly Micrognathia Clinodactyly Cognitive delay | ♂ 55 yr Pre- and postnatal growth failure Microcephaly Dysmorphic features Severe cognitive delay Deaf-mutism | ♂ 11 mo–9 yr Pre- and postnatal growth failure Microcephaly Non-dismorphic Clinodactyly Mild cognitive delay | ♂ 3.2–7.8 yr Pre- and postnatal growth failure Microcephaly Dysmorphic features Hyperactive behaviour Developmental delay | ♂ 2.3–8.4 yr Pre- and postnatal growth failure Microcephaly Micrognathia Clinodactyly Cognitive delay | ♂ 8.8 yr Severe postnatal growth failure Normal physical examination Normal cognitive development |
| Consanguinity | Yes | Yes | Yes | Yes | No | No |
| Birth weight (kg) | 1.4 (−3.9 SD) | 1.4 (−3.9 SDS) | 2.3 (−2.4 SDS) | 1.9 (−3.1 SDS) | 2.7 (−1.5 SDS) | 3.0 (−1.5 SDS) |
| Birth length (cm) | 37.8 (−5.4 SD) | 39 (−4.3 SDS) | 44 (−3.7 SDS) | 38 (−6.3 SDS) | 47.6 (−1.2 SDS) | 47 (−0.6 SDS) |
| Growth weight (kg) | 15.8 yr: 23 (−6.5 SD) | ND | 11 mo: 5.3 (−5.0 SDS) 2.8 yr: 7.0 (−7.0 SDS) | 3.2 yr: 6.1 (−5.1 SDS) 7.8 yr: 9.6 (−5.0 SDS) | 2.3 yr: 8.8 (−3.8 SDS) 8.4 yr: 21.9 (−1.5 SDS) | 8.8 yr: 21 (−2.1 SDS) |
| Growth height (cm) | 15.8 yr: 119.1 (−6.9 SD) | 55 yr: 117.8 (−8.5 SDS) | 11 mo: 64 (−3.7 SDS) 2.8 yr: 76 (−4.9 SDS) | 3.2 yr: 74 (−6.2 SDS) 7.8 yr: 90.2 (−6.5 SDS) | 2.3 yr: 77.5 (−3.1 SDS) 8.4 yr: 114.9 (−2.7 SDS) | 8.8 yr: 109 (−4.0 SDS) |
| Auditory function | Severe BHL (15.8 yr) | Severe BHL (55 yr) | Normal hearing (9 yr) | BHL (3.2 yr) | Normal hearing | Normal hearing |
| IGF-1 levels (ng/mL) | Undetectable 15 yr: <3 | Very high 55 yr: 606 (+7.3 SDS) | Low 2.7 yr: 11 (before GH treatment) | Variable 4 yr: 47 (−1.15 SDS) 6.4 yr: 206 (+2.95 SDS) | Low-normal. 2.3 yr: 43.7; 5.3 yr: 58.5 8.4 yr: 100 | Low-normal 9.3 yr: 115 (−2.2 SDS) (before GH treatment) |
| IGFBP-3 levels (mg/L) | Normal 15 yr: 3.3 | Normal 55 yr: 1.98 (+0.1 SDS) | Normal-high (after GH treatment) | Normal-high (−1.58 to +2.31 SDS) | Normal 5.3 yr: 4.3; 8.4 yr: 5.4 | Normal 9.3 yr: 2.4 (−1.2 SDS) (before GH) |
| ALS levels (mg/L) | Normal | High 55 yr: 28.9 (+3.4 SDS) | Normal-high (after GH treatment) | ND | Normal 5.3 yr: 10 | Normal-high 9.3 yr: 13 (before GH) |
| IGF-1 affinity for IGF1R | None | Extremely low 90-fold lower | Partially reduced 3.9-fold lower | Reduced | ND | ND |
4.2. Human Hearing Loss Syndromes and Involvement of GH
4.3. GH-IGF-1 Axis and Therapeutic Potential
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- LeRoith, D.; Holly, J.M.P.; Forbes, B.E. Insulin-like Growth Factors: Ligands, Binding Proteins, and Receptors. Mol. Metab. 2021, 101245. [Google Scholar] [CrossRef]
- Bach, L.A. IGF-Binding Proteins. J. Mol. Endocrinol. 2018, 61, T11–T28. [Google Scholar] [CrossRef] [Green Version]
- Allard, J.B.; Duan, C. IGF-Binding Proteins: Why Do They Exist and Why Are There So Many? Front. Endocrinol. 2018, 9, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguchi, G.M.; Huising, M.O. Integrating the Inputs That Shape Pancreatic Islet Hormone Release. Nat. Metab. 2019, 1, 1189–1201. [Google Scholar] [CrossRef] [Green Version]
- Müller, E.E.; Locatelli, V.; Cocchi, D. Neuroendocrine Control of Growth Hormone Secretion. Physiol. Rev. 1999, 79, 511–607. [Google Scholar] [CrossRef] [PubMed]
- Kelberman, D.; Dattani, M.T. The Role of Transcription Factors Implicated in Anterior Pituitary Development in the Aetiology of Congenital Hypopituitarism. Ann. Med. 2006, 38, 560–577. [Google Scholar] [CrossRef] [PubMed]
- Dehkhoda, F.; Lee, C.M.M.; Medina, J.; Brooks, A.J. The Growth Hormone Receptor: Mechanism of Receptor Activation, Cell Signaling, and Physiological Aspects. Front. Endocrinol. 2018, 9, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Calderon, H.; Rodriguez-de la Rosa, L.; Milo, M.; Pichel, J.G.; Holley, M.; Varela-Nieto, I. RNA Microarray Analysis in Prenatal Mouse Cochlea Reveals Novel IGF-I Target Genes: Implication of MEF2 and FOXM1 Transcription Factors. PLoS ONE 2010, 5, e8699. [Google Scholar] [CrossRef] [Green Version]
- Okano, T.; Xuan, S.; Kelley, M.W. Insulin-like Growth Factor Signaling Regulates the Timing of Sensory Cell Differentiation in the Mouse Cochlea. J. Neurosci. 2011, 31, 18104–18118. [Google Scholar] [CrossRef]
- Camarero, G.; Avendano, C.; Fernandez-Moreno, C.; Villar, A.; Contreras, J.; de Pablo, F.; Pichel, J.G.; Varela-Nieto, I. Delayed Inner Ear Maturation and Neuronal Loss in Postnatal Igf-1-Deficient Mice. J. Neurosci. 2001, 21, 7630–7641. [Google Scholar] [CrossRef] [Green Version]
