
The Crossroads between RAS and RHO Signaling Pathways in Cellular Transformation, Motility and Contraction



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
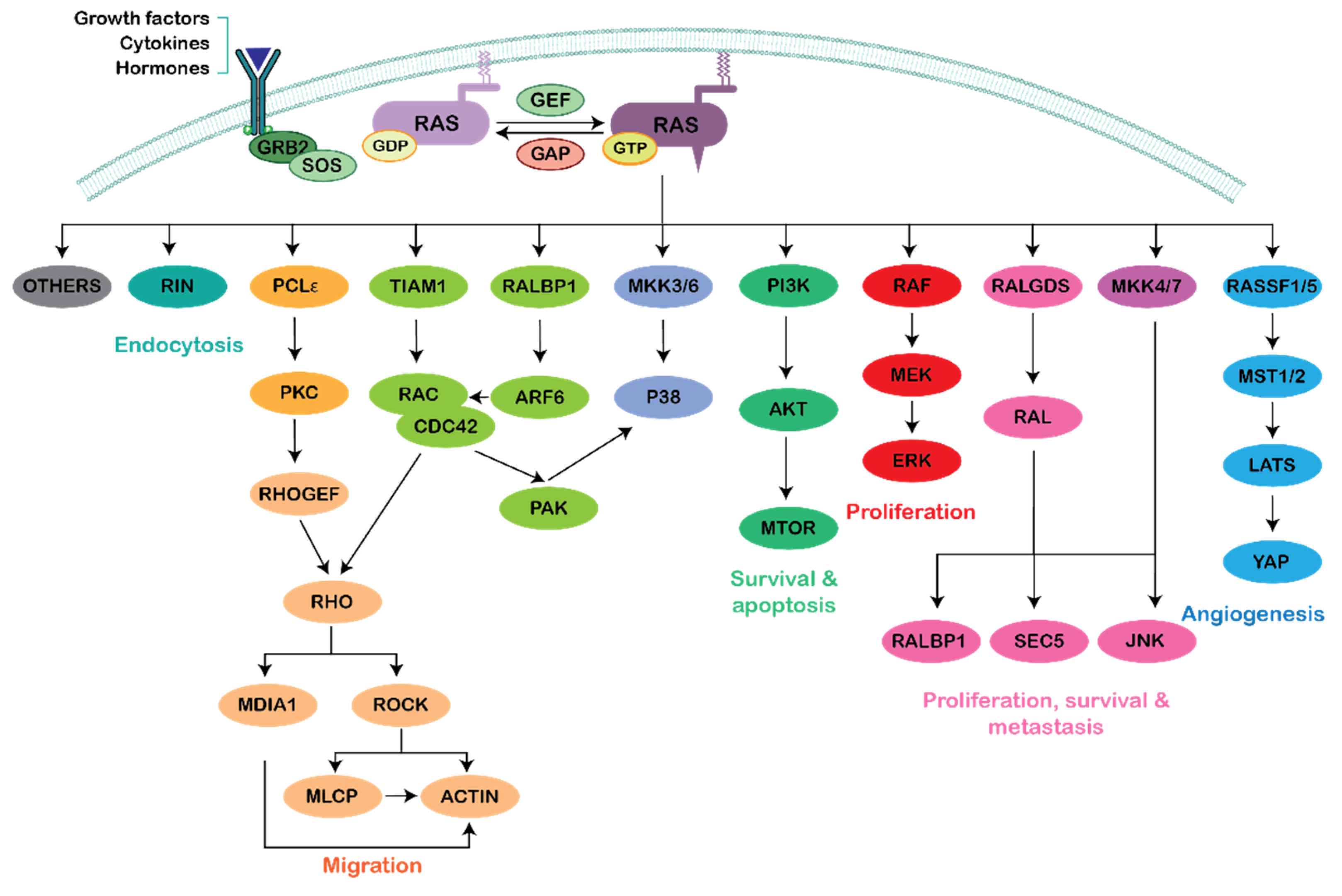
:1. Introduction
2. Ras–Rho Crosstalk through PI3K Activation
3. Ras–Rho Crosstalk through Raf/MEK/ERK Activation
4. Ras–Rho Crosstalk through GEFs and GAPs
4.1. GEFs
4.2. GAPs
5. Ras–Rho Crosstalk through Ral
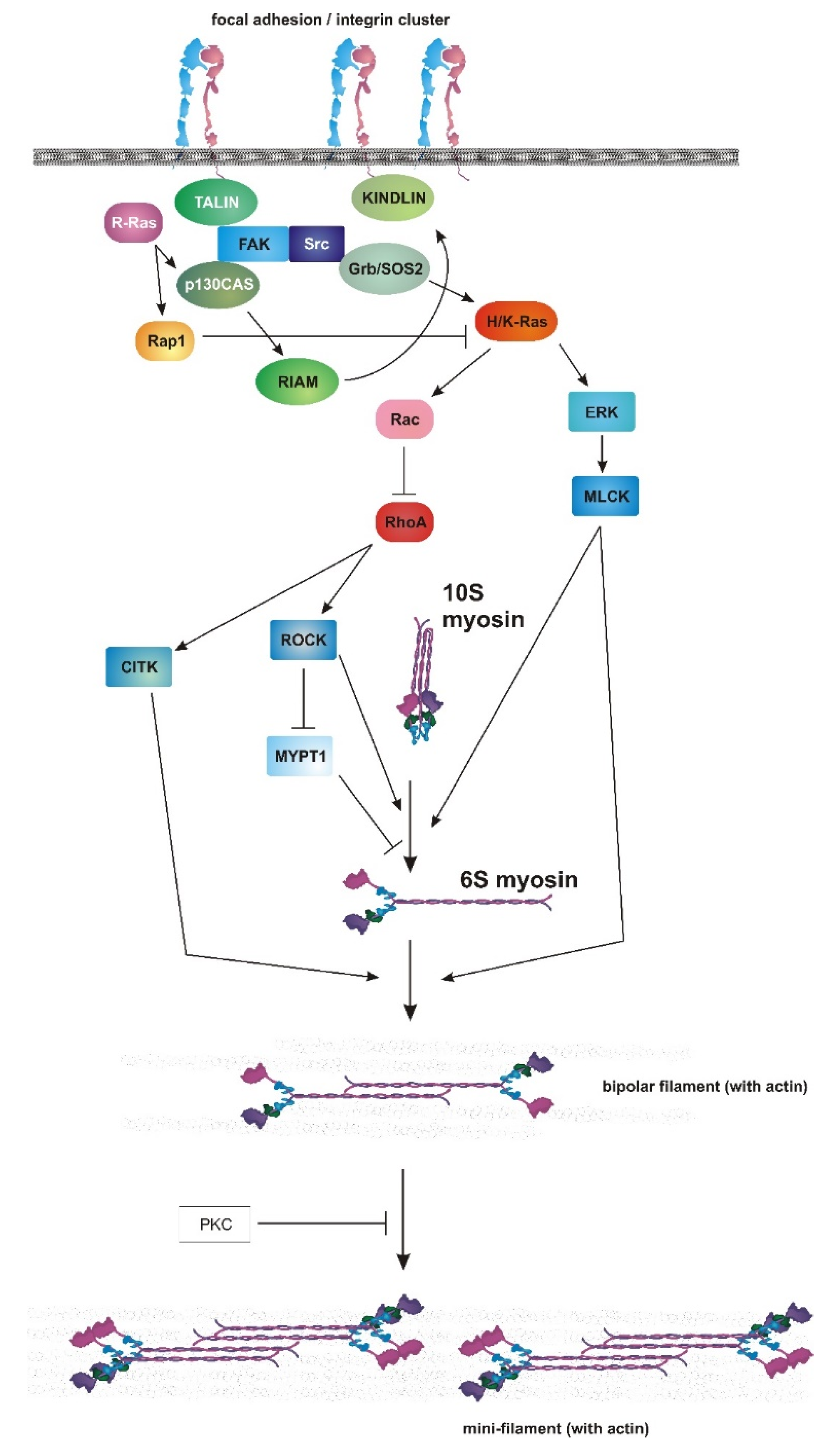
6. Ras, Rac and Mechanosensing
7. Myosin II as an Endpoint of Chemical and Mechanical Signaling
8. Role of RAS–MAPK in Myosin II Activation and Adhesion Dynamics
9. Connecting PI3K to Cellular Contractility
10. Mechanical Control of PI3K Activity
11. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| EGFR | epidermal growth factor receptor |
| GEFs | guanine nucleotide exchange-factors |
| GAPs | GTPase-activating proteins |
| MAPK | mitogen-activated protein kinase |
| PI3K | phosphoinositide 3-kinase |
| Cdc42 | Cell Division Cycle 42 |
| Nf1 | neurofibromin |
| ROCK | Rho-associated protein kinase |
| TGF-β | transforming growth factor beta |
| EMT | epithelial to mesenchymal transition |
| ERK | extracellular signal-regulated kinase |
| RBD | Ras-binding domain |
| EGF | epidermal growth factor |
| FGF2 | fibroblast growth factor 2 |
| MEF | mouse embryonic fibroblast |
| HGF | hepatocyte growth factor |
| PDGF | platelet-derived growth factor |
| PtdIns(3,4,5)P3 | phosphatidylinositol (3,4,5)-trisphosphate |
| PH domain | pleckstrin homology domain |
| Tiam-1 | T-cell lymphoma invasion and metastasis 1 protein |
| WRC | Wave Regulatory Complex |
| PTEN | phosphatidylinositol-3;4;5-trisphosphate 3-phosphatase |
| SHIP-1 | Src homology 2 domain containing inositol polyphosphate 5-phosphatase 1 |
| PtdIns(4,5)P2 | Phosphatidylinositol 4;5-bisphosphate |
| PtdIns(3,4)P2 | Phosphatidylinositol (3,4)-bisphosphate |
| PREX1 | phosphatidylinositol-3,4,5-trisphosphate-dependent Rac exchange factor |
| GPCR | G protein-coupled receptor |
| LPA | lipoprotein A |
| S1P | sphingosine-1-phosphate |
| DLC1 | deleted in liver cancer 1 |
| IGF-1 | insulin-like growth factor-1 |
| mTORC2 | mammalian target of rapamycin complex 2 |
| MEK | mitogen-activated protein kinase kinase |
| WAVE2 | Wiskott–Aldrich Syndrome Protein-family verprolin homologous protein 2 |
| RACK1 | receptor for activated C kinase 1 |
| CCL2 | chemokine (C–C motif) ligand 2 |
| CRD | cysteine-rich domain |
| TNF-α | tumor necrosis factor alpha |
| ARHGEF2 | Rho guanine nucleotide exchange factor 2 |
| Sos | son of sevenless |
| Grb2 | growth factor receptor-bound protein 2 |
| BCR | breakpoint cluster region protein |
| SH2 | Src homology 2 |
| SH3 | Src homology 3 |
| FAK | Focal Adhesion Kinase |
| RGL1 | Ral guanine nucleotide dissociation stimulator-like 1 |
| RGL2 | Ral guanine nucleotide dissociation stimulator-like 2 |
| YAP | yes-associated protein 1 |
| ECM | extracellular matrix |
| HER-2 | human epidermal growth factor receptor 2 |
| WT | wild-type |
References
- Reiner, D.J.; Lundquist, E.A. Small GTPases. WormBook 2018, 2018, 1–65. [Google Scholar] [CrossRef] [PubMed]
- Wennerberg, K.; Rossman, K.L.; Der, C.J. The Ras superfamily at a glance. J. Cell Sci. 2005, 118, 843–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef] [Green Version]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef]
- Fakih, M.; O’Neil, B.; Price, T.J.; Falchook, G.S.; Desai, J.; Kuo, J.; Govindan, R.; Rasmussen, E.; Morrow, P.K.; Ngang, J.; et al. Phase 1 study evaluating the safety, tolerability, pharmacokinetics (PK), and efficacy of AMG 510, a novel small molecule KRASG12C inhibitor, in advanced solid tumors. J. Clin. Oncol. 2019, 37, 3003. [Google Scholar] [CrossRef]
- Uprety, D.; Adjei, A.A. KRAS: From undruggable to a druggable Cancer Target. Cancer Treat. Rev. 2020, 89, 102070. [Google Scholar] [CrossRef]
- Cherfils, J.; Zeghouf, M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 2013, 93, 269–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. GEFs and GAPs: Critical elements in the control of small G proteins. Cell 2007, 129, 865–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiel, C.; Matallanas, D.; Kolch, W. The Ins and Outs of RAS Effector Complexes. Biomolecules 2021, 11, 236. [Google Scholar] [CrossRef]
- Krygowska, A.A.; Castellano, E. PI3K: A Crucial Piece in the RAS Signaling Puzzle. Cold Spring Harb. Perspect. Med. 2018, 8, a031450. [Google Scholar] [CrossRef]
- Terrell, E.M.; Morrison, D.K. Ras-Mediated Activation of the Raf Family Kinases. Cold Spring Harb. Perspect. Med. 2019, 9, a033746. [Google Scholar] [CrossRef]
- Madaule, P.; Axel, R. A novel ras-related gene family. Cell 1985, 41, 31–40. [Google Scholar] [CrossRef]
- Hodge, R.G.; Schaefer, A.; Howard, S.V.; Der, C.J. RAS and RHO family GTPase mutations in cancer: Twin sons of different mothers? Crit. Rev. Biochem. Mol. Biol. 2020, 55, 386–407. [Google Scholar] [CrossRef]
- Nobes, C.D.; Hall, A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 1995, 81, 53–62. [Google Scholar] [CrossRef] [Green Version]
- Nobes, C.D.; Hall, A. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J. Cell Biol. 1999, 144, 1235–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridley, A.J.; Paterson, H.F.; Johnston, C.L.; Diekmann, D.; Hall, A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 1992, 70, 401–410. [Google Scholar] [CrossRef]
- Bustelo, X.R. RHO GTPases in cancer: Known facts, open questions, and therapeutic challenges. Biochem. Soc. Trans. 2018, 46, 741–760. [Google Scholar] [CrossRef]
- Lawson, C.D.; Ridley, A.J. Rho GTPase signaling complexes in cell migration and invasion. J. Cell Biol. 2018, 217, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Marshall, C.J. RHO-GTPases and cancer. Nat. Rev. Cancer 2002, 2, 133–142. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Khosravi-Far, R.; Solski, P.A.; Kurzawa, H.; Lebowitz, P.F.; Der, C.J. Critical role of Rho in cell transformation by oncogenic Ras. Oncogene 1995, 10, 2289–2296. [Google Scholar]
- Qiu, R.G.; Chen, J.; McCormick, F.; Symons, M. A role for Rho in Ras transformation. Proc. Natl. Acad. Sci. USA 1995, 92, 11781–11785. [Google Scholar] [CrossRef] [Green Version]
- Sahai, E.; Olson, M.F.; Marshall, C.J. Cross-talk between Ras and Rho signalling pathways in transformation favours proliferation and increased motility. EMBO J. 2001, 20, 755–766. [Google Scholar] [CrossRef] [Green Version]
