
A Novel Splice-Site Variant in CACNA1F Causes a Phenotype Synonymous with Åland Island Eye Disease and Incomplete Congenital Stationary Night Blindness

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
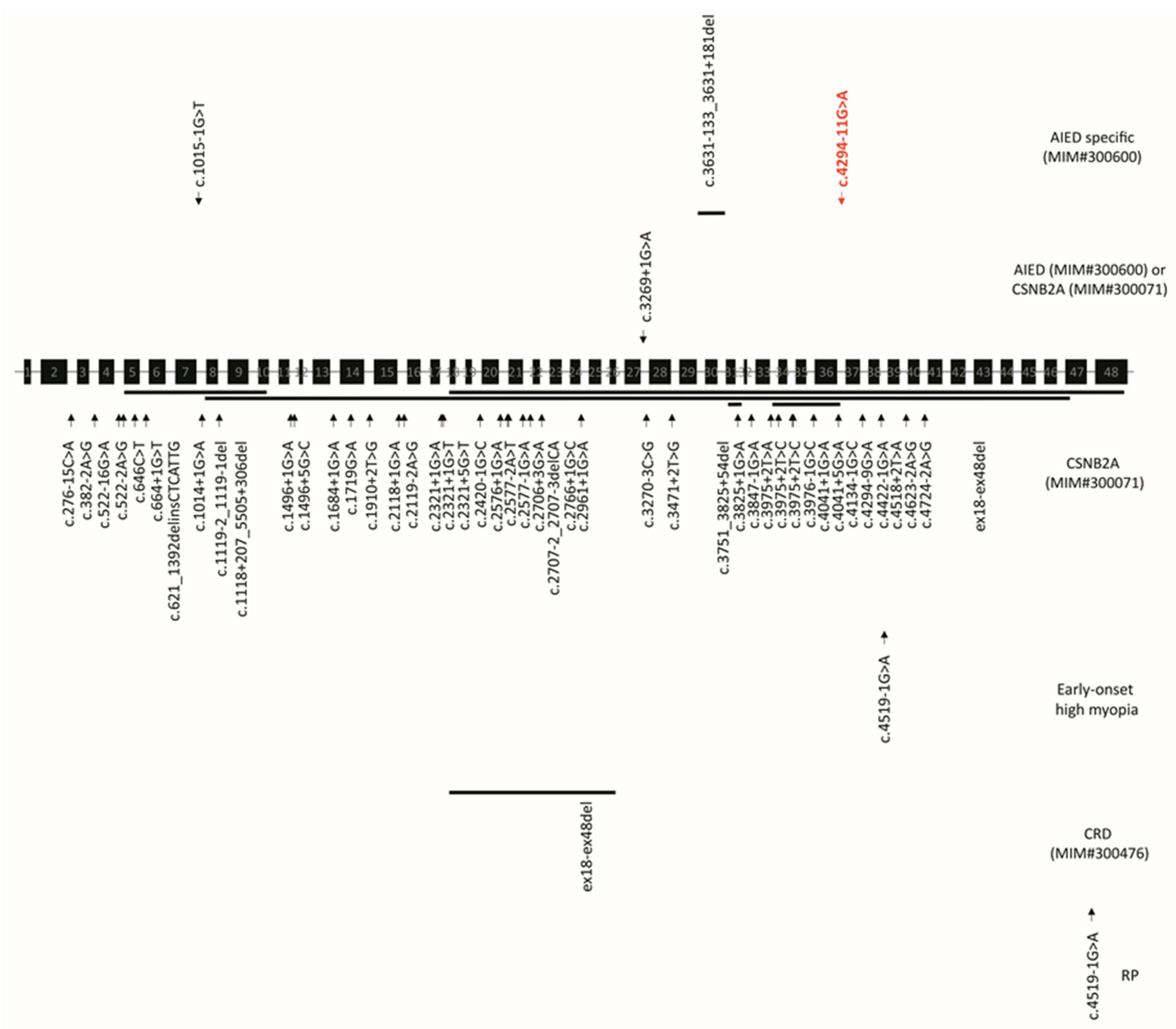
2. Case Description
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Forsius, H.; Eriksson, A.W. A new eye syndrome with x–chromosomal transmission. A family clan with fundus albinism, fovea hypoplasia, nystagmus, myopia, astigmatism and dyschromatopsia. Klin. Monatsbl. Augenheilkd 1964, 144, 447–457. [Google Scholar] [PubMed]
- Van Dorp, D.B.; Eriksson, A.W.; Delleman, J.W.; van Vliet, A.G.; Collewijn, H.; van Balen, A.T.; Forsius, H.R. Aland eye disease: No albino misrouting. Clin. Genet. 1985, 28, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Hawksworth, N.R.; Headland, S.; Good, P.; Thomas, N.S.; Clarke, A. Aland island eye disease: Clinical and electrophysiological studies of a Welsh family. Br. J. Ophthalmol. 1995, 79, 424–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, S.; Vesti, E.; Raitta, C.; Donner, M.; Eriksson, A.W.; Forsius, H. Clinical and electroretinographic comparison between Aland island eye disease and a newly found related disease with X-chromosomal inheritance. Acta Ophthalmol. (Copenh) 1991, 69, 703–710. [Google Scholar] [CrossRef]
- Waardenburg, P.J. Some notes on publications of Professor Arnold Sorsby and Åland Eye Disease (Forsius-Eriksson syndrome). J. Med. Genet. 1970, 7, 194–199. [Google Scholar] [CrossRef] [Green Version]
- Elenius, V.; Eriksson, A.; Forsius, H. ERG in a case of X-chromosomal pigment deficiency of fundus in combination with myopia, dyschromatopsia and defective dark adaptation. In The Clinical Value of Electroretinography, Proceedings of the 5th ISCERG Symposium, Ghent, Belgium; Francois, J., Ed.; Karger: Basel, Switzerland; New York, NY, USA, 1968; pp. 369–377. [Google Scholar]
- Van Vliet, A.G.; Waardenburg, P.J.; Forsius, H.; Eriksson, A.W. Nystagmographical studies in Aland eye disease. Acta Ophthalmol. (Copenh) 1973, 51, 782–790. [Google Scholar] [CrossRef]