- Gross, J.; Machulik, A.; Moller, R.; Fuchs, J.; Amarjargal, N.; Ungethüm, U.; Kuban, R.-J.; Szczepek, A.J.; Haupt, H.; Mazurek, B. MRNA Expression of Members of the IGF System in the Organ of Corti, the Modiolus and the Stria Vascularis of Newborn Rats. Growth Factors 2008, 26, 180–191. [Google Scholar] [CrossRef]
- Kolla, L.; Kelly, M.C.; Mann, Z.F.; Anaya-Rocha, A.; Ellis, K.; Lemons, A.; Palermo, A.T.; So, K.S.; Mays, J.C.; Orvis, J.; et al. Characterization of the Development of the Mouse Cochlear Epithelium at the Single Cell Level. Nat. Commun. 2020, 11, 2389. [Google Scholar] [CrossRef]
- De Iriarte Rodríguez, R.; Pulido, S.; Rodríguez-de la Rosa, L.; Magariños, M.; Varela-Nieto, I. Age-Regulated Function of Autophagy in the Mouse Inner Ear. Hear. Res. 2015, 330, 39–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celaya, A.M.; Rodríguez-de la Rosa, L.; Bermúdez-Muñoz, J.M.; Zubeldia, J.M.; Romá-Mateo, C.; Avendaño, C.; Pallardó, F.V.; Varela-Nieto, I. IGF-1 Haploinsufficiency Causes Age-Related Chronic Cochlear Inflammation and Increases Noise-Induced Hearing Loss. Cells 2021, 10, 1686. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chen, L.; Giffen, K.P.; Stringham, S.T.; Li, Y.; Judge, P.D.; Beisel, K.W.; He, D.Z.Z. Cell-Specific Transcriptome Analysis Shows That Adult Pillar and Deiters’ Cells Express Genes Encoding Machinery for Specializations of Cochlear Hair Cells. Front. Mol. Neurosci. 2018, 11, 356. [Google Scholar] [CrossRef] [PubMed]
- Okano, T.; Kelley, M.W. Expression of Insulin-like Growth Factor Binding Proteins during Mouse Cochlear Development. Dev. Dyn. 2013, 242, 1210–1221. [Google Scholar] [CrossRef]
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr. Rev. 2017, 38, 379–431. [Google Scholar] [CrossRef]
- Girnita, L.; Takahashi, S.-I.; Crudden, C.; Fukushima, T.; Worrall, C.; Furuta, H.; Yoshihara, H.; Hakuno, F.; Girnita, A. Chapter Seven—When Phosphorylation Encounters Ubiquitination: A Balanced Perspective on IGF-1R Signaling. Prog. Mol. Biol. Transl. Sci. 2016, 141, 277–311. [Google Scholar] [CrossRef]
- Hakuno, F.; Takahashi, S.-I. IGF1 Receptor Signaling Pathways. J. Mol. Endocrinol. 2018, 61, T69–T86. [Google Scholar] [CrossRef] [Green Version]
- El-Shewy, H.M.; Luttrell, L.M. Insulin-like Growth Factor-2/Mannose-6 Phosphate Receptors. Vitam. Horm. 2009, 80, 667–697. [Google Scholar] [CrossRef]
- Hawkes, C.; Jhamandas, J.H.; Harris, K.H.; Fu, W.; MacDonald, R.G.; Kar, S. Single Transmembrane Domain Insulin-like Growth Factor-II/Mannose-6-Phosphate Receptor Regulates Central Cholinergic Function by Activating a G-Protein-Sensitive, Protein Kinase C-Dependent Pathway. J. Neurosci. 2006, 26, 585–596. [Google Scholar] [CrossRef]
- Agbemenyah, H.Y.; Agis-Balboa, R.C.; Burkhardt, S.; Delalle, I.; Fischer, A. Insulin Growth Factor Binding Protein 7 Is a Novel Target to Treat Dementia. Neurobiol. Dis. 2014, 62, 135–143. [Google Scholar] [CrossRef]
- Girnita, L.; Worrall, C.; Takahashi, S.-I.; Seregard, S.; Girnita, A. Something Old, Something New and Something Borrowed: Emerging Paradigm of Insulin-like Growth Factor Type 1 Receptor (IGF-1R) Signaling Regulation. Cell. Mol. Life Sci. 2014, 71, 2403–2427. [Google Scholar] [CrossRef] [Green Version]
- Murillo-Cuesta, S.; Camarero, G.; González-Rodríguez, A.; De La Rosa, L.R.; Burks, D.J.; Avendaño, C.; Valverde, A.M.; Varela-Nieto, I. Insulin Receptor Substrate 2 (IRS2)-Deficient Mice Show Sensorineural Hearing Loss That Is Delayed by Concomitant Protein Tyrosine Phosphatase 1B (PTP1B) Loss of Function. Mol. Med. Camb. Mass 2012, 18, 260–269. [Google Scholar] [CrossRef] [Green Version]
- DeMambro, V.E.; Kawai, M.; Clemens, T.L.; Fulzele, K.; Maynard, J.A.; Marín de Evsikova, C.; Johnson, K.R.; Canalis, E.; Beamer, W.G.; Rosen, C.J.; et al. A Novel Spontaneous Mutation of Irs1 in Mice Results in Hyperinsulinemia, Reduced Growth, Low Bone Mass and Impaired Adipogenesis. J. Endocrinol. 2010, 204, 241–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baltensperger, K.; Kozma, L.M.; Cherniack, A.D.; Klarlund, J.K.; Chawla, A.; Banerjee, U.; Czech, M.P. Binding of the Ras Activator Son of Sevenless to Insulin Receptor Substrate-1 Signaling Complexes. Science 1993, 260, 1950–1952. [Google Scholar] [CrossRef]
- Lavoie, H.; Gagnon, J.; Therrien, M. ERK Signalling: A Master Regulator of Cell Behaviour, Life and Fate. Nat. Rev. Mol. Cell Biol. 2020, 21, 607–632. [Google Scholar] [CrossRef] [PubMed]
- Chitnis, M.M.; Yuen, J.S.P.; Protheroe, A.S.; Pollak, M.; Macaulay, V.M. The Type 1 Insulin-like Growth Factor Receptor Pathway. Clin. Cancer Res. 2008, 14, 6364–6370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Sehat, B.; Tofigh, A.; Lin, Y.; Trocmé, E.; Liljedahl, U.; Lagergren, J.; Larsson, O. SUMOylation Mediates the Nuclear Translocation and Signaling of the IGF-1 Receptor. Sci. Signal. 2010, 3, ra10. [Google Scholar] [CrossRef] [PubMed]