- Qiu, R.G.; Chen, J.; Kirn, D.; McCormick, F.; Symons, M. An essential role for Rac in Ras transformation. Nature 1995, 374, 457–459. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.F.; Paterson, H.F.; Marshall, C.J. Signals from Ras and Rho GTPases interact to regulate expression of p21Waf1/Cip1. Nature 1998, 394, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Danen, E.H.; Sonneveld, P.; Sonnenberg, A.; Yamada, K.M. Dual stimulation of Ras/mitogen-activated protein kinase and RhoA by cell adhesion to fibronectin supports growth factor-stimulated cell cycle progression. J. Cell Biol. 2000, 151, 1413–1422. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.M.; Lambert, Q.T.; Reuther, G.W.; Malliri, A.; Siderovski, D.P.; Sondek, J.; Collard, J.G.; Der, C.J. Tiam1 mediates Ras activation of Rac by a PI(3)K-independent mechanism. Nat. Cell Biol. 2002, 4, 621–625. [Google Scholar] [CrossRef]
- Bar-Sagi, D.; Feramisco, J.R. Induction of membrane ruffling and fluid-phase pinocytosis in quiescent fibroblasts by ras proteins. Science 1986, 233, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Walsh, A.B.; Bar-Sagi, D. Differential activation of the Rac pathway by Ha-Ras and K-Ras. J. Biol. Chem. 2001, 276, 15609–15615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joneson, T.; White, M.A.; Wigler, M.H.; Bar-Sagi, D. Stimulation of membrane ruffling and MAP kinase activation by distinct effectors of RAS. Science 1996, 271, 810–812. [Google Scholar] [CrossRef]
- Khosravi-Far, R.; Solski, P.A.; Clark, G.J.; Kinch, M.S.; Der, C.J. Activation of Rac1, RhoA, and mitogen-activated protein kinases is required for Ras transformation. Mol. Cell. Biol. 1995, 15, 6443–6453. [Google Scholar] [CrossRef] [Green Version]
- Kissil, J.L.; Walmsley, M.J.; Hanlon, L.; Haigis, K.M.; Bender Kim, C.F.; Sweet-Cordero, A.; Eckman, M.S.; Tuveson, D.A.; Capobianco, A.J.; Tybulewicz, V.L.; et al. Requirement for Rac1 in a K-ras induced lung cancer in the mouse. Cancer Res. 2007, 67, 8089–8094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heid, I.; Lubeseder-Martellato, C.; Sipos, B.; Mazur, P.K.; Lesina, M.; Schmid, R.M.; Siveke, J.T. Early requirement of Rac1 in a mouse model of pancreatic cancer. Gastroenterology 2011, 141, 719–730. [Google Scholar] [CrossRef]
- Samuel, M.S.; Lourenco, F.C.; Olson, M.F. K-Ras mediated murine epidermal tumorigenesis is dependent upon and associated with elevated Rac1 activity. PLoS ONE 2011, 6, e17143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lionarons, D.A.; Hancock, D.C.; Rana, S.; East, P.; Moore, C.; Murillo, M.M.; Carvalho, J.; Spencer-Dene, B.; Herbert, E.; Stamp, G.; et al. RAC1(P29S) Induces a Mesenchymal Phenotypic Switch via Serum Response Factor to Promote Melanoma Development and Therapy Resistance. Cancer Cell 2019, 36, 68–83.e69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollard, T.D. Actin and Actin-Binding Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a018226. [Google Scholar] [CrossRef] [Green Version]
- Vicente-Manzanares, M.; Ma, X.; Adelstein, R.S.; Horwitz, A.R. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat. Rev. Mol. Cell. Biol. 2009, 10, 778–790. [Google Scholar] [CrossRef] [Green Version]
- Devreotes, P.; Horwitz, A.R. Signaling networks that regulate cell migration. Cold Spring Harb. Perspect. Biol. 2015, 7, a005959. [Google Scholar] [CrossRef] [Green Version]
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell migration: Integrating signals from front to back. Science 2003, 302, 1704–1709. [Google Scholar] [CrossRef] [Green Version]
- Campellone, K.G.; Welch, M.D. A nucleator arms race: Cellular control of actin assembly. Nat. Rev. Mol. Cell. Biol. 2010, 11, 237–251. [Google Scholar] [CrossRef] [Green Version]
- Julian, L.; Olson, M.F. Rho-associated coiled-coil containing kinases (ROCK): Structure, regulation, and functions. Small GTPases 2014, 5, e29846. [Google Scholar] [CrossRef] [PubMed]
- Pankova, K.; Rosel, D.; Novotny, M.; Brabek, J. The molecular mechanisms of transition between mesenchymal and amoeboid invasiveness in tumor cells. Cell. Mol. Life Sci. 2010, 67, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giehl, K. Oncogenic Ras in tumour progression and metastasis. Biol. Chem. 2005, 386, 193–205. [Google Scholar] [CrossRef]
- Campbell, P.M.; Der, C.J. Oncogenic Ras and its role in tumor cell invasion and metastasis. Semin. Cancer Biol. 2004, 14, 105–114. [Google Scholar] [CrossRef]
- Ridley, A.J. Rho GTPase signalling in cell migration. Curr. Opin. Cell Biol. 2015, 36, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Drosten, M.; Dhawahir, A.; Sum, E.Y.; Urosevic, J.; Lechuga, C.G.; Esteban, L.M.; Castellano, E.; Guerra, C.; Santos, E.; Barbacid, M. Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival. EMBO J. 2010, 29, 1091–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, M.A.; Kraut, N.; Beug, H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr. Opin. Cell Biol. 2005, 17, 548–558. [Google Scholar] [CrossRef]
- Turley, E.A.; Veiseh, M.; Radisky, D.C.; Bissell, M.J. Mechanisms of disease: Epithelial-mesenchymal transition—Does cellular plasticity fuel neoplastic progression? Nat. Clin. Pract. Oncol. 2008, 5, 280–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makrodouli, E.; Oikonomou, E.; Koc, M.; Andera, L.; Sasazuki, T.; Shirasawa, S.; Pintzas, A. BRAF and RAS oncogenes regulate Rho GTPase pathways to mediate migration and invasion properties in human colon cancer cells: A comparative study. Mol. Cancer 2011, 10, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritsch, R.; de Krijger, I.; Fritsch, K.; George, R.; Reason, B.; Kumar, M.S.; Diefenbacher, M.; Stamp, G.; Downward, J. RAS and RHO families of GTPases directly regulate distinct phosphoinositide 3-kinase isoforms. Cell 2013, 153, 1050–1063. [Google Scholar] [CrossRef] [Green Version]
- Orme, M.H.; Alrubaie, S.; Bradley, G.L.; Walker, C.D.; Leevers, S.J. Input from Ras is required for maximal PI(3)K signalling in Drosophila. Nat. Cell Biol. 2006, 8, 1298–1302. [Google Scholar] [CrossRef]
- Suire, S.; Condliffe, A.M.; Ferguson, G.J.; Ellson, C.D.; Guillou, H.; Davidson, K.; Welch, H.; Coadwell, J.; Turner, M.; Chilvers, E.R.; et al. Gbetagammas and the Ras binding domain of p110gamma are both important regulators of PI(3)Kgamma signalling in neutrophils. Nat. Cell Biol. 2006, 8, 1303–1309. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Ramjaun, A.R.; Haiko, P.; Wang, Y.; Warne, P.H.; Nicke, B.; Nye, E.; Stamp, G.; Alitalo, K.; Downward, J. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell 2007, 129, 957–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellano, E.; Molina-Arcas, M.; Krygowska, A.A.; East, P.; Warne, P.; Nicol, A.; Downward, J. RAS signalling through PI3-Kinase controls cell migration via modulation of Reelin expression. Nat. Commun. 2016, 7, 11245. [Google Scholar] [CrossRef] [Green Version]
