Abstract
Inherited optic neuropathies, including Leber Hereditary Optic Neuropathy (LHON) and Dominant Optic Atrophy (DOA), are monogenetic diseases with a final common pathway of mitochondrial dysfunction leading to retinal ganglion cell (RGC) death and ultimately loss of vision. They are, therefore, excellent models with which to investigate this ubiquitous disease process—implicated in both common polygenetic ocular diseases (e.g., Glaucoma) and late-onset central nervous system neurodegenerative diseases (e.g., Parkinson disease). In recent years, cellular and animal models of LHON and DOA have matured in parallel with techniques (such as RNA-seq) to determine and analyze the transcriptomes of affected cells. This confluence leaves us at a particularly exciting time with the potential for the identification of novel pathogenic players and therapeutic targets. Here, we present a discussion of the importance of inherited optic neuropathies and how transcriptomic techniques can be exploited in the development of novel mutation-independent, neuroprotective therapies.
Keywords:
transcriptomics; RNA-seq; neuroprotection; DOA; OPA1; LHON; gene-therapy; mitochondrial; optic neuropathies 1. Introduction
1.1. Optic Neuropathies
Optic neuropathies are among the most common causes of blindness in the working age population [1], with inherited forms (including Leber Hereditary Optic Neuropathy (LHON) [2] and Dominant Optic Atrophy (DOA)) affecting about 1 in 10,000 of the population [3,4,5]. In this review, we will present a brief introduction to these conditions and how the availability of powerful emerging techniques, including transcriptomics, are quickly revolutionizing both diagnosis and development of novel therapies with potential applications beyond the eye.
Vision is ultimately lost in both LHON and DOA as retinal ganglion cells (RGCs) die secondary to mitochondrial dysfunction [6]. This specific susceptibility of RGCs to such dysfunction is not completely understood. However, the relatively large metabolic demand for these specialized cells and their unique anatomy are thought to be important contributory factors. RGCs have long axonal segments which lack myelin throughout their intraocular course but gain a myelin sheath on exiting the eye beyond the lamina cribosa [7,8]. As the only nervous tissue visible in vivo and with increasingly sophisticated cell culture techniques [9,10], RGCs present a powerful system in which to interrogate mitochondrial dysfunction and the pathways that ultimately lead to cell loss and disease development. Such dysfunction has been implicated in major neurodegenerative diseases, such as Parkinson’s disease [11], Alzheimer’s disease [12], and other forms of dementia [13], but the polygenetic inheritance and environmental contribution to these common conditions are particularly challenging when investigating their pathogenesis.
As monogenetic conditions where mitochondrial function is disturbed, both LHON and DOA can mitigate some of these challenges and act as useful model diseases of more complex neurodegenerative disease processes. LHON is a primary mitochondrial DNA (mtDNA) disorder, with the majority of cases caused by one of three point mutations—namely, m.3460G>A in the MT-ND1 gene, m.11778G>A in the MT-ND4 gene, and m.14484T>C in the MT-ND6 gene—all of which encode for essential subunits of mitochondrial complex I [3,7,14,15,16]. LHON is additionally interesting due to its predilection to manifest in males and its marked incomplete penetrance—both of which could perhaps have origins in transcriptomic differences.
In comparison, DOA is nuclear-encoded mitochondrial optic neuropathy caused by mutations in the OPA1 gene (3q28-q29), which encodes for a multimeric dynamin GTPase protein located within the mitochondrial inner membrane. OPA1 subserves a number of functions, including the regulation of mitochondrial fusion, the stability of the mitochondrial respiratory chain complexes and mitochondrial biogenesis, the sequestration of pro-apoptotic cytochrome c within the mitochondrial cristae, and mitochondrial turnover (mitophagy) [4,17,18,19,20].
1.2. “-Omics” Technologies as Applied to Inherited Optic Neuropathies
Our understanding of mitochondrial biology and disease has advanced greatly over recent years, not least due to the development and maturation of “-omics” technologies. These can be defined as “high-throughput technologies capable of detecting differences in a multitude of molecular constituents in organisms [21]”, with those that represent the three strata of central biological dogma (genomics, transcriptomics, and proteomics) being prominent. These fields deal with the detection of differences in DNA sequences, gene transcription, and proteins within tissue. Additionally, particularly relevant to mitochondrial disease is the developing field of metabolomics (centered on the comparison of levels of products of metabolism) [22,23] and lipidomics [24].
Whilst these disciplines are linked by their comparative nature—experimental plans often involve the contrast of different conditions (e.g., control and “diseased” states, or between different cell types)—the emerging field of multi-omics (or vertical -omics) focuses on complimentary comparisons across domains (Figure 1). For example, highlighting changes replicated across the transcriptome, the proteome and metabolome will carry particular significance [24,25], and this approach is already being used in mitochondrial research [26]. As these technologies and their complementary bioinformatic analysis techniques develop, the power of “-omics” investigations is likely to increase.
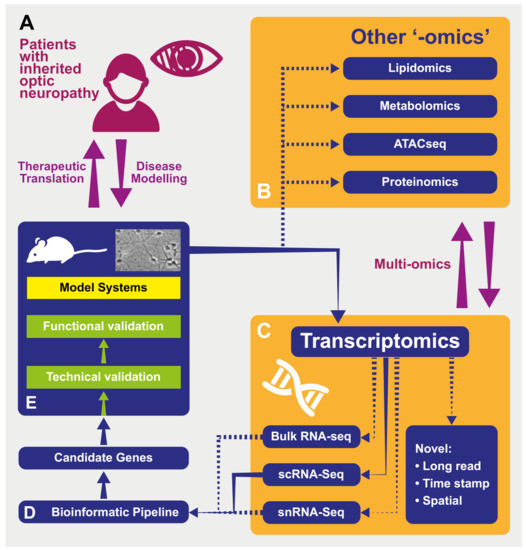
Figure 1.
A schematic representing the processes of modeling and investigating inherited optic neuropathies using “-omics” methods. (A) Model diseases such as DOA and LHON can be powerful tools in which to investigate the effects of mitochondrial dysfunction. The study of patients with inherited optic neuropathies is often a two-way process: in one direction, the characterization of their phenotype and genotype allows the development of useful disease models (for example, cell -and animal-based). These provide an efficient environment in which to increase our understanding of underlying disease processes as well as a testing ground for novel therapies before their return in the opposite direction, back into patients. (B) Tissues from model systems can be investigated using used in multiple “-omics” techniques—in some cases, these can be performed simultaneously and analyzed in vertical “multi-omics” experiments (see text). ATACseq—Assay for Transposase Accessible Chromatin—a method for assessing which areas of the chromatin superstructure are open and so likely available for active in transcription, is an example of the rapidly expanding field of epigenomics. (C) Multiple transcriptomic techniques are available and have particular strength in different experimental situations. “Novel”: Novel techniques are emerging that will further the resolution of these techniquesm including those with abilities to sequence longer transcripts in one read and those that integrate temporal and spatial information regarding transcripts (see text). RNA-seq—RNA-sequencing; scRNA-seq—single-cell RNAseq; snRNAseq—single nucleus RNAseq. (D) Transcriptomic techniques gain power from the large quantity of data that they produce. This necessitates adequately designed bioinformatic pipelines that are tailored to the exact scientific question being asked in order to produce a list of candidate genes for further investigation back in model systems. (E) Model systems of diseases can include those based on cultured patient cells or be animals carrying pathogenic variants, leading to phenocopies of human disease. Samples from these models used in transcriptomic analysis can include tissue (such as retinas) or cell cultures. As many transcriptomic experiments compare expression between conditions (e.g., disease and control) to produce lists of differentially expressed genes, the further technical and functional validation of these can be performed back in model systems in preparation for therapeutic translation in patients (see text).