- Aleksic, T.; Chitnis, M.M.; Perestenko, O.V.; Gao, S.; Thomas, P.H.; Turner, G.D.; Protheroe, A.S.; Howarth, M.; Macaulay, V.M. Type 1 Insulin-like Growth Factor Receptor Translocates to the Nucleus of Human Tumor Cells. Cancer Res. 2010, 70, 6412–6419. [Google Scholar] [CrossRef] [Green Version]
- Solomon-Zemler, R.; Sarfstein, R.; Werner, H. Nuclear Insulin-like Growth Factor-1 Receptor (IGF1R) Displays Proliferative and Regulatory Activities in Non-Malignant Cells. PLoS ONE 2017, 12, e0185164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poreba, E.; Durzynska, J. Nuclear Localization and Actions of the Insulin-like Growth Factor 1 (IGF-1) System Components: Transcriptional Regulation and DNA Damage Response. Mutat. Res. 2020, 784, 108307. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Sen, R.; Queipo, M.J.; Gil-Redondo, J.C.; Ortega, F.; Gómez-Villafuertes, R.; Miras-Portugal, M.T.; Delicado, E.G. Dual-Specificity Phosphatase Regulation in Neurons and Glial Cells. Int. J. Mol. Sci. 2019, 20, 1999. [Google Scholar] [CrossRef] [Green Version]
- Zarubin, T.; Han, J. Activation and Signaling of the P38 MAP Kinase Pathway. Cell Res. 2005, 15, 11–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Destrument, A.; Tournier, C. Physiological Roles of MKK4 and MKK7: Insights from Animal Models. Biochim. Biophys. Acta 2007, 1773, 1349–1357. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Cseh, B.; Doma, E.; Baccarini, M. “RAF” Neighborhood: Protein-Protein Interaction in the Raf/Mek/Erk Pathway. FEBS Lett. 2014, 588, 2398–2406. [Google Scholar] [CrossRef] [Green Version]
- De Iriarte Rodríguez, R.; Magariños, M.; Pfeiffer, V.; Rapp, U.R.; Varela-Nieto, I. C-Raf Deficiency Leads to Hearing Loss and Increased Noise Susceptibility. Cell. Mol. Life Sci. 2015, 72, 3983–3998. [Google Scholar] [CrossRef] [Green Version]
- Ishii, M.; Tateya, T.; Matsuda, M.; Hirashima, T. Retrograde ERK Activation Waves Drive Base-to-Apex Multicellular Flow in Murine Cochlear Duct Morphogenesis. eLife 2021, 10, e61092. [Google Scholar] [CrossRef]
- Maeda, Y.; Fukushima, K.; Omichi, R.; Kariya, S.; Nishizaki, K. Time Courses of Changes in Phospho- and Total- MAP Kinases in the Cochlea after Intense Noise Exposure. PLoS ONE 2013, 8, e58775. [Google Scholar] [CrossRef] [Green Version]
- Kurioka, T.; Matsunobu, T.; Satoh, Y.; Niwa, K.; Endo, S.; Fujioka, M.; Shiotani, A. ERK2 Mediates Inner Hair Cell Survival and Decreases Susceptibility to Noise-Induced Hearing Loss. Sci. Rep. 2015, 5, 16839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herranen, A.; Ikäheimo, K.; Virkkala, J.; Pirvola, U. The Stress Response in the Non-Sensory Cells of the Cochlea Under Pathological Conditions-Possible Role in Mediating Noise Vulnerability. J. Assoc. Res. Otolaryngol. 2018, 19, 637–652. [Google Scholar] [CrossRef]
- Yu, X.; Fan, Z.; Han, Y.; Zhang, D.; Xu, L.; Wang, M.; Yang, Q.; Li, H.; Zhou, M.; Zhang, L.; et al. Paeoniflorin Reduces Neomycin-Induced Ototoxicity in Hair Cells by Suppression of Reactive Oxygen Species Generation and Extracellularly Regulated Kinase Signalization. Toxicol. Lett. 2018, 285, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Celaya, A.M.; Sánchez-Pérez, I.; Bermúdez-Muñoz, J.M.; Rodríguez-de la Rosa, L.; Pintado-Berninches, L.; Perona, R.; Murillo-Cuesta, S.; Varela-Nieto, I. Deficit of Mitogen-Activated Protein Kinase Phosphatase 1 (DUSP1) Accelerates Progressive Hearing Loss. eLife 2019, 8, e39159. [Google Scholar] [CrossRef] [PubMed]
- Sha, S.-H.; Chen, F.-Q.; Schacht, J. Activation of Cell Death Pathways in the Inner Ear of the Aging CBA/J Mouse. Hear. Res. 2009, 254, 92–99. [Google Scholar] [CrossRef] [Green Version]
- Urness, L.D.; Li, C.; Wang, X.; Mansour, S.L. Expression of ERK Signaling Inhibitors Dusp6, Dusp7, and Dusp9 during Mouse Ear Development. Dev. Dyn. 2008, 237, 163–169. [Google Scholar] [CrossRef] [Green Version]
- Bermúdez-Muñoz, J.M.; Celaya, A.M.; García_Mato, A.; Muñoz-Espín, D.; Rodríguez-de la Rosa, L.; Serrano, M.; Varela-Nieto, I. Dual-Specificity Phosphatase 1 (DUSP1) Has a Central Role in Redox Homeostasis and Inflammation in the Mouse Cochlea. Antioxidants 2021, in press. [Google Scholar] [CrossRef]
- Alagramam, K.N.; Stepanyan, R.; Jamesdaniel, S.; Chen, D.H.-C.; Davis, R.R. Noise Exposure Immediately Activates Cochlear Mitogen-Activated Protein Kinase Signaling. Noise Health 2014, 16, 400–409. [Google Scholar] [CrossRef]