- Castellano, E.; Sheridan, C.; Thin, M.Z.; Nye, E.; Spencer-Dene, B.; Diefenbacher, M.E.; Moore, C.; Kumar, M.S.; Murillo, M.M.; Gronroos, E.; et al. Requirement for interaction of PI3-kinase p110alpha with RAS in lung tumor maintenance. Cancer Cell 2013, 24, 617–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.W.; Shin, M.G.; Lee, S.; Kim, J.R.; Park, W.S.; Cho, K.H.; Meyer, T.; Heo, W.D. Cooperative activation of PI3K by Ras and Rho family small GTPases. Mol. Cell 2012, 47, 281–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdrabou, A.; Brandwein, D.; Liu, C.; Wang, Z. Rac1 S71 Mediates the Interaction between Rac1 and 14-3-3 Proteins. Cells 2019, 8, 1006. [Google Scholar] [CrossRef] [Green Version]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Eva, A.; Aaronson, S.A. Isolation of a new human oncogene from a diffuse B-cell lymphoma. Nature 1985, 316, 273–275. [Google Scholar] [CrossRef]
- Hart, M.J.; Eva, A.; Evans, T.; Aaronson, S.A.; Cerione, R.A. Catalysis of guanine nucleotide exchange on the CDC42Hs protein by the dbl oncogene product. Nature 1991, 354, 311–314. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Ferguson, K.M.; Schlessinger, J. PH domains: Diverse sequences with a common fold recruit signaling molecules to the cell surface. Cell 1996, 85, 621–624. [Google Scholar] [CrossRef] [Green Version]
- Fleming, I.N.; Batty, I.H.; Prescott, A.R.; Gray, A.; Kular, G.S.; Stewart, H.; Downes, C.P. Inositol phospholipids regulate the guanine-nucleotide-exchange factor Tiam1 by facilitating its binding to the plasma membrane and regulating GDP/GTP exchange on Rac1. Biochem. J. 2004, 382, 857–865. [Google Scholar] [CrossRef]
- Derivery, E.; Gautreau, A. Generation of branched actin networks: Assembly and regulation of the N-WASP and WAVE molecular machines. Bioessays 2010, 32, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Gardel, M.L.; Schneider, I.C.; Aratyn-Schaus, Y.; Waterman, C.M. Mechanical integration of actin and adhesion dynamics in cell migration. Annu. Rev. Cell Dev. Biol. 2010, 26, 315–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haga, R.B.; Ridley, A.J. Rho GTPases: Regulation and roles in cancer cell biology. Small GTPases 2016, 7, 207–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, K.C.; Rivero, F.; Meili, R.; Lee, S.; Apone, F.; Firtel, R.A. Rac regulation of chemotaxis and morphogenesis in Dictyostelium. EMBO J. 2004, 23, 4177–4189. [Google Scholar] [CrossRef] [Green Version]
- Stephens, L.R.; Hughes, K.T.; Irvine, R.F. Pathway of phosphatidylinositol(3,4,5)-trisphosphate synthesis in activated neutrophils. Nature 1991, 351, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.T.; Eguinoa, A.; Qiu, R.G.; Stokoe, D.; Cooke, F.T.; Walters, R.; Wennstrom, S.; Claesson-Welsh, L.; Evans, T.; Symons, M.; et al. PDGF stimulates an increase in GTP-Rac via activation of phosphoinositide 3-kinase. Curr. Biol. 1995, 5, 393–403. [Google Scholar] [CrossRef] [Green Version]
- Wymann, M.; Arcaro, A. Platelet-derived growth factor-induced phosphatidylinositol 3-kinase activation mediates actin rearrangements in fibroblasts. Biochem. J. 1994, 298, 517–520. [Google Scholar] [CrossRef] [Green Version]
- Arcaro, A.; Wymann, M.P. Wortmannin is a potent phosphatidylinositol 3-kinase inhibitor: The role of phosphatidylinositol 3,4,5-trisphosphate in neutrophil responses. Biochem. J. 1993, 296, 297–301. [Google Scholar] [CrossRef] [Green Version]
- Heit, B.; Robbins, S.M.; Downey, C.M.; Guan, Z.; Colarusso, P.; Miller, B.J.; Jirik, F.R.; Kubes, P. PTEN functions to ‘prioritize’ chemotactic cues and prevent ‘distraction’ in migrating neutrophils. Nat. Immunol. 2008, 9, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Nishio, M.; Watanabe, K.; Sasaki, J.; Taya, C.; Takasuga, S.; Iizuka, R.; Balla, T.; Yamazaki, M.; Watanabe, H.; Itoh, R.; et al. Control of cell polarity and motility by the PtdIns(3,4,5)P3 phosphatase SHIP1. Nat. Cell Biol. 2007, 9, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Damen, J.E.; Liu, L.; Rosten, P.; Humphries, R.K.; Jefferson, A.B.; Majerus, P.W.; Krystal, G. The 145-kDa protein induced to associate with Shc by multiple cytokines is an inositol tetraphosphate and phosphatidylinositol 3,4,5-triphosphate 5-phosphatase. Proc. Natl. Acad. Sci. USA 1996, 93, 1689–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maehama, T.; Dixon, J.E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondal, S.; Subramanian, K.K.; Sakai, J.; Bajrami, B.; Luo, H.R. Phosphoinositide lipid phosphatase SHIP1 and PTEN coordinate to regulate cell migration and adhesion. Mol. Biol. Cell 2012, 23, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Cote, J.F.; Motoyama, A.B.; Bush, J.A.; Vuori, K. A novel and evolutionarily conserved PtdIns(3,4,5)P3-binding domain is necessary for DOCK180 signalling. Nat. Cell Biol. 2005, 7, 797–807. [Google Scholar] [CrossRef]
- Fleming, I.N.; Gray, A.; Downes, C.P. Regulation of the Rac1-specific exchange factor Tiam1 involves both phosphoinositide 3-kinase-dependent and -independent components. Biochem. J. 2000, 351, 173–182. [Google Scholar] [CrossRef]
- Han, J.; Luby-Phelps, K.; Das, B.; Shu, X.; Xia, Y.; Mosteller, R.D.; Krishna, U.M.; Falck, J.R.; White, M.A.; Broek, D. Role of substrates and products of PI 3-kinase in regulating activation of Rac-related guanosine triphosphatases by Vav. Science 1998, 279, 558–560. [Google Scholar] [CrossRef]
- Weiner, O.D.; Neilsen, P.O.; Prestwich, G.D.; Kirschner, M.W.; Cantley, L.C.; Bourne, H.R. A PtdInsP(3)- and Rho GTPase-mediated positive feedback loop regulates neutrophil polarity. Nat. Cell Biol. 2002, 4, 509–513. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.Y.; Guan, M.; Vigil, D.; Der, C.J.; Lowy, D.R.; Popescu, N.C. p120Ras-GAP binds the DLC1 Rho-GAP tumor suppressor protein and inhibits its RhoA GTPase and growth-suppressing activities. Oncogene 2009, 28, 1401–1409. [Google Scholar] [CrossRef] [Green Version]
- Ridley, A.J. Life at the leading edge. Cell 2011, 145, 1012–1022. [Google Scholar] [CrossRef] [Green Version]