1.3. Transcriptomics
Specifically, “the transcriptome” refers to the RNA transcribed within a cell, or group of cells, often with a particular focus on mRNA (both coding and non-coding). Several methods to quantify mRNA have been developed (see below) and are applied to genetic eye diseases. For example, assessing transcribed features in a particular sample can be used to compare changes in gene expression either over time or between control and diseased states (such as optic neuropathies) [27]. Features showing differential expression may be implicated in the disease process, highlighting areas for further investigation to uncover aetiologic pathways, novel biomarkers, and therapeutic targets.
2. Transcriptomics in Inherited Optic Neuropathies
2.1. Applications of Transcriptomics in Optic Neuropathies
Several factors make transcriptomics a particularly suitable methodology for the investigation of optic neuropathies. Whilst the anatomy and physiology of the retina and optic nerve are relatively well understood compared with other areas of the central nervous system [28], our understanding of pathophysiology in these structures can be less comprehensive. This is particularly true for inherited optic neuropathies such as LHON and DOA, where the genomic determinant of the disease in many patients is readily identifiable as a single gene (monogenic disorder) [14,18,19]. However, less is known regarding how this translates into the clinical phenotype of RGC death as well as other as yet unexplained facets of these model diseases (such as the incomplete penetrance in LHON when the pathogenic mitochondrial DNA mutation is present in the homoplastic state in both affected patients and carriers). Thus, this presents an unmet need to identify the novel pathways and genes involved to which comparative transcriptomics is particularly suited. Whilst direct access to RGCs and patient tissues is limited, cellular and mouse model systems [7,29,30] have developed in recent years into powerful platforms with which to perform transcriptomic studies (and, more importantly, validate and investigate their findings). For example, in vivo neuro-retinal tissue can easily be visualized (if not directly sampled) at the cellular level with techniques such as optical coherence tomography (OCT), and there are well defined metrics of RGC function at the behavioral (acuity) [31], reflex (pupillary) [32], and electrophysiological levels [33]. To compliment this, induced pluripotent stem cell (iPSC) RGC models derived from LHON and DOA patient tissues have proved invaluable for molecular investigations [7]
2.2. Disadvantages of Transcriptomics in Optic Neuropathies
Despite the suitability of inherited optic neuropathy investigations, the limitations of transcriptomics must be borne in mind when considering data from such studies. In isolation, transcriptomics gives us no direct information on protein dynamics. The mRNA expression level of a particular gene may correspond to increased protein levels, increased protein turnover, or indeed changes to post translational protein modifications. Therefore, the validation of transcriptomic findings at the protein or functional level is required if conclusions are to be drawn regarding the downstream effects of mRNA changes. Planning this can present further challenges—for example, when comparing transcriptomes in conditions (such as optic neuropathies) with changes dramatic enough to lead to cell death, large numbers of differentially expressed genes (DEGs) are likely. Therefore, methods to prioritize which DEG to validate while minimizing bias have been developed, and these are discussed further below and reviewed elsewhere [34].
Whilst preparing tissue for transcriptomic studies, careful tissue handling is equally important in reducing bias, and indices of extracted RNA quality (such as RNA Integrity Number “RIN” [35]) can be used to assess this. Additionally, and especially in a highly cellular, complex tissue such as the retina, it is essential to ensure that the identity of cells undergoing processing is known (for example, photoreceptors and bipolar cells have an interdigitated synapse that can make them difficult to dissociate and isolate [36]. Additionally, the cells isolated must be viable. It is well established that the dissection of retina from mouse models requires the cutting of the optic nerve (and therefore the transection of RGC axons), so processing should be as expedient as possible to minimize the stress response recorded. Indeed, many of these limitations have been addressed as isolation methods have been developed.
3. Transcriptomic Methodologies
3.1. Tissue Selection and Sample Preparation
One methodological aspect that has been particularly refined is that of tissue selection and preparation. Earlier studies made use of tissues dissected by hand from animal models [37] and dissociated into suspension or collected from cultured cells [38] in bulk. This has the advantage of providing a large quantity of RNA for further processing. In addition, the processing of tissue in this way is relatively easy and expedient, reducing postmortem alterations in expression—particularly relevant to RGCs following the truncation of their axon during enucleation. Indeed, while improvements have been made to tissue preparation processes (see below), the basic processes remain constant: RNA is extracted, isolated, and reverse-transcribed into complementary DNA (cDNA) representing a library of the transcripts present [27]. Libraries are sequenced after undergoing amplification [39] or enrichment steps for a particular form of RNA—for example, selecting for the poly-adenosine tail of transcribed mRNA (of either nuclear or mitochondrial origin) [40] or depleting ribosomal RNA before reverse transcription [41] in order to focus on protein coding mRNA.
3.2. Quantifying Expression
3.2.1. Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
Once the library has been prepared, it is necessary to quantify the expression of transcripts. In the most basic form, qRT-PCR is an accurate technique to interrogate a small number of genes [42,43]; however, it is limited by its need for oligo-primers requiring the a priori selection of candidate genes. This technique remains in use as a confirmatory tool (for example, to confirm sample purity) due to its ease, ubiquity, and economy. However, the inherent amplification of error in the technique [44,45] and the concurrent improvement in the technical reliability of microarray and RNA-seq techniques has seen it widely replaced by other assays in validation experiments.
3.2.2. RNA-Seq
Whilst the development of microarray assays [46] represented a paradigm shift and the beginning of high-throughput transcriptomics, their requirement to define which genes to investigate a priori, as well as technical limitations [47,48], have led to their replacement in most applications by RNA-seq techniques. RNA-seq refers to techniques where cDNA is directly sequenced using high-throughput sequencing techniques [27]. The superior flexibility and unbiased nature of this approach (not requiring the a priori definition of probes) has seen it supersede microarrays in many areas—particularly comparative transcriptomics. Indeed, due to these advantages RNAseq has become an integral technique in clinical and laboratory science and is the the gold standard in multiple disciplines, most notably in oncology [49] but also more generally (reviewed elegantly here [50]).