- Jalkanen, R.; Tobias, R.; Mäntyjärvi, M.; De La Chapelle, A.; Bech-Hansen, N.T.; Sankila, E.-M.; Forsius, H.; Alitalo, T. A novel CACNA1F gene mutation causes Åland island eye disease. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2498–2502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strom, T.M.; Nyakatura, G.; Apfelstedt-Sylla, E.; Hellebrand, H.; Lorenz, B.; Weber, B.H.F.; Wutz, K.; Gutwillinger, N.; Rüther, K.; Drescher, B.; et al. An L-type calcium-channel gene mutated in incomplete X-linked congenital stationary night blindness. Nat. Genet. 1998, 19, 260–263. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.; Wright, T.; Day, M.A.; Westall, C.A.; Héon, E. A novel p.Gly603Arg mutation in CACNA1F causes Åland island eye disease and incomplete congenital stationary night blindness phenotypes in a family. Mol. Vis. 2011, 17, 3262–3270. [Google Scholar] [PubMed]
- Wutz, K.; Sauer, C.; Zrenner, E.; Lorenz, B.; Alitalo, T.; Broghammer, M.; Hergersberg, M.; De La Chapelle, A.; Weber, B.H.F.; Wissinger, B.; et al. Thirty distinct CACNA1F mutations in 33 families with incomplete type of XLCSNB and Cacna1f expression profiling I mouse retina. Eur. J. Hum. Genet. 2002, 10, 449–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDonald, I.M.; Hoang, S.; Tuupanen, S. X-linked congenital stationary night blindness. In GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 2019; (Updated 3 July 2019). [Google Scholar]
- Naylor, M.J.; Rancourt, D.E.; Bech-Hansen, N. Isolation and characterization of a calcium channel gene, Cacnaf1, the murine orthologue of the gene for incomplete X-linked congenital stationary night blindness. Genomics 2000, 66, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ellinor, P.T.; Sather, W.A.; Zhang, J.-F.; Tsien, R.W. Molecular determinants of Ca2+ selectivity and ion permeation in L-type Ca2+ channels. Nature 1993, 366, 158–161. [Google Scholar] [CrossRef] [PubMed]
- Zeitz, C.; Michiels, C.; Nauille, M.; Friedburg, C.; Condroyer, C.; Boyard, F.; Antonio, A.; Bouzidi, N.; Milicevic, D.; Veaux, R.; et al. Where are the missing gene defects in inherited retinal disorders? Intronic and synonymous variants contribute at least to 4% of CACNA1F-mediated inherited retinal disorders. Hum. Mutat. 2019, 40, 765–787. [Google Scholar] [CrossRef] [PubMed]