- Raftopoulou, M.; Hall, A. Cell migration: Rho GTPases lead the way. Dev. Biol. 2004, 265, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, S.; Wang, F.; Glavas, S.; Ott, A.; Hofmann, F.; Aktories, K.; Kalman, D.; Bourne, H.R. Rac and Cdc42 play distinct roles in regulating PI(3,4,5)P3 and polarity during neutrophil chemotaxis. J. Cell Biol. 2003, 160, 375–385. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Herzmark, P.; Weiner, O.D.; Srinivasan, S.; Servant, G.; Bourne, H.R. Lipid products of PI(3)Ks maintain persistent cell polarity and directed motility in neutrophils. Nat. Cell Biol. 2002, 4, 513–518. [Google Scholar] [CrossRef]
- Servant, G.; Weiner, O.D.; Herzmark, P.; Balla, T.; Sedat, J.W.; Bourne, H.R. Polarization of chemoattractant receptor signaling during neutrophil chemotaxis. Science 2000, 287, 1037–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peyrollier, K.; Hajduch, E.; Gray, A.; Litherland, G.J.; Prescott, A.R.; Leslie, N.R.; Hundal, H.S. A role for the actin cytoskeleton in the hormonal and growth-factor-mediated activation of protein kinase B. Biochem. J. 2000, 352, 617–622. [Google Scholar] [CrossRef]
- Rodriguez-Viciana, P.; Warne, P.H.; Vanhaesebroeck, B.; Waterfield, M.D.; Downward, J. Activation of phosphoinositide 3-kinase by interaction with Ras and by point mutation. EMBO J. 1996, 15, 2442–2451. [Google Scholar] [CrossRef]
- Sasaki, A.T.; Firtel, R.A. Regulation of chemotaxis by the orchestrated activation of Ras, PI3K, and TOR. Eur. J. Cell Biol. 2006, 85, 873–895. [Google Scholar] [CrossRef] [PubMed]
- Bar-Sagi, D.; Hall, A. Ras and Rho GTPases: A family reunion. Cell 2000, 103, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Kawai, K.; Iwamae, Y.; Yamaga, M.; Kiyota, M.; Ishii, H.; Hirata, H.; Homma, Y.; Yagisawa, H. Focal adhesion-localization of START-GAP1/DLC1 is essential for cell motility and morphology. Genes Cells 2009, 14, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, B.K.; Grant, T.; Qian, X.; Zhou, M.; Mertins, P.; Wang, D.; Papageorge, A.G.; Tarasov, S.G.; Hunter, K.W.; Carr, S.A.; et al. Receptor tyrosine kinase activation of RhoA is mediated by AKT phosphorylation of DLC1. J. Cell Biol. 2017, 216, 4255–4270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senoo, H.; Wai, M.; Matsubayashi, H.T.; Sesaki, H.; Iijima, M. Hetero-oligomerization of Rho and Ras GTPases Connects GPCR Activation to mTORC2-AKT Signaling. Cell Rep. 2020, 33, 108427. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Parent, C.A. Review series: TOR kinase complexes and cell migration. J. Cell Biol. 2011, 194, 815–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.; Fingar, D.C. Growing knowledge of the mTOR signaling network. Semin. Cell Dev. Biol. 2014, 36, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Stahle, M.; Veit, C.; Bachfischer, U.; Schierling, K.; Skripczynski, B.; Hall, A.; Gierschik, P.; Giehl, K. Mechanisms in LPA-induced tumor cell migration: Critical role of phosphorylated ERK. J. Cell Sci. 2003, 116, 3835–3846. [Google Scholar] [CrossRef] [Green Version]
- Bian, D.; Su, S.; Mahanivong, C.; Cheng, R.K.; Han, Q.; Pan, Z.K.; Sun, P.; Huang, S. Lysophosphatidic Acid Stimulates Ovarian Cancer Cell Migration via a Ras-MEK Kinase 1 Pathway. Cancer Res. 2004, 64, 4209–4217. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, M.C.; Er, E.E.; Zhang, W.; Ballif, B.A.; Elliott, H.L.; Danuser, G.; Blenis, J. ERK-MAPK drives lamellipodia protrusion by activating the WAVE2 regulatory complex. Mol. Cell 2011, 41, 661–671. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, M.C.; Vilela, M.; Juarez, J.E.; Blenis, J.; Danuser, G. ERK reinforces actin polymerization to power persistent edge protrusion during motility. Sci. Signal. 2015, 8, ra47. [Google Scholar] [CrossRef] [Green Version]
- Boeckeler, K.; Rosse, C.; Howell, M.; Parker, P.J. Manipulating signal delivery—Plasma-membrane ERK activation in aPKC-dependent migration. J. Cell Sci. 2010, 123, 2725–2732. [Google Scholar] [CrossRef] [Green Version]
- Webb, D.J.; Donais, K.; Whitmore, L.A.; Thomas, S.M.; Turner, C.E.; Parsons, J.T.; Horwitz, A.F. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell Biol. 2004, 6, 154–161. [Google Scholar] [CrossRef]
- Cheresh, D.A.; Leng, J.; Klemke, R.L. Regulation of cell contraction and membrane ruffling by distinct signals in migratory cells. J. Cell Biol. 1999, 146, 1107–1116. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.H.; Catling, A.D.; Webb, D.J.; Sankovic, M.; Walker, L.A.; Somlyo, A.V.; Weber, M.J.; Gonias, S.L. Myosin light chain kinase functions downstream of Ras/ERK to promote migration of urokinase-type plasminogen activator-stimulated cells in an integrin-selective manner. J. Cell Biol. 1999, 146, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Samson, S.C.; Elliott, A.; Mueller, B.D.; Kim, Y.; Carney, K.R.; Bergman, J.P.; Blenis, J.; Mendoza, M.C. p90 ribosomal S6 kinase (RSK) phosphorylates myosin phosphatase and thereby controls edge dynamics during cell migration. J. Biol. Chem. 2019, 294, 10846–10862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vomastek, T.; Iwanicki, M.P.; Schaeffer, H.J.; Tarcsafalvi, A.; Parsons, J.T.; Weber, M.J. RACK1 targets the extracellular signal-regulated kinase/mitogen-activated protein kinase pathway to link integrin engagement with focal adhesion disassembly and cell motility. Mol. Cell. Biol. 2007, 27, 8296–8305. [Google Scholar] [CrossRef] [Green Version]
- Fincham, V.J.; James, M.; Frame, M.C.; Winder, S.J. Active ERK/MAP kinase is targeted to newly forming cell-matrix adhesions by integrin engagement and v-Src. EMBO J. 2000, 19, 2911–2923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slack-Davis, J.K.; Eblen, S.T.; Zecevic, M.; Boerner, S.A.; Tarcsafalvi, A.; Diaz, H.B.; Marshall, M.S.; Weber, M.J.; Parsons, J.T.; Catling, A.D. PAK1 phosphorylation of MEK1 regulates fibronectin-stimulated MAPK activation. J. Cell Biol. 2003, 162, 281–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klimova, Z.; Braborec, V.; Maninova, M.; Caslavsky, J.; Weber, M.J.; Vomastek, T. Symmetry breaking in spreading RAT2 fibroblasts requires the MAPK/ERK pathway scaffold RACK1 that integrates FAK, p190A-RhoGAP and ERK2 signaling. Biochim. Biophys. Acta 2016, 1863, 2189–2200. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.C.; Zhuang, S.; Nguyen, T.H.; Boss, G.R.; Pilz, R.B. Oncogenic Ras leads to Rho activation by activating the mitogen-activated protein kinase pathway and decreasing Rho-GTPase-activating protein activity. J. Biol. Chem. 2003, 278, 2807–2818. [Google Scholar] [CrossRef] [Green Version]