Several modifications of this technique are particularly relevant to the study of RGCs and optic neuropathies. Given the diversity of RGC cell types [51,52,53] and their differential response to disease [54], techniques that are able to prepare libraries from and sequence the transcriptomes of individual cells (single cell "scRNA-seq” [55,56] or single-nuclei snRNA-seq [57]), are particularly useful. These techniques overcome difficulties with “bulk” RNAseq techniques in resolving changes in gene expression between subpopulations of cells. This is achieved by labelling individual transcripts as being from a particularly labelled cell: this vastly increases resolution by allowing analysis at the level of individual cells, but concurrently increases the complexity of analysis and the resources required.
In such methods, single cells are isolated (for example, using a microwell plate or in individual droplets using microfluidics) and combined with reaction substrate and a barcoded bead (see [58] for a review of individual methods). This allows library preparation and sequencing on a cell by cell basis, potentially uncovering changes masked by techniques dealing with bulk batches of cells [59]. Single-cell sequencing has also been useful when used with clustering techniques to group similar single-cell transcriptomes from individual RGCs in the search for novel molecular markers correlating with anatomical and functional characteristics for subpopulations of RGCs [53].
3.2.3. Future Directions in Quantifying Expression
This characterization of RGC subpopulations and their role in optic neuropathies is likely to particularly benefit from rapidly developing new technologies within RNA-seq. Improvements in the resolution of the single-cell “long read” sequencing [60] (as opposed to short reads focusing on the 3′ end of transcripts) and RNA timestamping [61] (which details temporal changes in mRNA transcripts) have enhanced our ability to understand how differing transcripts interact and turn over. At the tissue level, the emergence of technology to allow single-cell multiomics simultaneously on the same cells [62] will further increase the resolution of our understanding of these cells in optic neuropathies.
Perhaps most excitingly, however, is the emerging technique of spatial transcriptomics [63], in which the molecular labelling of individually sequenced cells can preserve information regarding their anatomical location. If this can be developed in conjunction with multiple electrode array neurophysiology [64], which similarly preserves retinal topology, there is potential for a technique to obtain both single-cell transcriptomic and single-cell functional data from the same cells, allowing direct validation.
3.3. Analysis of RNA-Seq Data
As RNA-seq techniques have developed over the past few years, so too have the bioinformatic approaches required to make sense of the often vast quantities of data produced. Indeed, there is no “gold-standard” pipeline or set of processes for either bulk [65] or scRNA-seq [66] analysis, and the approaches have to be tailored to the individual experiment and the scientific question being addressed by a particular study. However, general stages of processing do tend to be applied, even if their details vary (Figure 1, and reviewed here [27,55,67,68,69]). This variability and the great pace of advancement in bioinformatic methodologies make it essential to precisely document software versions and the settings, as these can greatly alter results [55]. Indeed, replicating notable results using different software packages may add to the power of conclusions.
4. Clinical and Research Applications of Transcriptomics
4.1. Diagnostic
Mirroring this rapid advancement in data analysis and software techniques in RNA-seq has been a concurrent improvement in our diagnostic capabilities in genetic disease. In recent years, approaches to attaining molecular diagnoses in optic neuropathies and in genetic disease more generally have changed with the continually improving technologies. The traditional approach of testing for a particular pathogenic variant in a single gene based on a characteristic clinical presentation has been extended with the testing of “panels” of genes associated with a particular phenotype (optic atrophy, for example). This has the advantage of combining clinical prior probability to provide a high diagnostic confidence when positive, but limits diagnoses to those pre-determined variants tested for on the panels. While whole-exome (WES) and whole-genome sequencing (WGS) have increased diagnostic yields up to as much as 50% of cases [70] by allowing the identification of novel variants, one of their disadvantages is not providing direct information on the pathogenicity of variants [71].
In this situation, sequencing transcribed RNA (RNA-seq) can be hugely useful in indicating the downstream effects of variants—for example, by detecting expression levels outside the normal physiological range (up or down), splicing-related errors, posttranscriptional modifications, or mono-allelic expression [72]. This is especially useful where little is known regarding a gene, its role in the target tissue, or variants in non-coding regions. In combination with WGS, RNA-seq has been used to characterize novel disease-causing genes in mitochondrial disease [73].
One potential drawback to diagnostic RNA-seq in optic neuropathies lies in its need to be performed on biopsied disease tissues where the target genes are expressed, which is not feasible for the optic nerve. A muscle biopsy has proven to be a useful surrogate in mitochondrial disease, given its high energy demand [74]. However, it is emerging that many genes can be investigated using less invasive techniques, such as blood sampling [74]. With the licensing of gene replacement therapies, the need for molecular diagnosis is more important than ever for individual patients in order to select the most appropriate gene therapy as these options emerge. To this end, RNA-seq is an important addition to our armamentarium by expanding our diagnostic ability.
4.2. Disease Mechanisms
4.2.1. Exploring Disease Mechanisms Using Transcriptomics
As discussed above, transcriptomics is invaluable to the identification of differentially expressed genes between samples, in particular the comparison of healthy and diseased tissues. This has great potential for inherited optic neuropathies (see Table 1) where the mechanisms connecting pathogenic genomic variants and eventual RGC death are poorly understood. Genes expressed at significantly higher or lower levels in diseased tissues compared to healthy tissues are obvious candidate players involved in the underlying disease process [37]. However, with the scale of transcriptomic data, this list can extend to many hundreds of genes, and processes are required to prioritize the most biologically plausible candidates for further investigation.

Table 1.
Studies in the literature performing transcriptomic techniques in models of inherited optic neuropathy. Studies in glaucoma and mitochondrial disease in general have been reviewed [82]. LHON—Leber Hereditary Optic Neuropathy; OPA1-DOA—Dominant Optic Atrophy caused by OPA1 mutations; RGC—Retinal Ganglion Cells; iPSC—induced pluripotent stem cells; KO—Knock Out; AAV—Adeno associated virus; GABA—γ-aminobutyric acid.
A fruitful approach to this has been to interrogate lists of differentially expressed genes (DEGs) for connected sets of genes. With the development of microarray and RNA-seq technology [75], increasingly sophisticated methods, both those relying on the intrinsic properties of DEG data sets (“unbiased”) and those drawing from databases of gene and protein function and interactions [76,77,78,79] (introducing “biological insight”) have been developed. These include overrepresentation analysis, where lists of DEGs are compared to annotated databases (such as Gene Ontology [80] categories) to highlight ontologies that are seen particularly often in the list [76]. Gene Set Enrichment Analysis (GSEA) [81] provides another strategy where lists of DEG are interrogated for the overrepresentation of groups of genes that are connected to particular biological functions. Ultimately, however, the decision on which DEGs to further investigate in order to best answer their individual research question lies with the investigator, armed with the objective prioritization that these techniques allow (Figure 1).
4.2.2. Technical Validation
Having prioritized candidate DEGs for further investigation, initial investigations are needed to validate the transcriptomic findings at the technical level [90]. This can involve techniques to confirm sample purity, such as the qRT-PCR of genes that are known to be expressed (or not) in the sampled tissue. For example, in a sample of RGCs this involves checking that known RGC-specific genes (such as THY1) are expressed and those known to be specific to other, potentially contaminating, cell types (such as RHO from rods) are absent [36,44,45]. In addition, confirming that proteins relating to candidate genes are present in target tissues (for example, by immunohistochemistry, Western blotting, or similar) is a helpful approach to relate findings at the transcriptomic to the effector (protein) level if the appropriate a priori knowledge is available.