- Brand, Y.; Levano, S.; Radojevic, V.; Naldi, A.M.; Setz, C.; Ryan, A.F.; Pak, K.; Hemmings, B.A.; Bodmer, D. All Akt Isoforms (Akt1, Akt2, Akt3) Are Involved in Normal Hearing, but Only Akt2 and Akt3 Are Involved in Auditory Hair Cell Survival in the Mammalian Inner Ear. PLoS ONE 2015, 10, e0121599. [Google Scholar] [CrossRef] [PubMed]
- Sha, S.-H.; Chen, F.-Q.; Schacht, J. PTEN Attenuates PIP3/Akt Signaling in the Cochlea of the Aging CBA/J Mouse. Hear. Res. 2010, 264, 86–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Sha, S.-H.; Schacht, J. Kanamycin Alters Cytoplasmic and Nuclear Phosphoinositide Signaling in the Organ of Corti in Vivo. J. Neurochem. 2006, 99, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.; Li, W.; Hu, P.; Xie, D.; Wen, J. Role of Hsp90/Akt Pathway in the Pathogenesis of Gentamicin-Induced Hearing Loss. Int. J. Clin. Exp. Pathol. 2018, 11, 4431–4438. [Google Scholar] [PubMed]
- Ellis, K.; Driver, E.C.; Okano, T.; Lemons, A.; Kelley, M.W. GSK3 Regulates Hair Cell Fate in the Developing Mammalian Cochlea. Dev. Biol. 2019, 453, 191–205. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and MTOR Regulate Autophagy through Direct Phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Leitmeyer, K.; Glutz, A.; Radojevic, V.; Setz, C.; Huerzeler, N.; Bumann, H.; Bodmer, D.; Brand, Y. Inhibition of MTOR by Rapamycin Results in Auditory Hair Cell Damage and Decreased Spiral Ganglion Neuron Outgrowth and Neurite Formation In Vitro. BioMed Res. Int. 2015, 2015, 925890. [Google Scholar] [CrossRef] [Green Version]
- Varela-Nieto, I.; Palmero, I.; Magariños, M. Complementary and Distinct Roles of Autophagy, Apoptosis and Senescence during Early Inner Ear Development. Hear. Res. 2019, 376, 86–96. [Google Scholar] [CrossRef] [Green Version]
- Lam, E.W.-F.; Brosens, J.J.; Gomes, A.R.; Koo, C.-Y. Forkhead Box Proteins: Tuning Forks for Transcriptional Harmony. Nat. Rev. Cancer 2013, 13, 482–495. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, X.; Sun, M.; Xu, T.; Wang, A. FoxO3a Plays a Key Role in the Protective Effects of Pomegranate Peel Extract against Amikacin-Induced Ototoxicity. Int. J. Mol. Med. 2017, 40, 175–181. [Google Scholar] [CrossRef] [Green Version]
- Oesterle, E.C.; Chien, W.-M.; Campbell, S.; Nellimarla, P.; Fero, M.L. P27(Kip1) Is Required to Maintain Proliferative Quiescence in the Adult Cochlea and Pituitary. Cell Cycle 2011, 10, 1237–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardisty-Hughes, R.E.; Parker, A.; Brown, S.D.M. A Hearing and Vestibular Phenotyping Pipeline to Identify Mouse Mutants with Hearing Impairment. Nat. Protoc. 2010, 5, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Mustapha, M.; Fang, Q.; Gong, T.-W.; Dolan, D.F.; Raphael, Y.; Camper, S.A.; Duncan, R.K. Deafness and Permanently Reduced Potassium Channel Gene Expression and Function in Hypothyroid Pit1dw Mutants. J. Neurosci. 2009, 29, 1212–1223. [Google Scholar] [CrossRef] [Green Version]
- Karolyi, I.J.; Dootz, G.A.; Halsey, K.; Beyer, L.; Probst, F.J.; Johnson, K.R.; Parlow, A.F.; Raphael, Y.; Dolan, D.F.; Camper, S.A. Dietary Thyroid Hormone Replacement Ameliorates Hearing Deficits in Hypothyroid Mice. Mamm. Genome 2007, 18, 596–608. [Google Scholar] [CrossRef]
- Fang, Q.; Giordimaina, A.M.; Dolan, D.F.; Camper, S.A.; Mustapha, M. Genetic Background of Prop1(Df) Mutants Provides Remarkable Protection against Hypothyroidism-Induced Hearing Impairment. J. Assoc. Res. Otolaryngol. 2012, 13, 173–184. [Google Scholar] [CrossRef] [Green Version]
- Prado-Barreto, V.M.; Salvatori, R.; Santos Júnior, R.C.; Brandão-Martins, M.B.; Correa, E.A.; Garcez, F.B.; Valença, E.H.O.; Souza, A.H.O.; Pereira, R.M.C.; Nunes, M.A.P.; et al. Hearing Status in Adult Individuals with Lifetime, Untreated Isolated Growth Hormone Deficiency. Otolaryngol.-Head Neck Surg. 2014, 150, 464–471. [Google Scholar] [CrossRef]
- Santos-Carvalho, H.A.; Aguiar-Oliveira, M.H.; Salvatori, R.; Valença, E.H.O.; Andrade-Guimarães, A.L.; Palanch-Repeke, C.E.; Moreira-Cândido, L.P.; Araújo-Daniel, C.R.; de Oliveira-Barreto, A.C.; Andrade, B.M.R.; et al. Vestibular Function in Severe GH Deficiency Due to an Inactivating Mutation in the GH-Releasing Hormone Receptor Gene. Endocrine 2020, 67, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Lin, C.-H.; Smith, M.E. Growth Hormone Promotes Hair Cell Regeneration in the Zebrafish (Danio Rerio) Inner Ear Following Acoustic Trauma. PLoS ONE 2011, 6, e28372. [Google Scholar] [CrossRef] [Green Version]