- Klein, R.M.; Spofford, L.S.; Abel, E.V.; Ortiz, A.; Aplin, A.E. B-RAF regulation of Rnd3 participates in actin cytoskeletal and focal adhesion organization. Mol. Biol. Cell 2008, 19, 498–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrenreiter, K.; Piazzolla, D.; Velamoor, V.; Sobczak, I.; Small, J.V.; Takeda, J.; Leung, T.; Baccarini, M. Raf-1 regulates Rho signaling and cell migration. J. Cell Biol. 2005, 168, 955–964. [Google Scholar] [CrossRef] [Green Version]
- Niault, T.; Sobczak, I.; Meissl, K.; Weitsman, G.; Piazzolla, D.; Maurer, G.; Kern, F.; Ehrenreiter, K.; Hamerl, M.; Moarefi, I.; et al. From autoinhibition to inhibition in trans: The Raf-1 regulatory domain inhibits Rok-alpha kinase activity. J. Cell Biol. 2009, 187, 335–342. [Google Scholar] [CrossRef] [Green Version]
- Wimmer, R.; Cseh, B.; Maier, B.; Scherrer, K.; Baccarini, M. Angiogenic sprouting requires the fine tuning of endothelial cell cohesion by the Raf-1/Rok-alpha complex. Dev. Cell 2012, 22, 158–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrenreiter, K.; Kern, F.; Velamoor, V.; Meissl, K.; Galabova-Kovacs, G.; Sibilia, M.; Baccarini, M. Raf-1 addiction in Ras-induced skin carcinogenesis. Cancer Cell 2009, 16, 149–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doma, E.; Rupp, C.; Varga, A.; Kern, F.; Riegler, B.; Baccarini, M. Skin tumorigenesis stimulated by Raf inhibitors relies upon Raf functions that are dependent and independent of ERK. Cancer Res. 2013, 73, 6926–6937. [Google Scholar] [CrossRef] [Green Version]
- Kern, F.; Doma, E.; Rupp, C.; Niault, T.; Baccarini, M. Essential, non-redundant roles of B-Raf and Raf-1 in Ras-driven skin tumorigenesis. Oncogene 2013, 32, 2483–2492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorard, C.; Cseh, B.; Ehrenreiter, K.; Wimmer, R.; Varga, A.; Hirschmugl, T.; Maier, B.; Kramer, K.; Furlinger, S.; Doma, E.; et al. RAF dimers control vascular permeability and cytoskeletal rearrangements at endothelial cell-cell junctions. FEBS J. 2019, 286, 2277–2294. [Google Scholar] [CrossRef] [Green Version]
- Varga, A.; Ehrenreiter, K.; Aschenbrenner, B.; Kocieniewski, P.; Kochanczyk, M.; Lipniacki, T.; Baccarini, M. RAF1/BRAF dimerization integrates the signal from RAS to ERK and ROKalpha. Sci. Signal. 2017, 10, eaai8482. [Google Scholar] [CrossRef]
- Noble, C.; Mercer, K.; Hussain, J.; Carragher, L.; Giblett, S.; Hayward, R.; Patterson, C.; Marais, R.; Pritchard, C.A. CRAF autophosphorylation of serine 621 is required to prevent its proteasome-mediated degradation. Mol. Cell 2008, 31, 862–872. [Google Scholar] [CrossRef] [Green Version]
- Mavria, G.; Vercoulen, Y.; Yeo, M.; Paterson, H.; Karasarides, M.; Marais, R.; Bird, D.; Marshall, C.J. ERK-MAPK signaling opposes Rho-kinase to promote endothelial cell survival and sprouting during angiogenesis. Cancer Cell 2006, 9, 33–44. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.; Hall, P.; Santos, L.L.; Gregory, J.L.; Fingerle-Rowson, G.; Bucala, R.; Morand, E.F.; Hickey, M.J. Macrophage migration inhibitory factor and CD74 regulate macrophage chemotactic responses via MAPK and Rho GTPase. J. Immunol. 2011, 186, 4915–4924. [Google Scholar] [CrossRef] [Green Version]
- Pollock, C.B.; Shirasawa, S.; Sasazuki, T.; Kolch, W.; Dhillon, A.S. Oncogenic K-RAS is required to maintain changes in cytoskeletal organization, adhesion, and motility in colon cancer cells. Cancer Res. 2005, 65, 1244–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vial, E.; Sahai, E.; Marshall, C.J. ERK-MAPK signaling coordinately regulates activity of Rac1 and RhoA for tumor cell motility. Cancer Cell 2003, 4, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, B.K.; Anderman, M.F.; Qian, X.; Zhou, M.; Wang, D.; Papageorge, A.G.; Lowy, D.R. SRC and ERK cooperatively phosphorylate DLC1 and attenuate its Rho-GAP and tumor suppressor functions. J. Cell Biol. 2019, 218, 3060–3076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujishiro, S.H.; Tanimura, S.; Mure, S.; Kashimoto, Y.; Watanabe, K.; Kohno, M. ERK1/2 phosphorylate GEF-H1 to enhance its guanine nucleotide exchange activity toward RhoA. Biochem. Biophys. Res. Commun. 2008, 368, 162–167. [Google Scholar] [CrossRef]
- Kakiashvili, E.; Dan, Q.; Vandermeer, M.; Zhang, Y.; Waheed, F.; Pham, M.; Szaszi, K. The epidermal growth factor receptor mediates tumor necrosis factor-alpha-induced activation of the ERK/GEF-H1/RhoA pathway in tubular epithelium. J. Biol. Chem. 2011, 286, 9268–9279. [Google Scholar] [CrossRef] [Green Version]
- von Thun, A.; Preisinger, C.; Rath, O.; Schwarz, J.P.; Ward, C.; Monsefi, N.; Rodriguez, J.; Garcia-Munoz, A.; Birtwistle, M.; Bienvenut, W.; et al. Extracellular signal-regulated kinase regulates RhoA activation and tumor cell plasticity by inhibiting guanine exchange factor H1 activity. Mol. Cell. Biol. 2013, 33, 4526–4537. [Google Scholar] [CrossRef] [Green Version]
- Tong, J.; Li, L.; Ballermann, B.; Wang, Z. Phosphorylation of Rac1 T108 by extracellular signal-regulated kinase in response to epidermal growth factor: A novel mechanism to regulate Rac1 function. Mol. Cell. Biol. 2013, 33, 4538–4551. [Google Scholar] [CrossRef] [Green Version]
- Tong, J.; Li, L.; Ballermann, B.; Wang, Z. Phosphorylation and Activation of RhoA by ERK in Response to Epidermal Growth Factor Stimulation. PLoS ONE 2016, 11, e0147103. [Google Scholar] [CrossRef]
- Mitin, N.; Rossman, K.L.; Der, C.J. Signaling interplay in Ras superfamily function. Curr. Biol. 2005, 15, R563–R574. [Google Scholar] [CrossRef] [Green Version]
- Murillo, M.M.; Rana, S.; Spencer-Dene, B.; Nye, E.; Stamp, G.; Downward, J. Disruption of the Interaction of RAS with PI 3-Kinase Induces Regression of EGFR-Mutant-Driven Lung Cancer. Cell Rep. 2018, 25, 3545–3553. [Google Scholar] [CrossRef] [Green Version]
- Sosa, M.S.; Lopez-Haber, C.; Yang, C.; Wang, H.; Lemmon, M.A.; Busillo, J.M.; Luo, J.; Benovic, J.L.; Klein-Szanto, A.; Yagi, H.; et al. Identification of the Rac-GEF P-Rex1 as an essential mediator of ErbB signaling in breast cancer. Mol. Cell 2010, 40, 877–892. [Google Scholar] [CrossRef] [Green Version]
- Baltanas, F.C.; Zarich, N.; Rojas-Cabaneros, J.M.; Santos, E. SOS GEFs in health and disease. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188445. [Google Scholar] [CrossRef]
- Nickerson, S.; Joy, S.T.; Arora, P.S.; Bar-Sagi, D. An orthosteric inhibitor of the RAS-SOS interaction. Enzymes 2013, 34, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J. Rho family proteins: Coordinating cell responses. Trends Cell Biol. 2001, 11, 471–477. [Google Scholar] [CrossRef]