4.2.3. Functional Validation
When investigating potential disease mechanisms in optic neuropathies, the validation of the functional role of DEGs is paramount. Do changes identified at the transcriptomic level result in an alteration in cellular function relative to disease progression which could represent a therapeutic target? As direct access to retinal and optic nerve tissues from patients with inherited optic neuropathies is not practical, such work has relied heavily on preclinical models. These have been established from patient-derived cells (blood cells, fibroblasts, lymphoblasts, and cybrid cell lines) which allow the faithful replication of some cellular disease processes found in mutation-carrying cells, but outside of the RGCs themselves (extensively reviewed elsewhere [10]). Over the last decade, technological advances have allowed the generation of RGC cell lines [9] engineered from patient fibroblasts (via induced pluripotent stem cell technology) and animal disease models [7] that faithfully replicate the environment of the RGC in the diseased human retina (reviewed in [7]).
Within model systems, the overexpression or inhibition of candidate genes can be fruitful approaches to validation. In the context of a promising therapeutic target, the manipulation of levels of gene expression in the model should lead to in changes in the metrics of the disease process (as in [59]). For an inherited optic neuropathy, the parameters can be anatomical, such as the rate of RGC survival, or relate to a known pathological process contributing to disease (e.g., mitochondrial oxidative phosphorylation or reactive oxygen species turnover [91]).
The increasing variety and sophistication of in vitro and in vivo disease models for inherited optic neuropathies offer differing advantages that could be effectively harnessed in future transcriptomic studies to form a hierarchical validation pathway. For example, a large number of prioritized DEGs could be screened in relatively inexpensive and efficient traditional cell lines. Promising candidates from this work could then be investigated in more complex iPSC-RGC models to identify a small number of candidates for validation in animal models with a view towards translation. Furthermore, by using clinically demonstrated vectors (such as the adeno-associated virus [92]) candidate genes could be manipulated in vivo, with the advantage of being able to investigate meaningful effects at an organism level—for example, by quantifying visual acuity [93] or electrophysiological measures [33].
4.3. Therapeutic Development
4.3.1. Personalized Medicine
The clinical application of transcriptomic techniques has increased our ability to provide a confirmed molecular diagnosis to patients with a suspected inherited optic neuropathy. In recent years, this has become particularly important as gene replacement therapies have advanced to human clinical trials, as in LHON [94,95]. Clinical transcriptomics are already in use to personalize therapy regimes in areas such as oncology [96], and its future application to inherited optic neuropathies represents an exciting development in the field.
4.3.2. Transferable Neuroprotective Strategies
Transcriptomics has the great potential to identify targets for generalizable, mutation-independent neuroprotective strategies for inherited optic neuropathies. These paradigms of mitochondrial disease can provide a platform from which to identify and develop novel therapeutics for other ocular and neurodegenerative diseases in which mitochondrial dysfunction has been implicated. This is particularly timely, given not only the ongoing maturation of models with which to functionally validate transcriptomic findings in optic neuropathy, but also the concurrent explosion in innovative ocular gene therapy approaches and viral vector delivery [97,98,99]. Such techniques which are already licensed for clinical use in other conditions could be used to quickly develop therapies based on in vitro work to directly manipulate the intended therapeutic targets [92]. For example, “gene replacement” therapy has been successfully used to express wild-type copies of genes with homozygous null or haploinsufficient heterozygous pathogenic variants in vivo [92,100,101], with similar techniques also used to express neuroprotective genes such as brain-derived neurotrophic factor (BDNF) in animal models [98]. Additionally, techniques to downregulate genes with a dominant negative effect have also been developed and successfully applied to various inherited retinal disorders [102,103,104,105].
One of the reasons that in vivo ocular gene therapy has been so successful is the accessibility of the target cells, with RGCs being the first cell type encountered by therapeutic agents injected intravitreally (be it a viral vector, small molecule compound [106], or other biologic therapy [97,107,108]). Clearly, the extrapolation of potential neuroprotective strategies from an ocular model to more generalized central nervous system disorders will pose additional challenges. Here, direct access to tissue will not be as straightforward and much larger vector doses may be required, with an increased risk of adverse off-target effects [109], which will require close monitoring.
5. Conclusions
In this review, we have critically appraised how powerful transcriptomic techniques have the potential to facilitate the diagnosis of patients with inherited optic neuropathies and, crucially, achieve a better understanding of the pathological processes that contribute to RGC loss. The insight gained can then be exploited to develop targeted therapies to enhance RGC survival, which could have much broader relevance for other neurodegenerative diseases characterized by disturbed mitochondrial function. Presently, an unprecedented confluent maturation of complementary techniques is occurring in single-cell transcriptomics, bioinformatics, relevant disease models, and clinically translatable effector techniques. The potential for therapeutic advances in the eye, and beyond, is an exciting proposition for the coming decade.
Author Contributions
M.J.G. planned and wrote article, critically reviewed article, and approved it for publication; N.O., M.M., and P.Y.W.M. critically reviewed article and approved it for publication. All authors have read and agreed to the published version of the manuscript.