- Mansergh, F.C.; Orton, N.C.; Vessey, J.P.; LaLonde, M.R.; Stell, W.K.; Tremblay, F.; Barnes, S.; Rancourt, D.E.; Bech-Hansen, N.T. Mutation of the calcium channel gene Cacna1f disrupts calcium signaling, synaptic transmission and cellular organization in mouse retina. Hum. Mol. Genet. 2005, 14, 3035–3046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyake, Y.; Yagasaki, K.; Horiguchi, M.; Kawase, Y.; Kanda, T. Congenital stationary night blindness with negative electroretinogram. A new classification. Arch. Ophthalmol. 1986, 104, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Ito, S.; Piao, C.H.; Terasaki, H.; Miyake, Y. Retinal and optic disc atrophy associated with a CACNA1F mutation in a Japanese family. Arch. Ophthalmol. 2003, 121, 1028–1033. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, T.; Schwartz, M.; Simonsen, S.E. Åland eye disease (Forsius-Eriksson-Miyake syndrome) with probability established in a Danish family. Acta Ophthalmol. (Copenh) 1990, 68, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Zeitz, C.; Robson, A.G.; Audo, I. Congenital stationary night blindness: An analysis and update of genotype-phenotype correlations and pathogenic mechanisms. Prog. Retin. Eye Res. 2015, 45, 58–110. [Google Scholar] [CrossRef] [PubMed]
- Boycott, K.M.; Pearce, W.G.; Bech-Hansen, N.T. Clinical variability among patients with incomplete X-linked congenital stationary night blindness and a founder mutation in CACNA1F. Can. J. Ophthalmol. 2000, 35, 204–213. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahmood, U.; Méjécase, C.; Ali, S.M.A.; Moosajee, M.; Kozak, I. A Novel Splice-Site Variant in CACNA1F Causes a Phenotype Synonymous with Åland Island Eye Disease and Incomplete Congenital Stationary Night Blindness. Genes 2021, 12, 171. https://doi.org/10.3390/genes12020171
Mahmood U, Méjécase C, Ali SMA, Moosajee M, Kozak I. A Novel Splice-Site Variant in CACNA1F Causes a Phenotype Synonymous with Åland Island Eye Disease and Incomplete Congenital Stationary Night Blindness. Genes. 2021; 12(2):171. https://doi.org/10.3390/genes12020171
Chicago/Turabian StyleMahmood, Usman, Cécile Méjécase, Syed M. A. Ali, Mariya Moosajee, and Igor Kozak. 2021. "A Novel Splice-Site Variant in CACNA1F Causes a Phenotype Synonymous with Åland Island Eye Disease and Incomplete Congenital Stationary Night Blindness" Genes 12, no. 2: 171. https://doi.org/10.3390/genes12020171