- Di Fiore, P.P.; Scita, G. Eps8 in the midst of GTPases. Int. J. Biochem. Cell Biol. 2002, 34, 1178–1183. [Google Scholar] [CrossRef]
- Pierre, S.; Bats, A.S.; Coumoul, X. Understanding SOS (Son of Sevenless). Biochem. Pharmacol. 2011, 82, 1049–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Innocenti, M.; Tenca, P.; Frittoli, E.; Faretta, M.; Tocchetti, A.; Di Fiore, P.P.; Scita, G. Mechanisms through which Sos-1 coordinates the activation of Ras and Rac. J. Cell Biol. 2002, 156, 125–136. [Google Scholar] [CrossRef] [Green Version]
- Scita, G.; Nordstrom, J.; Carbone, R.; Tenca, P.; Giardina, G.; Gutkind, S.; Bjarnegard, M.; Betsholtz, C.; Di Fiore, P.P. EPS8 and E3B1 transduce signals from Ras to Rac. Nature 1999, 401, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Innocenti, M.; Frittoli, E.; Ponzanelli, I.; Falck, J.R.; Brachmann, S.M.; Di Fiore, P.P.; Scita, G. Phosphoinositide 3-kinase activates Rac by entering in a complex with Eps8, Abi1, and Sos-1. J. Cell Biol. 2003, 160, 17–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanday, F.A.; Santhanam, L.; Kasuno, K.; Yamamori, T.; Naqvi, A.; Dericco, J.; Bugayenko, A.; Mattagajasingh, I.; Disanza, A.; Scita, G.; et al. Sos-mediated activation of rac1 by p66shc. J. Cell Biol. 2006, 172, 817–822. [Google Scholar] [CrossRef] [Green Version]
- Gerboth, S.; Frittoli, E.; Palamidessi, A.; Baltanas, F.C.; Salek, M.; Rappsilber, J.; Giuliani, C.; Troglio, F.; Rolland, Y.; Pruneri, G.; et al. Phosphorylation of SOS1 on tyrosine 1196 promotes its RAC GEF activity and contributes to BCR-ABL leukemogenesis. Leukemia 2018, 32, 820–827. [Google Scholar] [CrossRef]
- Takacs, T.; Kudlik, G.; Kurilla, A.; Szeder, B.; Buday, L.; Vas, V. The effects of mutant Ras proteins on the cell signalome. Cancer Metastasis Rev. 2020, 39, 1051–1065. [Google Scholar] [CrossRef]
- Boissier, P.; Huynh-Do, U. The guanine nucleotide exchange factor Tiam1: A Janus-faced molecule in cellular signaling. Cell Signal. 2014, 26, 483–491. [Google Scholar] [CrossRef]
- Malliri, A.; van der Kammen, R.A.; Clark, K.; van der Valk, M.; Michiels, F.; Collard, J.G. Mice deficient in the Rac activator Tiam1 are resistant to Ras-induced skin tumours. Nature 2002, 417, 867–871. [Google Scholar] [CrossRef]
- Mierke, C.T.; Puder, S.; Aermes, C.; Fischer, T.; Kunschmann, T. Effect of PAK Inhibition on Cell Mechanics Depends on Rac1. Front. Cell Dev. Biol. 2020, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- Semenova, G.; Stepanova, D.S.; Dubyk, C.; Handorf, E.; Deyev, S.M.; Lazar, A.J.; Chernoff, J. Targeting group I p21-activated kinases to control malignant peripheral nerve sheath tumor growth and metastasis. Oncogene 2017, 36, 5421–5431. [Google Scholar] [CrossRef] [Green Version]
- Woodcock, S.A.; Rooney, C.; Liontos, M.; Connolly, Y.; Zoumpourlis, V.; Whetton, A.D.; Gorgoulis, V.G.; Malliri, A. SRC-induced disassembly of adherens junctions requires localized phosphorylation and degradation of the rac activator tiam1. Mol. Cell 2009, 33, 639–653. [Google Scholar] [CrossRef]
- Ren, Y.; Li, R.; Zheng, Y.; Busch, H. Cloning and characterization of GEF-H1, a microtubule-associated guanine nucleotide exchange factor for Rac and Rho GTPases. J. Biol. Chem. 1998, 273, 34954–34960. [Google Scholar] [CrossRef] [Green Version]
- Cullis, J.; Meiri, D.; Sandi, M.J.; Radulovich, N.; Kent, O.A.; Medrano, M.; Mokady, D.; Normand, J.; Larose, J.; Marcotte, R.; et al. The RhoGEF GEF-H1 is required for oncogenic RAS signaling via KSR-1. Cancer Cell 2014, 25, 181–195. [Google Scholar] [CrossRef] [Green Version]
- Brecht, M.; Steenvoorden, A.C.; Collard, J.G.; Luf, S.; Erz, D.; Bartram, C.R.; Janssen, J.W. Activation of gef-h1, a guanine nucleotide exchange factor for RhoA, by DNA transfection. Int. J. Cancer 2005, 113, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Khoo, P.; Allan, K.; Willoughby, L.; Brumby, A.M.; Richardson, H.E. In Drosophila, RhoGEF2 cooperates with activated Ras in tumorigenesis through a pathway involving Rho1-Rok-Myosin-II and JNK signalling. Dis. Models Mech. 2013, 6, 661–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, O.A.; Sandi, M.J.; Burston, H.E.; Brown, K.R.; Rottapel, R. An oncogenic KRAS transcription program activates the RHOGEF ARHGEF2 to mediate transformed phenotypes in pancreatic cancer. Oncotarget 2017, 8, 4484–4500. [Google Scholar] [CrossRef] [Green Version]
- Kent, O.A.; Sandi, M.J.; Rottapel, R. Co-dependency between KRAS addiction and ARHGEF2 promotes an adaptive escape from MAPK pathway inhibition. Small GTPases 2019, 10, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Scheffzek, K.; Ahmadian, M.R.; Wittinghofer, A. GTPase-activating proteins: Helping hands to complement an active site. Trends Biochem. Sci. 1998, 23, 257–262. [Google Scholar] [CrossRef]
- Camonis, J.H.; White, M.A. Ral GTPases: Corrupting the exocyst in cancer cells. Trends Cell Biol. 2005, 15, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.; Moon, S.Y.; Zheng, Y. p200 RhoGAP promotes cell proliferation by mediating cross-talk between Ras and Rho signaling pathways. J. Biol. Chem. 2007, 282, 8801–8811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pamonsinlapatham, P.; Hadj-Slimane, R.; Lepelletier, Y.; Allain, B.; Toccafondi, M.; Garbay, C.; Raynaud, F. p120-Ras GTPase activating protein (RasGAP): A multi-interacting protein in downstream signaling. Biochimie 2009, 91, 320–328. [Google Scholar] [CrossRef]
- Scheffzek, K.; Ahmadian, M.R.; Kabsch, W.; Wiesmuller, L.; Lautwein, A.; Schmitz, F.; Wittinghofer, A. The Ras-RasGAP complex: Structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science 1997, 277, 333–338. [Google Scholar] [CrossRef] [Green Version]
- Ahmadian, M.R.; Stege, P.; Scheffzek, K.; Wittinghofer, A. Confirmation of the arginine-finger hypothesis for the GAP-stimulated GTP-hydrolysis reaction of Ras. Nat. Struct. Biol. 1997, 4, 686–689. [Google Scholar] [CrossRef]
- Leblanc, V.; Tocque, B.; Delumeau, I. Ras-GAP controls Rho-mediated cytoskeletal reorganization through its SH3 domain. Mol. Cell. Biol. 1998, 18, 5567–5578. [Google Scholar] [CrossRef] [Green Version]
- Chan, P.C.; Chen, H.C. p120RasGAP-mediated activation of c-Src is critical for oncogenic Ras to induce tumor invasion. Cancer Res. 2012, 72, 2405–2415. [Google Scholar] [CrossRef] [Green Version]