Funding
M.J.G. is supported by a National Institute of Health Research (NIHR) Academic Clinical Lectureship and also receives funding from Moorfields Eye Charity, the Academy of Medical Sciences, and the National Eye Research Centre (UK). N.O. and M.M. are kindly supported by the Wellcome Trust (grant no. 205174/Z/16/Z). P.Y.W.M. is supported by a Clinician Scientist Fellowship Award (G1002570) from the Medical Research Council (UK), and also receives funding from Fight for Sight (UK), the Isaac Newton Trust (UK), Moorfields Eye Charity, the Addenbrooke’s Charitable Trust, the National Eye Research Centre (UK), the UK National Institute of Health Research (NIHR) as part of the Rare Diseases Translational Research Collaboration, and the NIHR Biomedical Research Centre based at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
UCL Health Creatives for professional production of Figure 1.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Liew, G.; Michaelides, M.; Bunce, C. A comparison of the causes of blindness certifications in England and Wales in working age adults (16–64 years), 1999–2000 with 2009–2010. BMJ Open 2014, 4, e004015. [Google Scholar] [CrossRef] [PubMed]
- Newman, N.J.; Carelli, V.; Taiel, M.; Yu-Wai-Man, P. Visual Outcomes in Leber Hereditary Optic Neuropathy Patients with the m.11778G>A (MTND4) Mitochondrial DNA Mutation. J. Neuro-Ophthalmol. 2020, 40, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Griffiths, P.G.; Brown, D.T.; Howell, N.; Turnbull, D.M.; Chinnery, P.F. The Epidemiology of Leber Hereditary Optic Neuropathy in the North East of England. Am. J. Hum. Genet. 2003, 72, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Griffiths, P.G.; Burke, A.; Sellar, P.W.; Clarke, M.P.; Gnanaraj, L.; Ah-Kine, D.; Hudson, G.; Czermin, B.; Taylor, R.W.; et al. The Prevalence and Natural History of Dominant Optic Atrophy Due to OPA1 Mutations. Ophthalmology 2010, 117, 1538–1546.e1. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Griffiths, P.G.; Chinnery, P.F. Mitochondrial optic neuropathies—Disease mechanisms and therapeutic strategies. Prog. Retin. Eye Res. 2011, 30, 81–114. [Google Scholar] [CrossRef] [PubMed]
- Carelli, V.; Ross-Cisneros, F.N.; Sadun, A.A. Mitochondrial dysfunction as a cause of optic neuropathies. Prog. Retin. Eye Res. 2004, 23, 53–89. [Google Scholar] [CrossRef]
- Bahr, T.; Welburn, K.; Donnelly, J.; Bai, Y. Emerging model systems and treatment approaches for Leber’s hereditary optic neuropathy: Challenges and opportunities. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2020, 1866, 165743. [Google Scholar] [CrossRef]
- Coussa, R.G.; Merat, P.; Levin, L.A. Propagation and Selectivity of Axonal Loss in Leber Hereditary Optic Neuropathy. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Gill, K.P.; Hung, S.S.C.; Sharov, A.; Lo, C.Y.; Needham, K.; Lidgerwood, G.E.; Jackson, S.; Crombie, D.E.; Nayagam, B.A.; Cook, A.L.; et al. Enriched retinal ganglion cells derived from human embryonic stem cells. Sci. Rep. 2016, 6, 30552. [Google Scholar] [CrossRef]
- Jankauskaitė, E.; Bartnik, E.; Kodroń, A. Investigating Leber’s hereditary optic neuropathy: Cell models and future perspectives. Mitochondrion 2017, 32, 19–26. [Google Scholar] [CrossRef]
- Rani, L.; Mondal, A.C. Emerging concepts of mitochondrial dysfunction in Parkinson’s disease progression: Pathogenic and therapeutic implications. Mitochondrion 2020, 50, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Burté, F.; Carelli, V.; Chinnery, P.F.; Yu-Wai-Man, P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 2015, 11, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Kageyama, Y.; Serre, V.; Moore, D.G.; et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 2014, 137, 2329–2345. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Singh, G.; Lott, M.T.; Hodge, J.; Schurr, T.G.; Lezza, A.M.; Elsas, L.J.; Nikoskelainen, E.K. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988, 242, 1427–1430. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, Y.; Luo, H.; Zhang, K. Leber hereditary optic neuropathy and oxidative stress. Proc. Natl. Acad. Sci. USA 2012, 109, 19882–19883. [Google Scholar] [CrossRef]
- Wallace, D.C.; Lott, M.T. Leber Hereditary Optic Neuropathy: Exemplar of an mtDNA Disease. In Pharmacology of Mitochondria; Singh, H., Sheu, S.-S., Eds.; Springer International: Cham, Switzerland, 2017; pp. 339–376. [Google Scholar] [CrossRef]
- Liao, C.; Ashley, N.; Diot, A.; Morten, K.; Phadwal, K.; Williams, A.; Fearnley, I.; Rosser, L.; Lowndes, J.; Fratter, C.; et al. Dysregulated mitophagy and mitochondrial organization in optic atrophy due to OPA1 mutations. Neurology 2017, 88, 131–142. [Google Scholar] [CrossRef]
- Alexander, C.; Votruba, M.; Pesch, U.E.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef]
- Delettre, C.; Lenaers, G.; Griffoin, J.-M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef]
- Ham, M.; Han, J.; Osann, K.; Smith, M.; Kimonis, V. Meta-analysis of genotype-phenotype analysis of OPA1 mutations in autosomal dominant optic atrophy. Mitochondrion 2019, 46, 262–269. [Google Scholar] [CrossRef]
- Rahman, J.; Rahman, S. Mitochondrial medicine in the omics era. Lancet 2018, 391, 2560–2574. [Google Scholar] [CrossRef]
- Zhang, A.; Sun, H.; Yan, G.; Wang, P.; Wang, X. Mass spectrometry-based metabolomics: Applications to biomarker and metabolic pathway research. Biomed. Chromatogr. 2016, 30, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Esterhuizen, K.; Van Der Westhuizen, F.H.; Louw, R. Metabolomics of mitochondrial disease. Mitochondrion 2017, 35, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Kappler, L.; Lehmann, R. Mass-spectrometric multi-omics linked to function—State-of-the-art investigations of mitochondria in systems medicine. TrAC Trends Anal. Chem. 2019, 119, 115635. [Google Scholar] [CrossRef]
- Hasin, Y.; Seldin, M.; Lusis, A.J. Multi-omics approaches to disease. Genome Biol. 2017, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Quirós, P.M.; Prado, M.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef] [PubMed]
- Owen, N.; Moosajee, M. RNA-sequencing in ophthalmology research: Considerations for experimental design and analysis. Ther. Adv. Ophthalmol. 2019, 11, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Masland, R.H. The Neuronal Organization of the Retina. Neuron 2012, 76, 266–280. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Riazifar, H.; Minxin, G.; Huang, T. Modeling autosomal dominant optic atrophy using induced pluripotent stem cells and identifying potential therapeutic targets. Stem Cell Res. Ther. 2016, 7, 1–14. [Google Scholar] [CrossRef]
- Davies, V.J.; Hollins, A.J.; Piechota, M.J.; Yip, W.; Davies, J.R.; White, K.E.; Nicols, P.P.; Boulton, M.E.; Votruba, M. OPA1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum. Mol. Genet. 2007, 16, 1307–1318. [Google Scholar] [CrossRef]