- Attias, J.; Zarchi, O.; Nageris, B.I.; Laron, Z. Cochlear Hearing Loss in Patients with Laron Syndrome. Eur. Arch. Oto-Rhino-Laryngol. 2012, 269, 461–466. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, B.C.; Maheshwari, H.G.; He, L.; Reed, M.; Lozykowski, M.; Okada, S.; Cataldo, L.; Coschigamo, K.; Wagner, T.E.; et al. A Mammalian Model for Laron Syndrome Produced by Targeted Disruption of the Mouse Growth Hormone Receptor/Binding Protein Gene (the Laron Mouse). Proc. Natl. Acad. Sci. USA 1997, 94, 13215–13220. [Google Scholar] [CrossRef] [Green Version]
- Wilson, T.; Omelchenko, I.; Foster, S.; Zhang, Y.; Shi, X.; Nuttall, A.L. JAK2/STAT3 Inhibition Attenuates Noise-Induced Hearing Loss. PLoS ONE 2014, 9, e108276. [Google Scholar] [CrossRef]
- Teng, Z.-P.; Tian, R.; Xing, F.-L.; Tang, H.; Xu, J.-J.; Zhang, B.-W.; Qi, J.-W. An Association of Type 1 Diabetes Mellitus with Auditory Dysfunction: A Systematic Review and Meta-Analysis. Laryngoscope 2017, 127, 1689–1697. [Google Scholar] [CrossRef] [PubMed]
- Lomedico, P.; Rosenthal, N.; Efstratidadis, A.; Gilbert, W.; Kolodner, R.; Tizard, R. The Structure and Evolution of the Two Nonallelic Rat Preproinsulin Genes. Cell 1979, 18, 545–558. [Google Scholar] [CrossRef]
- Huerzeler, N.; Petkovic, V.; Sekulic-Jablanovic, M.; Kucharava, K.; Wright, M.B.; Bodmer, D. Insulin Receptor and Glucose Transporters in the Mammalian Cochlea. Audiol. Neurootol. 2019, 24, 65–76. [Google Scholar] [CrossRef]
- Wang, S.; Schacht, J. Insulin Stimulates Protein Synthesis and Phospholipid Signaling Systems but Does Not Regulate Glucose Uptake in the Inner Ear. Hear. Res. 1990, 47, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Camarero, G.; Villar, M.A.; Contreras, J.; Fernández-Moreno, C.; Pichel, J.G.; Avendaño, C.; Varela-Nieto, I. Cochlear Abnormalities in Insulin-like Growth Factor-1 Mouse Mutants. Hear. Res. 2002, 170, 2–11. [Google Scholar] [CrossRef]
- Cediel, R.; Riquelme, R.; Contreras, J.; Díaz, A.; Varela-Nieto, I. Sensorineural Hearing Loss in Insulin-like Growth Factor I-Null Mice: A New Model of Human Deafness. Eur. J. Neurosci. 2006, 23, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Woods, K.A.; Camacho-Hübner, C.; Savage, M.O.; Clark, A.J. Intrauterine Growth Retardation and Postnatal Growth Failure Associated with Deletion of the Insulin-like Growth Factor I Gene. N. Engl. J. Med. 1996, 335, 1363–1367. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-de la Rosa, L.; Lassaletta, L.; Calvino, M.; Murillo-Cuesta, S.; Varela-Nieto, I. The Role of Insulin-Like Growth Factor 1 in the Progression of Age-Related Hearing Loss. Front. Aging Neurosci. 2017, 9, 411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López, I.P.; Rodriguez-de la Rosa, L.; Pais, R.S.; Piñeiro-Hermida, S.; Torrens, R.; Contreras, J.; Varela-Nieto, I.; Pichel, J.G. Differential Organ Phenotypes after Postnatal Igf1r Gene Conditional Deletion Induced by Tamoxifen in UBC-CreERT2; Igf1r Fl/Fl Double Transgenic Mice. Transgenic Res. 2015, 24, 279–294. [Google Scholar] [CrossRef]
- Cardoso, S.; López, I.P.; Piñeiro-Hermida, S.; Pichel, J.G.; Moreira, P.I. IGF1R Deficiency Modulates Brain Signaling Pathways and Disturbs Mitochondria and Redox Homeostasis. Biomedicines 2021, 9, 158. [Google Scholar] [CrossRef] [PubMed]
- Holzenberger, M.; Dupont, J.; Ducos, B.; Leneuve, P.; Géloën, A.; Even, P.C.; Cervera, P.; Le Bouc, Y. IGF-1 Receptor Regulates Lifespan and Resistance to Oxidative Stress in Mice. Nature 2003, 421, 182–187. [Google Scholar] [CrossRef]
- Ludwig, T.; Eggenschwiler, J.; Fisher, P.; D’Ercole, A.J.; Davenport, M.L.; Efstratiadis, A. Mouse Mutants Lacking the Type 2 IGF Receptor (IGF2R) Are Rescued from Perinatal Lethality in Igf2 and Igf1r Null Backgrounds. Dev. Biol. 1996, 177, 517–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, A.A.; LeRoith, D. Minireview: Tissue-Specific versus Generalized Gene Targeting of the Igf1 and Igf1r Genes and Their Roles in Insulin-like Growth Factor Physiology. Endocrinology 2001, 142, 1685–1688. [Google Scholar] [CrossRef]
- Chen, J.; Yuan, H.; Talaska, A.E.; Hill, K.; Sha, S.-H. Increased Sensitivity to Noise-Induced Hearing Loss by Blockade of Endogenous PI3K/Akt Signaling. J. Assoc. Res. Otolaryngol. 2015, 16, 347–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Zhao, J.; Jin, Y.; Hou, C.; Zong, W.; Lu, T.; Li, H.; Gao, J. PTEN Regulation of the Proliferation and Differentiation of Auditory Progenitors through the PTEN/PI3K/Akt-Signaling Pathway in Mice. Neuroreport 2014, 25, 177–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.; Sun, X.; Zhang, L.; Jin, Y.; Chai, R.; Yang, L.; Zhang, A.; Liu, X.; Bai, X.; Li, J.; et al. Tuberous Sclerosis Complex-Mediated MTORC1 Overactivation Promotes Age-Related Hearing Loss. J. Clin. Investig. 2018, 128, 4938–4955. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Xu, N.; Chen, P.; Liu, Y.; Qi, X.; Liu, S.; Li, C.; Tang, J. Rapamycin Protects Spiral Ganglion Neurons from Gentamicin-Induced Degeneration In Vitro. J. Assoc. Res. Otolaryngol. 2019, 20, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-J.; Kim, H.-J.; Bae, G.-S.; Seo, S.-W.; Kim, D.-Y.; Jung, W.-S.; Kim, M.-S.; Song, M.-Y.; Kim, E.-K.; Kwon, K.-B.; et al. Selective GSK-3beta Inhibitors Attenuate the Cisplatin-Induced Cytotoxicity of Auditory Cells. Hear. Res. 2009, 257, 53–62. [Google Scholar] [CrossRef]