- Clark, G.J.; Der, C.J. Aberrant function of the Ras signal transduction pathway in human breast cancer. Breast Cancer Res. Treat. 1995, 35, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Clark, G.J.; Westwick, J.K.; Der, C.J. p120 GAP modulates Ras activation of Jun kinases and transformation. J. Biol. Chem. 1997, 272, 1677–1681. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, M.; Dvorsky, R.; Amin, E.; Risse, S.L.; Fansa, E.K.; Zhang, S.C.; Taha, M.S.; Gauhar, A.R.; Nakhaei-Rad, S.; Kordes, C.; et al. Functional cross-talk between ras and rho pathways: A Ras-specific GTPase-activating protein (p120RasGAP) competitively inhibits the RhoGAP activity of deleted in liver cancer (DLC) tumor suppressor by masking the catalytic arginine finger. J. Biol. Chem. 2014, 289, 6839–6849. [Google Scholar] [CrossRef] [Green Version]
- Asnaghi, L.; Vass, W.C.; Quadri, R.; Day, P.M.; Qian, X.; Braverman, R.; Papageorge, A.G.; Lowy, D.R. E-cadherin negatively regulates neoplastic growth in non-small cell lung cancer: Role of Rho GTPases. Oncogene 2010, 29, 2760–2771. [Google Scholar] [CrossRef] [Green Version]
- Herbrand, U.; Ahmadian, M.R. p190-RhoGAP as an integral component of the Tiam1/Rac1-induced downregulation of Rho. Biol. Chem. 2006, 387, 311–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Tung, P.S.; Moran, M.F. Association of p120 ras GAP with endocytic components and colocalization with epidermal growth factor (EGF) receptor in response to EGF stimulation. Cell Growth Differ. 1996, 7, 123–133. [Google Scholar]
- Hu, K.Q.; Settleman, J. Tandem SH2 binding sites mediate the RasGAP-RhoGAP interaction: A conformational mechanism for SH3 domain regulation. EMBO J. 1997, 16, 473–483. [Google Scholar] [CrossRef] [Green Version]
- Jaber Chehayeb, R.; Stiegler, A.L.; Boggon, T.J. Crystal structures of p120RasGAP N-terminal SH2 domain in its apo form and in complex with a p190RhoGAP phosphotyrosine peptide. PLoS ONE 2019, 14, e0226113. [Google Scholar] [CrossRef] [Green Version]
- Vallee, B.; Doudeau, M.; Godin, F.; Gombault, A.; Tchalikian, A.; de Tauzia, M.L.; Benedetti, H. Nf1 RasGAP inhibition of LIMK2 mediates a new cross-talk between Ras and Rho pathways. PLoS ONE 2012, 7, e47283. [Google Scholar] [CrossRef] [Green Version]
- Ozawa, T.; Araki, N.; Yunoue, S.; Tokuo, H.; Feng, L.; Patrakitkomjorn, S.; Hara, T.; Ichikawa, Y.; Matsumoto, K.; Fujii, K.; et al. The neurofibromatosis type 1 gene product neurofibromin enhances cell motility by regulating actin filament dynamics via the Rho-ROCK-LIMK2-cofilin pathway. J. Biol. Chem. 2005, 280, 39524–39533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starinsky-Elbaz, S.; Faigenbloom, L.; Friedman, E.; Stein, R.; Kloog, Y. The pre-GAP-related domain of neurofibromin regulates cell migration through the LIM kinase/cofilin pathway. Mol. Cell. Neurosci. 2009, 42, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D.; Borisy, G.G. Cellular motility driven by assembly and disassembly of actin filaments. Cell 2003, 112, 453–465. [Google Scholar] [CrossRef] [Green Version]
- Scott, R.W.; Olson, M.F. LIM kinases: Function, regulation and association with human disease. J. Mol. Med. 2007, 85, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Corral, T.; Jimenez, M.; Hernandez-Munoz, I.; Perez de Castro, I.; Pellicer, A. NF1 modulates the effects of Ras oncogenes: Evidence of other NF1 function besides its GAP activity. J. Cell. Physiol. 2003, 197, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Kweh, F.; Zheng, M.; Kurenova, E.; Wallace, M.; Golubovskaya, V.; Cance, W.G. Neurofibromin physically interacts with the N-terminal domain of focal adhesion kinase. Mol. Carcinog. 2009, 48, 1005–1017. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.L.; Lei, Y.T.; Hong, C.J.; Hsueh, Y.P. Syndecan-2 induces filopodia and dendritic spine formation via the neurofibromin-PKA-Ena/VASP pathway. J. Cell Biol. 2007, 177, 829–841. [Google Scholar] [CrossRef] [Green Version]
- Shirakawa, R.; Horiuchi, H. Ral GTPases: Crucial mediators of exocytosis and tumourigenesis. J. Biochem. 2015, 157, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Gentry, L.R.; Martin, T.D.; Reiner, D.J.; Der, C.J. Ral small GTPase signaling and oncogenesis: More than just 15minutes of fame. Biochim. Biophys. Acta 2014, 1843, 2976–2988. [Google Scholar] [CrossRef] [Green Version]
- Kashatus, D.F. Ral GTPases in tumorigenesis: Emerging from the shadows. Exp. Cell Res. 2013, 319, 2337–2342. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.; Feig, L.; Montell, D.J. Two distinct roles for Ras in a developmentally regulated cell migration. Development 1996, 122, 409–418. [Google Scholar] [CrossRef]
- Takaya, A.; Ohba, Y.; Kurokawa, K.; Matsuda, M. RalA activation at nascent lamellipodia of epidermal growth factor-stimulated Cos7 cells and migrating Madin-Darby canine kidney cells. Mol. Biol. Cell 2004, 15, 2549–2557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosse, C.; Hatzoglou, A.; Parrini, M.C.; White, M.A.; Chavrier, P.; Camonis, J. RalB mobilizes the exocyst to drive cell migration. Mol. Cell. Biol. 2006, 26, 727–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oxford, G.; Owens, C.R.; Titus, B.J.; Foreman, T.L.; Herlevsen, M.C.; Smith, S.C.; Theodorescu, D. RalA and RalB: Antagonistic relatives in cancer cell migration. Cancer Res. 2005, 65, 7111–7120. [Google Scholar] [CrossRef] [Green Version]
- Neel, N.F.; Rossman, K.L.; Martin, T.D.; Hayes, T.K.; Yeh, J.J.; Der, C.J. The RalB small GTPase mediates formation of invadopodia through a GTPase-activating protein-independent function of the RalBP1/RLIP76 effector. Mol. Cell. Biol. 2012, 32, 1374–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zago, G.; Biondini, M.; Camonis, J.; Parrini, M.C. A family affair: A Ral-exocyst-centered network links Ras, Rac, Rho signaling to control cell migration. Small GTPases 2019, 10, 323–330. [Google Scholar] [CrossRef]
- de Gorter, D.J.; Reijmers, R.M.; Beuling, E.A.; Naber, H.P.; Kuil, A.; Kersten, M.J.; Pals, S.T.; Spaargaren, M. The small GTPase Ral mediates SDF-1-induced migration of B cells and multiple myeloma cells. Blood 2008, 111, 3364–3372. [Google Scholar] [CrossRef] [Green Version]
- Spiczka, K.S.; Yeaman, C. Ral-regulated interaction between Sec5 and paxillin targets Exocyst to focal complexes during cell migration. J. Cell Sci. 2008, 121, 2880–2891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, A.L.; Bement, W.M. Regulation of cytokinesis by Rho GTPase flux. Nat. Cell Biol. 2009, 11, 71–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrini, M.C.; Camonis, J. Cell motility: The necessity of Rac1 GDP/GTP flux. Commun. Integr. Biol. 2011, 4, 772–774. [Google Scholar] [CrossRef]