- Prusky, G.T.; West, P.W.; Douglas, R.M. Behavioral assessment of visual acuity in mice and rats. Vis. Res. 2000, 40, 2201–2209. [Google Scholar] [CrossRef]
- Jagannath, A.; Hughes, S.; Abdelgany, A.; Pothecary, C.A.; Di Pretoro, S.; Pires, S.S.; Vachtsevanos, A.; Pilorz, V.; Brown, L.A.; Hossbach, M.; et al. Isoforms of Melanopsin Mediate Different Behavioral Responses to Light. Curr. Biol. 2015, 25, 2430–2434. [Google Scholar] [CrossRef] [PubMed]
- Barnard, A.R.; Issa, P.C.; Perganta, G.; Williams, P.A.; Davies, V.J.; Sekaran, S.; Votruba, M.; MacLaren, R.E. Specific deficits in visual electrophysiology in a mouse model of dominant optic atrophy. Exp. Eye Res. 2011, 93, 771–777. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.-M.; Shafi, A.; Nguyen, T.; Draghici, S. Identifying significantly impacted pathways: A comprehensive review and assessment. Genome Biol. 2019, 20, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, A.; Mueller, O.; Stocker, S.; Salowsky, R.; Leiber, M.; Gassmann, M.; Lightfoot, S.; Menzel, W.; Granzow, M.; Ragg, T. The RIN: An RNA integrity number for assigning integrity values to RNA measurements. BMC Mol. Biol. 2006, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Gilhooley, M.J.; Hickey, D.G.; Hughes, S.; Hankins, M.W. Retinal bipolar cell gene changes in the rd1 model of inherited retinal degeneration. In Proceedings of ARVO Annual Meeting Abstract, Honolulu, HI, USA, 29 April 2018; Investigative Ophthalmology & Visual Science: Washington, DC, USA, 2018; Volume 59, 1007p. [Google Scholar]
- Lozano, D.C.; Choi, D.; Jayaram, H.; Morrison, J.C.; Johnson, E.C. Utilizing RNA-Seq to Identify Differentially Expressed Genes in Glaucoma Model Tissues, Such as the Rodent Optic Nerve Head. In Advanced Structural Safety Studies; Springer: Singapore, 2017; Volume 1695, pp. 299–310. [Google Scholar]
- Yu-Wai-Man, C.; Owen, N.; Lees, J.; Tagalakis, A.D.; Hart, S.L.; Webster, A.R.; Orengo, C.A.; Khaw, P.T. Genome-wide RNA-Sequencing analysis identifies a distinct fibrosis gene signature in the conjunctiva after glaucoma surgery. Sci. Rep. 2017, 7, 5644. [Google Scholar] [CrossRef] [PubMed]
- Parekh, S.; Ziegenhain, C.; Vieth, B.; Enard, W.; Hellmann, I. The impact of amplification on differential expression analyses by RNA-seq. Sci. Rep. 2016, 6, 25533. [Google Scholar] [CrossRef]
- Zhao, W.; He, X.; Hoadley, K.A.; Parker, J.S.; Hayes, D.N.; Perou, C.M. Comparison of RNA-Seq by poly(A) capture, ribosomal RNA depletion, and DNA microarray for expression profiling. BMC Genom. 2014, 15, 1–11. [Google Scholar] [CrossRef]
- O’Neil, D.; Glowatz, H.; Schlumpberger, M. Ribosomal RNA Depletion for Efficient Use of RNA-Seq Capacity. Curr. Protoc. Mol. Biol. 2013, 103, 1–8. [Google Scholar] [CrossRef]
- Peirson, S.N. Quantitative analysis of ocular gene expression. In Real-Time PCR, 1st ed.; Taylor & Francis: Abingdon, UK, 2007; pp. 135–154. [Google Scholar]
- Wang, W.; Mcnatt, L.G.; Shepard, A.; Jacobson, N.; Nishimura, D.; Stone, E.; Sheffield, V.; Clark, A. Optimal procedure for extracting RNA from human ocular tissues and expression profiling of the congenital glaucoma gene FOXC1 using quantitative RT-PCR. Mol. Vis. 2001, 7, 89–94. [Google Scholar]
- Peirson, S.N.; Butler, J.N.; Foster, R.G. Experimental validation of novel and conventional approaches to quantitative real-time PCR data analysis. Nucleic Acids Res. 2003, 31, e73. [Google Scholar] [CrossRef]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, research0034.1. [Google Scholar] [CrossRef] [PubMed]
- Schena, M.; Shalon, D.; Davis, R.W.; Brown, P.O. Quantitative Monitoring of Gene Expression Patterns with a Complementary DNA Microarray. Science 1995, 270, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Shendure, J. The beginning of the end for microarrays? Nat. Methods 2008, 5, 585–587. [Google Scholar] [CrossRef] [PubMed]
- Bumgarner, R. Overview of DNA Microarrays: Types, Applications, and Their Future. In Current Protocols in Molecular Biology; Wiley-Blackwell: Hoboken, NJ, USA, 2013; Volume 101. [Google Scholar] [CrossRef]
- Cieślik, M.; Chinnaiyan, A.M. Cancer transcriptome profiling at the juncture of clinical translation. Nat. Rev. Genet. 2018, 19, 93–109. [Google Scholar] [CrossRef]
- Byron, S.A.; Van Keuren-Jensen, K.R.; Engelthaler, D.M.; Carpten, J.D.; Craig, D.W. Translating RNA sequencing into clinical diagnostics: Opportunities and challenges. Nat. Rev. Genet. 2016, 17, 257–271. [Google Scholar] [CrossRef]
- Martersteck, E.M.; Hirokawa, K.E.; Evarts, M.; Bernard, A.; Duan, X.; Li, Y.; Ng, L.; Oh, S.W.; Ouellette, B.; Royall, J.J.; et al. Diverse Central Projection Patterns of Retinal Ganglion Cells. Cell Rep. 2017, 18, 2058–2072. [Google Scholar] [CrossRef]
- Baden, T.; Berens, P.; Franke, K.J.; Rosón, M.R.; Bethge, M.; Euler, T. The functional diversity of retinal ganglion cells in the mouse. Nature 2016, 529, 345–350. [Google Scholar] [CrossRef]
- Rheaume, B.A.; Jereen, A.; Bolisetty, M.; Sajid, M.S.; Yang, Y.; Renna, K.; Sun, L.; Robson, P.; Trakhtenberg, E.F. Single cell transcriptome profiling of retinal ganglion cells identifies cellular subtypes. Nat. Commun. 2018, 9, 2759. [Google Scholar] [CrossRef]
- Cui, Q.; Ren, C.; Sollars, P.J.; Pickard, G.E.; So, K.-F. The injury resistant ability of melanopsin-expressing intrinsically photosensitive retinal ganglion cells. Neuroscience 2015, 284, 845–853. [Google Scholar] [CrossRef]
- Kulkarni, A.; Anderson, A.G.; Merullo, D.P.; Konopka, G. Beyond bulk: A review of single cell transcriptomics methodologies and applications. Curr. Opin. Biotechnol. 2019, 58, 129–136. [Google Scholar] [CrossRef]
- Aldridge, S.; Teichmann, S.A. Single cell transcriptomics comes of age. Nat. Commun. 2020, 11, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Lake, B.B.; Codeluppi, S.; Yung, Y.C.; Gao, D.; Chun, J.; Kharchenko, P.; Linnarsson, S.; Zhang, K. A comparative strategy for single-nucleus and single-cell transcriptomes confirms accuracy in predicted cell-type expression from nuclear RNA. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kashima, Y.; Sakamoto, Y.; Kaneko, K.; Seki, M.; Suzuki, Y.; Suzuki, A. Single-cell sequencing techniques from individual to multiomics analyses. Exp. Mol. Med. 2020, 52, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.M.; Shekhar, K.; Whitney, I.E.; Jacobi, A.; Benhar, I.; Hong, G.; Yan, W.; Adiconis, X.; Arnold, M.E.; Lee, J.M.; et al. Single-Cell Profiles of Retinal Ganglion Cells Differing in Resilience to Injury Reveal Neuroprotective Genes. Neuron 2019, 104, 1039–1055.e12. [Google Scholar] [CrossRef] [PubMed]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 1–16. [Google Scholar] [CrossRef]