- Liu, T.; Zong, S.; Luo, P.; Qu, Y.; Wen, Y.; Du, P.; Xiao, H. Enhancing Autophagy by Down-Regulating GSK-3β Alleviates Cisplatin-Induced Ototoxicity in Vivo and in Vitro. Toxicol. Lett. 2019, 313, 11–18. [Google Scholar] [CrossRef]
- Laine, H.; Sulg, M.; Kirjavainen, A.; Pirvola, U. Cell Cycle Regulation in the Inner Ear Sensory Epithelia: Role of Cyclin D1 and Cyclin-Dependent Kinase Inhibitors. Dev. Biol. 2010, 337, 134–146. [Google Scholar] [CrossRef] [Green Version]
- Saxton, T.M.; Cheng, A.M.; Ong, S.H.; Lu, Y.; Sakai, R.; Cross, J.C.; Pawson, T. Gene Dosage-Dependent Functions for Phosphotyrosine-Grb2 Signaling during Mammalian Tissue Morphogenesis. Curr. Biol. 2001, 11, 662–670. [Google Scholar] [CrossRef] [Green Version]
- Battaglia, A.; Pak, K.; Brors, D.; Bodmer, D.; Frangos, J.A.; Ryan, A.F. Involvement of Ras Activation in Toxic Hair Cell Damage of the Mammalian Cochlea. Neuroscience 2003, 122, 1025–1035. [Google Scholar] [CrossRef] [Green Version]
- Powell, M.B.; Hyman, P.; Bell, O.D.; Balmain, A.; Brown, K.; Alberts, D.; Bowden, G.T. Hyperpigmentation and Melanocytic Hyperplasia in Transgenic Mice Expressing the Human T24 Ha-Ras Gene Regulated by a Mouse Tyrosinase Promoter. Mol. Carcinog. 1995, 12, 82–90. [Google Scholar] [CrossRef]
- Wojnowski, L.; Stancato, L.F.; Zimmer, A.M.; Hahn, H.; Beck, T.W.; Larner, A.C.; Rapp, U.R.; Zimmer, A. Craf-1 Protein Kinase Is Essential for Mouse Development. Mech. Dev. 1998, 76, 141–149. [Google Scholar] [CrossRef]
- Walenkamp, M.J.E.; Karperien, M.; Pereira, A.M.; Hilhorst-Hofstee, Y.; van Doorn, J.; Chen, J.W.; Mohan, S.; Denley, A.; Forbes, B.; van Duyvenvoorde, H.A.; et al. Homozygous and Heterozygous Expression of a Novel Insulin-like Growth Factor-I Mutation. J. Clin. Endocrinol. Metab. 2005, 90, 2855–2864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keselman, A.C.; Martin, A.; Scaglia, P.A.; Sanguineti, N.M.; Armando, R.; Gutiérrez, M.; Braslavsky, D.; Ballerini, M.G.; Ropelato, M.G.; Ramirez, L.; et al. A Homozygous Mutation in the Highly Conserved Tyr60 of the Mature IGF1 Peptide Broadens the Spectrum of IGF1 Deficiency. Eur. J. Endocrinol. 2019, 181, K43–K53. [Google Scholar] [CrossRef] [PubMed]
- Netchine, I.; Azzi, S.; Houang, M.; Seurin, D.; Perin, L.; Ricort, J.-M.; Daubas, C.; Legay, C.; Mester, J.; Herich, R.; et al. Partial Primary Deficiency of Insulin-like Growth Factor (IGF)-I Activity Associated with IGF1 Mutation Demonstrates Its Critical Role in Growth and Brain Development. J. Clin. Endocrinol. Metab. 2009, 94, 3913–3921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batey, L.; Moon, J.E.; Yu, Y.; Wu, B.; Hirschhorn, J.N.; Shen, Y.; Dauber, A. A Novel Deletion of IGF1 in a Patient with Idiopathic Short Stature Provides Insight Into IGF1 Haploinsufficiency. J. Clin. Endocrinol. Metab. 2014, 99, E153–E159. [Google Scholar] [CrossRef]
- Fuqua, J.S.; Derr, M.; Rosenfeld, R.G.; Hwa, V. Identification of a Novel Heterozygous IGF1 Splicing Mutation in a Large Kindred with Familial Short Stature. Horm. Res. Paediatr. 2012, 78, 59–66. [Google Scholar] [CrossRef]
- Van Duyvenvoorde, H.A.; van Doorn, J.; Koenig, J.; Gauguin, L.; Oostdijk, W.; Wade, J.D.; Karperien, M.; Ruivenkamp, C.A.L.; Losekoot, M.; van Setten, P.A.; et al. The Severe Short Stature in Two Siblings with a Heterozygous IGF1 Mutation Is Not Caused by a Dominant Negative Effect of the Putative Truncated Protein. Growth Horm. IGF Res. 2011, 21, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Van Duyvenvoorde, H.A.; van Setten, P.A.; Walenkamp, M.J.E.; van Doorn, J.; Koenig, J.; Gauguin, L.; Oostdijk, W.; Ruivenkamp, C.A.L.; Losekoot, M.; Wade, J.D.; et al. Short Stature Associated with a Novel Heterozygous Mutation in the Insulin-like Growth Factor 1 Gene. J. Clin. Endocrinol. Metab. 2010, 95, E363–E367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walenkamp, M.J.E.; Robers, J.M.L.; Wit, J.M.; Zandwijken, G.R.J.; van Duyvenvoorde, H.A.; Oostdijk, W.; Hokken-Koelega, A.C.S.; Kant, S.G.; Losekoot, M. Phenotypic Features and Response to GH Treatment of Patients With a Molecular Defect of the IGF-1 Receptor. J. Clin. Endocrinol. Metab. 2019, 104, 3157–3171. [Google Scholar] [CrossRef] [PubMed]