- Rodriques, S.G.; Chen, L.M.; Liu, S.; Zhong, E.D.; Scherrer, J.R.; Boyden, E.S.; Chen, F. RNA timestamps identify the age of single molecules in RNA sequencing. Nat. Biotechnol. 2020, 1–6. [Google Scholar] [CrossRef]
- Lee, J.; Hyeon, D.Y.; Hwang, D. Single-cell multiomics: Technologies and data analysis methods. Exp. Mol. Med. 2020, 52, 1428–1442. [Google Scholar] [CrossRef]
- Casasent, A.K.; Schalck, A.; Gao, R.; Sei, E.; Long, A.; Pangburn, W.; Casasent, T.; Meric-Bernstam, F.; Edgerton, M.E.; Navin, E.N. Multiclonal Invasion in Breast Tumors Identified by Topographic Single Cell Sequencing. Cell 2018, 172, 205–217.e12. [Google Scholar] [CrossRef]
- Reinhard, K.; Tikidji-Hamburyan, A.; Seitter, H.; Idrees, S.; Mutter, M.; Benkner, B.; Münch, T.A. Step-By-Step Instructions for Retina Recordings with Perforated Multi Electrode Arrays. PLoS ONE 2014, 9, e106148. [Google Scholar] [CrossRef]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 1–19. [Google Scholar] [CrossRef]
- Vieth, B.; Parekh, S.; Ziegenhain, C.; Enard, W.; Hellmann, I. A systematic evaluation of single cell RNA-seq analysis pipelines. Nat. Commun. 2019, 10, 4667. [Google Scholar] [CrossRef] [PubMed]
- Hwang, B.; Lee, J.H.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Costa-Silva, J.; Domingues, D.; Lopes, F.M. RNA-Seq differential expression analysis: An extended review and a software tool. PLoS ONE 2017, 12, e0190152. [Google Scholar] [CrossRef] [PubMed]
- McDermaid, A.; Monier, B.; Zhao, J.; Liu, B.; Ma, Q. Interpretation of differential gene expression results of RNA-seq data: Review and integration. Brief. Bioinform. 2019, 20, 2044–2054. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.J. Precision Medicine in Practice: Molecular Diagnosis Enabling Precision Therapies. Clin. Lab. Med. 2020, 40, 113–230. [Google Scholar] [CrossRef]
- Thompson, K.; Collier, J.J.; Glasgow, R.I.C.; Robertson, F.M.; Pyle, A.; Blakely, E.L.; Alston, C.L.; Oláhová, M.; McFarland, R.; Taylor, R.W. Recent advances in understanding the molecular genetic basis of mitochondrial disease. J. Inherit. Metab. Dis. 2020, 43, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Stenton, S.L.; Prokisch, H. Advancing genomic approaches to the molecular diagnosis of mitochondrial disease. Essays Biochem. 2018, 62, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Kremer, L.S.; Bader, D.M.; Mertes, C.; Kopajtich, R.; Pichler, G.; Iuso, A.; Haack, T.B.; Graf, E.; Schwarzmayr, T.; Terrile, C.; et al. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat. Commun. 2017, 8, 15824. [Google Scholar] [CrossRef]
- Frésard, L.; Smail, C.; Ferraro, N.M.; Teran, N.A.; Li, X.; Smith, K.S.; Bonner, D.; Kernohan, K.D.; Marwaha, S.; Zappala, Z.; et al. Identification of rare-disease genes using blood transcriptome sequencing and large control cohorts. Nat. Med. 2019, 25, 911–919. [Google Scholar] [CrossRef]
- Oshlack, A.; Robinson, M.D.; Young, M.D. From RNA-seq reads to differential expression results. Genome Biol. 2010, 11, 1–10. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14–R12. [Google Scholar] [CrossRef] [PubMed]
- Rahmatallah, Y.; Emmert-Streib, F.; Glazko, G. Gene set analysis approaches for RNA-seq data: Performance evaluation and application guideline. Brief. Bioinform. 2015, 17, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Mathur, R.; Rotroff, D.; Ma, J.; Shojaie, A.; Motsinger-Reif, A. Gene set analysis methods: A systematic comparison. BioData Min. 2018, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology Consortium. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Elstner, M.; Turnbull, D.M. Transcriptome analysis in mitochondrial disorders. Brain Res. Bull. 2012, 88, 285–293. [Google Scholar] [CrossRef]
- Danielson, S.R.; Carelli, V.; Tan, G.; Martinuzzi, A.; Schapira, A.H.V.; Savontaus, M.-L.; Cortopassi, G. Isolation of transcriptomal changes attributable to LHON mutations and the cybridization process. Brain 2005, 128, 1026–1037. [Google Scholar] [CrossRef]
- Cortopassi, G.; Danielson, S.; Alemi, M.; Zhan, S.S.; Tong, W.; Carelli, V.; Martinuzzi, A.; Marzuki, S.; Majamaa, K.; Wong, A. Mitochondrial disease activates transcripts of the unfolded protein response and cell cycle and inhibits vesicular secretion and oligodendrocyte-specific transcripts. Mitochondrion 2006, 6, 161–175. [Google Scholar] [CrossRef]
- Yu, A.K.; Song, L.; Murray, K.D.; Van Der List, D.; Sun, C.; Shen, Y.; Xia, Z.; Cortopassi, G. Mitochondrial complex I deficiency leads to inflammation and retinal ganglion cell death in the Ndufs4 mouse. Hum. Mol. Genet. 2015, 24, 2848–2860. [Google Scholar] [CrossRef]
- Cheng, J.; Liu, M.; Kaushik, A.M.; Chang, X.; Duan, Y.; Chen, L.; Wang, J.; Berlinicke, C.; Zack, D.J. Single-Cell Transcriptome Profiling of Human Stem Cell-Derived Retinal Ganglion Cells in a Dominant Optic Atrophy Model. In Proceedings of ARVO Annual Meeting Abstract, Honolulu, HI, USA, 29 April 2018; Investigative Ophthalmology & Visual Science: Washington, DC, USA, 2018; Volume 59, 1998p. [Google Scholar]
- Wu, Y.-R.; Wang, A.-G.; Chen, Y.-T.; Yarmishyn, A.A.; Buddhakosai, W.; Yang, T.-C.; Hwang, D.-K.; Yang, Y.-P.; Shen, C.-N.; Lee, H.-C.; et al. Bioactivity and gene expression profiles of hiPSC-generated retinal ganglion cells in MT-ND4 mutated Leber’s hereditary optic neuropathy. Exp. Cell Res. 2018, 363, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Caglayan, S.; Hashim, A.; Cieslar-Pobuda, A.; Jensen, V.; Behringer, S.; Talug, B.; Chu, D.T.; Pecquet, C.; Rogne, M.; Brech, A.; et al. Optic Atrophy 1 Controls Human Neuronal Development by Preventing Aberrant Nuclear DNA Methylation. iScience 2020, 23, 101154. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Sant, D.W.; Wang, G.; Guy, J. Mitochondrial Transfer of the Mutant Human ND6T14484C Gene Causes Visual Loss and Optic Neuropathy. Transl. Vis. Sci. Technol. 2020, 9, 1. [Google Scholar] [CrossRef]
- Fang, Z.; Cui, X. Design and validation issues in RNA-seq experiments. Brief. Bioinform. 2011, 12, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Lanza, I.R.; Nair, K.S. Mitochondrial metabolic function assessed in vivo and in vitro. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.R.; Bennett, J.; Wellman, J.; Do, D.C.C.; Yu, Z.-F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Douglas, R.M.; Alam, N.M.; Silver, B.D.; McGill, T.J.; Tschetter, W.W.; Prusky, G.T. Independent visual threshold measurements in the two eyes of freely moving rats and mice using a virtual-reality optokinetic system. Vis. Neurosci. 2005, 22, 677–684. [Google Scholar] [CrossRef]
- Safety Evaluation of Gene Therapy in Leber Hereditary Optic Neuropathy (LHON) Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT02064569 (accessed on 22 January 2021).