- Gannagé-Yared, M.-H.; Klammt, J.; Chouery, E.; Corbani, S.; Mégarbané, H.; Abou Ghoch, J.; Choucair, N.; Pfäffle, R.; Mégarbané, A. Homozygous Mutation of the IGF1 Receptor Gene in a Patient with Severe Pre- and Postnatal Growth Failure and Congenital Malformations. Eur. J. Endocrinol. 2013, 168, K1–K7. [Google Scholar] [CrossRef]
- Prontera, P.; Micale, L.; Verrotti, A.; Napolioni, V.; Stangoni, G.; Merla, G. A New Homozygous IGF1R Variant Defines a Clinically Recognizable Incomplete Dominant Form of SHORT Syndrome. Hum. Mutat. 2015, 36, 1043–1047. [Google Scholar] [CrossRef]
- Ester, W.A.; van Duyvenvoorde, H.A.; de Wit, C.C.; Broekman, A.J.; Ruivenkamp, C.A.L.; Govaerts, L.C.P.; Wit, J.M.; Hokken-Koelega, A.C.S.; Losekoot, M. Two Short Children Born Small for Gestational Age with Insulin-like Growth Factor 1 Receptor Haploinsufficiency Illustrate the Heterogeneity of Its Phenotype. J. Clin. Endocrinol. Metab. 2009, 94, 4717–4727. [Google Scholar] [CrossRef] [Green Version]
- Begemann, M.; Zirn, B.; Santen, G.; Wirthgen, E.; Soellner, L.; Büttel, H.-M.; Schweizer, R.; van Workum, W.; Binder, G.; Eggermann, T. Paternally Inherited IGF2 Mutation and Growth Restriction. N. Engl. J. Med. 2015, 373, 349–356. [Google Scholar] [CrossRef] [Green Version]
- Yamoto, K.; Saitsu, H.; Nakagawa, N.; Nakajima, H.; Hasegawa, T.; Fujisawa, Y.; Kagami, M.; Fukami, M.; Ogata, T. De Novo IGF2 Mutation on the Paternal Allele in a Patient with Silver-Russell Syndrome and Ectrodactyly. Hum. Mutat. 2017, 38, 953–958. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wang, Y.; Yang, X.-A.; Liu, D. De Novo Mutation of Paternal IGF2 Gene Causing Silver-Russell Syndrome in a Sporadic Patient. Front. Genet. 2017, 8, 105. [Google Scholar] [CrossRef] [Green Version]
- Rockstroh, D.; Pfäffle, H.; Le Duc, D.; Rößler, F.; Schlensog-Schuster, F.; Heiker, J.T.; Kratzsch, J.; Kiess, W.; Lemke, J.R.; Abou Jamra, R.; et al. A New p.(Ile66Serfs*93) IGF2 Variant Is Associated with Pre- and Postnatal Growth Retardation. Eur. J. Endocrinol. 2019, 180, K1–K13. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Hao, S.; Zhang, Q.; Liu, F.; Zhou, B.; Xuan, F.; Xing, W.; Chen, X.; Wang, Y.; Ma, P.; et al. Maternal UPD of Chromosome 7 in a Patient with Silver-Russell Syndrome and Pendred Syndrome. J. Clin. Lab. Anal. 2020, 34, e23407. [Google Scholar] [CrossRef]
- Dyment, D.A.; Smith, A.C.; Alcantara, D.; Schwartzentruber, J.A.; Basel-Vanagaite, L.; Curry, C.J.; Temple, I.K.; Reardon, W.; Mansour, S.; Haq, M.R.; et al. Mutations in PIK3R1 Cause SHORT Syndrome. Am. J. Hum. Genet. 2013, 93, 158–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motta, M.; Pannone, L.; Pantaleoni, F.; Bocchinfuso, G.; Radio, F.C.; Cecchetti, S.; Ciolfi, A.; Di Rocco, M.; Elting, M.W.; Brilstra, E.H.; et al. Enhanced MAPK1 Function Causes a Neurodevelopmental Disorder within the RASopathy Clinical Spectrum. Am. J. Hum. Genet. 2020, 107, 499–513. [Google Scholar] [CrossRef]
- Van Trier, D.C.; van Nierop, J.; Draaisma, J.M.T.; van der Burgt, I.; Kunst, H.; Croonen, E.A.; Admiraal, R.J.C. External Ear Anomalies and Hearing Impairment in Noonan Syndrome. Int. J. Pediatr. Otorhinolaryngol. 2015, 79, 874–878. [Google Scholar] [CrossRef]
- Barrenäs, M.; Landin-Wilhelmsen, K.; Hanson, C. Ear and Hearing in Relation to Genotype and Growth in Turner Syndrome. Hear. Res. 2000, 144, 21–28. [Google Scholar] [CrossRef]
- Gómez, J.G.; Devesa, J. Growth Hormone and the Auditory Pathway: Neuromodulation and Neuroregeneration. Int. J. Mol. Sci. 2021, 22, 2829. [Google Scholar] [CrossRef]
- Laron, Z.; Werner, H. Laron Syndrome—A Historical Perspective. Rev. Endocr. Metab. Disord. 2021, 22, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Brisset, S.; Slamova, Z.; Dusatkova, P.; Briand-Suleau, A.; Milcent, K.; Metay, C.; Simandlova, M.; Sumnik, Z.; Tosca, L.; Goossens, M.; et al. Anophthalmia, Hearing Loss, Abnormal Pituitary Development and Response to Growth Hormone Therapy in Three Children with Microdeletions of 14q22q23. Mol. Cytogenet. 2014, 7, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, M.; Gertosio, C.; Pagani, S.; Meazza, C.; Fusco, I.; Bozzola, E.; Bozzola, M. A 5.8 Mb Interstitial Deletion on Chromosome Xq21.1 in a Boy with Intellectual Disability, Cleft Palate, Hearing Impairment and Combined Growth Hormone Deficiency. BMC Med. Genet. 2015, 16, 74. [Google Scholar] [CrossRef] [Green Version]
- Muus, J.S.; Weir, F.W.; Kreicher, K.L.; Bowlby, D.A.; Discolo, C.M.; Meyer, T.A. Hearing Loss in Children with Growth Hormone Deficiency. Int. J. Pediatr. Otorhinolaryngol. 2017, 100, 107–113. [Google Scholar] [CrossRef]
- Williamson, T.T.; Zhu, X.; Pineros, J.; Ding, B.; Frisina, R.D. Understanding Hormone and Hormone Therapies’ Impact on the Auditory System. J. Neurosci. Res. 2020, 98, 1721–1730. [Google Scholar] [CrossRef]
- Romano, S.; Maffei, P.; Bettini, V.; Milan, G.; Favaretto, F.; Gardiman, M.; Marshall, J.D.; Greggio, N.A.; Pozzan, G.B.; Collin, G.B.; et al. Alström Syndrome Is Associated with Short Stature and Reduced GH Reserve. Clin. Endocrinol. 2013, 79, 529–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girard, D.; Petrovsky, N. Alström Syndrome: Insights into the Pathogenesis of Metabolic Disorders. Nat. Rev. Endocrinol. 2011, 7, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Hearn, T. ALMS1 and Alström Syndrome: A Recessive Form of Metabolic, Neurosensory and Cardiac Deficits. J. Mol. Med. 2019, 97, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Dijk, D.R.; Bocca, G.; van Ravenswaaij-Arts, C.M. Growth in CHARGE Syndrome: Optimizing Care with a Multidisciplinary Approach. J. Multidiscip. Healthc. 2019, 12, 607–620. [Google Scholar] [CrossRef] [Green Version]
- Bonnard, Å.; Bark, R.; Hederstierna, C. Clinical Update on Sensorineural Hearing Loss in Turner Syndrome and the X-Chromosome. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 18–24. [Google Scholar] [CrossRef]
- Teixeira, L.S.; de Oliveira Silva, I.B.; Sampaio, A.L.L.; de Oliveira, C.A.P.; Bahamad Júnior, F. Hearing Loss in Acromegaly—A Review. Int. Arch. Otorhinolaryngol. 2018, 22, 313–316. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Nakagawa, T. Insulin-like Growth Factor 1: Role in the Auditory System and Therapeutic Potential in Otology. Curr. Opin. Otolaryngol. Head Neck Surg. 2020, 28, 286–290. [Google Scholar] [CrossRef]
- Nakagawa, T.; Yamamoto, M.; Kumakawa, K.; Usami, S.-I.; Hato, N.; Tabuchi, K.; Takahashi, M.; Fujiwara, K.; Sasaki, A.; Komune, S.; et al. Prognostic Impact of Salvage Treatment on Hearing Recovery in Patients with Sudden Sensorineural Hearing Loss Refractory to Systemic Corticosteroids: A Retrospective Observational Study. Auris. Nasus. Larynx 2016, 43, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Chern, A.; Dagi Glass, L.R.; Gudis, D.A. Thyroid Eye Disease, Teprotumumab, and Hearing Loss: An Evolving Role for Otolaryngologists. Otolaryngol.-Head Neck Surg. 2021, 1945998211004240. [Google Scholar] [CrossRef]
- Warnecke, A.; Prenzler, N.K.; Schmitt, H.; Daemen, K.; Keil, J.; Dursin, M.; Lenarz, T.; Falk, C.S. Defining the Inflammatory Microenvironment in the Human Cochlea by Perilymph Analysis: Toward Liquid Biopsy of the Cochlea. Front. Neurol. 2019, 10, 665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breil, T.; Kneppo, C.; Bettendorf, M.; German IGF-I Deficiency Study Group; Müller, H.L.; Kapelari, K.; Schnabel, D.; Woelfle, J. Sequential Measurements of IGF-I Serum Concentrations in Adolescents with Laron Syndrome Treated with Recombinant Human IGF-I (RhIGF-I). J. Pediatr. Endocrinol. Metab. 2018, 31, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Guerra, J.; Devesa, A.; Llorente, D.; Mouro, R.; Alonso, A.; García-Cancela, J.; Devesa, J. Early Treatment with Growth Hormone (GH) and Rehabilitation Recovers Hearing in a Child with Cerebral Palsy. Reports 2019, 2, 4. [Google Scholar] [CrossRef] [Green Version]
- Davenport, M.L.; Roush, J.; Liu, C.; Zagar, A.J.; Eugster, E.; Travers, S.; Fechner, P.Y.; Quigley, C.A. Growth Hormone Treatment Does Not Affect Incidences of Middle Ear Disease or Hearing Loss in Infants and Toddlers with Turner Syndrome. Horm. Res. Paediatr. 2010, 74, 23–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostberg, J.E.; Beckman, A.; Cadge, B.; Conway, G.S. Oestrogen Deficiency and Growth Hormone Treatment in Childhood Are Not Associated with Hearing in Adults with Turner Syndrome. Horm. Res. 2004, 62, 182–186. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Mato, Á.; Cervantes, B.; Murillo-Cuesta, S.; Rodríguez-de la Rosa, L.; Varela-Nieto, I. Insulin-like Growth Factor 1 Signaling in Mammalian Hearing. Genes 2021, 12, 1553. https://doi.org/10.3390/genes12101553
García-Mato Á, Cervantes B, Murillo-Cuesta S, Rodríguez-de la Rosa L, Varela-Nieto I. Insulin-like Growth Factor 1 Signaling in Mammalian Hearing. Genes. 2021; 12(10):1553. https://doi.org/10.3390/genes12101553
Chicago/Turabian StyleGarcía-Mato, Ángela, Blanca Cervantes, Silvia Murillo-Cuesta, Lourdes Rodríguez-de la Rosa, and Isabel Varela-Nieto. 2021. "Insulin-like Growth Factor 1 Signaling in Mammalian Hearing" Genes 12, no. 10: 1553. https://doi.org/10.3390/genes12101553