- Yu-Wai-Man, P.; Newman, N.J.; Carelli, V.; Moster, M.L.; Biousse, V.; Sadun, A.A.; Klopstock, T.; Vignal-Clermont, C.; Sergott, R.C.; Rudolph, G.; et al. Bilateral visual improvement with unilateral gene therapy for Leber hereditary optic neuropathy. Sci. Transl. Med. 2020, 12, eaaz7423. [Google Scholar] [CrossRef]
- Rodon, J.; Soria, J.-C.; Berger, R.; Miller, W.H.; Rubin, E.; Kugel, A.; Tsimberidou, A.; Saintigny, P.; Ackerstein, A.; Braña, I.; et al. Genomic and transcriptomic profiling expands precision cancer medicine: The WINTHER trial. Nat. Med. 2019, 25, 751–758. [Google Scholar] [CrossRef]
- Ratican, S.E.; Osborne, A.; Martin, K.R. Progress in Gene Therapy to Prevent Retinal Ganglion Cell Loss in Glaucoma and Leber’s Hereditary Optic Neuropathy. Neural Plast. 2018, 2018, 1–11. [Google Scholar] [CrossRef]
- Osborne, A.; Wang, A.X.; Tassoni, A.; Widdowson, P.S.; Martin, K.R. Design of a Novel Gene Therapy Construct to Achieve Sustained Brain-Derived Neurotrophic Factor Signaling in Neurons. Hum. Gene Ther. 2018, 29, 828–841. [Google Scholar] [CrossRef] [PubMed]
- de Silva, S.R.; McClements, M.E.; Hankins, M.W.; MacLaren, R.E. Adeno-Associated Viral Gene Therapy for Retinal Disorders. In Gene Delivery and Therapy for Neurological Disorders; Bo, X., Verhaagen, J., Eds.; Springer: New York, NY, USA, 2015; Volume 98, pp. 203–228. [Google Scholar]
- Moster, M.; Sadun, A.; Klopstock, T.; Newman, N.; Vignal-Clermont, C.; Carelli, V.; Yu-Wai-Man, P.; Biousse, V.; Sergott, R.; Katz, B.; et al. rAAV2/2-ND4 for the Treatment of Leber Hereditary Optic Neuropathy (LHON): Final Results from the RESCUE and REVERSE Phase III Clinical Trials and Experimental Data in Nonhuman Primates to Support a Bilateral Effect (2339). Neurology 2020, 94, 2339. [Google Scholar]
- Yu-Wai-Man, P.; Newman, N.J.; Carelli, V.; Biousse, V.; Sadun, A.A.; Moster, M.L.; Vignal-Clermont, C.; Sergott, R.C.; Klopstock, T.; Blouin, L.; et al. Bilateral Visual Improvement with Unilateral Gene Therapy for Leber Hereditary Optic Neuropathy (LHON). Invest. Ophthalmol. Vis. Sci. 2020, 61, 5181. [Google Scholar]
- Rossmiller, B.; Mao, H.; Lewin, A.S. Gene therapy in animal models of autosomal dominant retinitis pigmentosa. Mol. Vis. 2012, 18, 2479–2496. [Google Scholar] [PubMed]
- Orlans, H.O.; McClements, M.E.; Barnard, A.R.; Martinez-Fernandez, d.C.; MacLaren, R.E. Mirtron gene therapy for the treatment of rhodopsin-related dominant retinitis pigmentosa. In Proceedings of ARVO Annual Meeting Abstract, Vancouver, BC, Canada, 30 April 2019; Investigative Ophthalmology & Visual Science: Wahington, DC, USA, 2019; Volume 60. [Google Scholar]
- Cideciyan, A.V.; Sudharsan, R.; Dufour, V.L.; Massengill, M.T.; Iwabe, S.; Swider, M.; Lisi, B.; Sumaroka, A.; Marinho, L.F.; Appelbaum, T.; et al. Mutation-independent rhodopsin gene therapy by knockdown and replacement with a single AAV vector. Proc. Natl. Acad. Sci. USA 2018, 115, E8547–E8556. [Google Scholar] [CrossRef]
- Massengill, M.T.; Young, B.M.; Lewin, A.S.; Ildefonso, C.J. Co-Delivery of a Short-Hairpin RNA and a shRNA-Resistant Replacement Gene with Adeno-Associated Virus: An Allele-Independent Strategy for Autosomal-Dominant Retinal Disorders. In Methods in Molecular Biology; Springer: New York, NY, USA, 2019; pp. 235–258. [Google Scholar] [CrossRef]
- Tribble, J.R.; Otmani, A.; Sun, S.; Ellis, S.A.; Cimaglia, G.; Vohra, R.; Joe, M.; Lardner, E.; Venkataraman, A.P.; Dominguez-Vicent, A.; et al. Nicotinamide provides neuroprotection in glaucoma by protecting against mitochondrial and metabolic dysfunction. bioRxiv 2020, 2020.10.21.348250. Available online: https://www.biorxiv.org/content/10.1101/2020.10.21.348250v1 (accessed on 22 January 2021).
- Fu, L.; Kwok, S.S.; Chan, Y.-K.; Lai, J.S.M.; Pan, W.; Nie, L.; Shih, K.C. Therapeutic Strategies for Attenuation of Retinal Ganglion Cell Injury in Optic Neuropathies: Concepts in Translational Research and Therapeutic Implications. BioMed Res. Int. 2019, 2019, 1–10. [Google Scholar] [CrossRef]
- Martin, K.; Martin, K.R. Neuroprotection in Glaucoma: Towards Clinical Trials and Precision Medicine. Curr. Eye Res. 2020, 45, 327–338. [Google Scholar] [CrossRef]
- Pasi, K.J.; Rangarajan, S.; Mitchell, N.; Lester, W.; Symington, E.; Madan, B.; Laffan, M.; Russell, C.B.; Li, M.; Pierce, G.F.; et al. Multiyear Follow-up of AAV5-hFVIII-SQ Gene Therapy for Hemophilia A. N. Engl. J. Med. 2020, 382, 29–40. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).