- Biondini, M.; Sadou-Dubourgnoux, A.; Paul-Gilloteaux, P.; Zago, G.; Arslanhan, M.D.; Waharte, F.; Formstecher, E.; Hertzog, M.; Yu, J.; Guerois, R.; et al. Direct interaction between exocyst and Wave complexes promotes cell protrusions and motility. J. Cell Sci. 2016, 129, 3756–3769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Zhao, Y.; Sun, Y.; He, B.; Yang, C.; Svitkina, T.; Goldman, Y.E.; Guo, W. Exo70 stimulates the Arp2/3 complex for lamellipodia formation and directional cell migration. Curr. Biol. 2012, 22, 1510–1515. [Google Scholar] [CrossRef] [Green Version]
- Castro-Castro, A.; Ojeda, V.; Barreira, M.; Sauzeau, V.; Navarro-Lerida, I.; Muriel, O.; Couceiro, J.R.; Pimentel-Muinos, F.X.; Del Pozo, M.A.; Bustelo, X.R. Coronin 1A promotes a cytoskeletal-based feedback loop that facilitates Rac1 translocation and activation. EMBO J. 2011, 30, 3913–3927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunida, K.; Matsuda, M.; Aoki, K. FRET imaging and statistical signal processing reveal positive and negative feedback loops regulating the morphology of randomly migrating HT-1080 cells. J. Cell Sci. 2012, 125, 2381–2392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, M.A.; Nicolette, C.; Minden, A.; Polverino, A.; Van Aelst, L.; Karin, M.; Wigler, M.H. Multiple Ras functions can contribute to mammalian cell transformation. Cell 1995, 80, 533–541. [Google Scholar] [CrossRef] [Green Version]
- Ward, Y.; Wang, W.; Woodhouse, E.; Linnoila, I.; Liotta, L.; Kelly, K. Signal pathways which promote invasion and metastasis: Critical and distinct contributions of extracellular signal-regulated kinase and Ral-specific guanine exchange factor pathways. Mol. Cell. Biol. 2001, 21, 5958–5969. [Google Scholar] [CrossRef] [Green Version]
- Biondini, M.; Duclos, G.; Meyer-Schaller, N.; Silberzan, P.; Camonis, J.; Parrini, M.C. RalB regulates contractility-driven cancer dissemination upon TGFbeta stimulation via the RhoGEF GEF-H1. Sci. Rep. 2015, 5, 11759. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.H.; O’Hayer, K.; Adam, S.J.; Kendall, S.D.; Campbell, P.M.; Der, C.J.; Counter, C.M. Divergent roles for RalA and RalB in malignant growth of human pancreatic carcinoma cells. Curr. Biol. 2006, 16, 2385–2394. [Google Scholar] [CrossRef] [Green Version]
- Rybko, V.A.; Knizhnik, A.V.; Komelkov, A.V.; Aushev, V.N.; Trukhanova, L.S.; Tchevkina, E.M. Different metastasis promotive potency of small G-proteins RalA and RalB in in vivo hamster tumor model. Cancer Cell Int. 2011, 11, 22. [Google Scholar] [CrossRef] [Green Version]
- Zago, G.; Veith, I.; Singh, M.K.; Fuhrmann, L.; De Beco, S.; Remorino, A.; Takaoka, S.; Palmeri, M.; Berger, F.; Brandon, N.; et al. RalB directly triggers invasion downstream Ras by mobilizing the Wave complex. Elife 2018, 7, e40474. [Google Scholar] [CrossRef]
- Panciera, T.; Citron, A.; Di Biagio, D.; Battilana, G.; Gandin, A.; Giulitti, S.; Forcato, M.; Bicciato, S.; Panzetta, V.; Fusco, S.; et al. Reprogramming normal cells into tumour precursors requires ECM stiffness and oncogene-mediated changes of cell mechanical properties. Nat. Mater. 2020, 19, 797–806. [Google Scholar] [CrossRef]
- Kazanietz, M.G.; Caloca, M.J. The Rac GTPase in Cancer: From Old Concepts to New Paradigms. Cancer Res. 2017, 77, 5445–5451. [Google Scholar] [CrossRef] [Green Version]
- Kuipers, D.; Mehonic, A.; Kajita, M.; Peter, L.; Fujita, Y.; Duke, T.; Charras, G.; Gale, J.E. Epithelial repair is a two-stage process driven first by dying cells and then by their neighbours. J. Cell Sci. 2014, 127, 1229–1241. [Google Scholar] [CrossRef] [Green Version]
- Rosenblatt, J.; Raff, M.C.; Cramer, L.P. An epithelial cell destined for apoptosis signals its neighbors to extrude it by an actin- and myosin-dependent mechanism. Curr. Biol. 2001, 11, 1847–1857. [Google Scholar] [CrossRef] [Green Version]
- Chagnon-Lessard, S.; Jean-Ruel, H.; Godin, M.; Pelling, A.E. Mechanotransduction of Strain Regulates an Invasive Phenotype in Newly Transformed Epithelial Cells. Front. Phys. 2021. [Google Scholar] [CrossRef]
- Matthews, H.K.; Ganguli, S.; Plak, K.; Taubenberger, A.V.; Win, Z.; Williamson, M.; Piel, M.; Guck, J.; Baum, B. Oncogenic Signaling Alters Cell Shape and Mechanics to Facilitate Cell Division under Confinement. Dev. Cell 2020, 52, 563–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messal, H.A.; Alt, S.; Ferreira, R.M.M.; Gribben, C.; Wang, V.M.; Cotoi, C.G.; Salbreux, G.; Behrens, A. Tissue curvature and apicobasal mechanical tension imbalance instruct cancer morphogenesis. Nature 2019, 566, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Schafer, C.; Mohan, A.; Burford, W.; Driscoll, M.K.; Ludlow, A.T.; Wright, W.E.; Shay, J.W.; Danuser, G. Differential Kras(V12) protein levels control a switch regulating lung cancer cell morphology and motility. Converg. Sci. Phys. Oncol. 2016, 2, 035004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, C.; Helfman, D.M. The Ras-ERK pathway modulates cytoskeleton organization, cell motility and lung metastasis signature genes in MDA-MB-231 LM2. Oncogene 2014, 33, 3668–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bugyi, B.; Carlier, M.F. Control of actin filament treadmilling in cell motility. Annu. Rev. Biophys. 2010, 39, 449–470. [Google Scholar] [CrossRef]
- Small, J.V.; Resch, G.P. The comings and goings of actin: Coupling protrusion and retraction in cell motility. Curr. Opin. Cell Biol. 2005, 17, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Craig, R.; Smith, R.; Kendrick-Jones, J. Light-chain phosphorylation controls the conformation of vertebrate non-muscle and smooth muscle myosin molecules. Nature 1983, 302, 436–439. [Google Scholar] [CrossRef]
- Adelstein, R.S.; Conti, M.A. Phosphorylation of platelet myosin increases actin-activated myosin ATPase activity. Nature 1975, 256, 597–598. [Google Scholar] [CrossRef]
- Ridley, A.J.; Hall, A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 1992, 70, 389–399. [Google Scholar] [CrossRef]
- Chrzanowska-Wodnicka, M.; Burridge, K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J. Cell Biol. 1996, 133, 1403–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amano, M.; Ito, M.; Kimura, K.; Fukata, Y.; Chihara, K.; Nakano, T.; Matsuura, Y.; Kaibuchi, K. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J. Biol. Chem. 1996, 271, 20246–20249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, K.; Ito, M.; Amano, M.; Chihara, K.; Fukata, Y.; Nakafuku, M.; Yamamori, B.; Feng, J.; Nakano, T.; Okawa, K.; et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 1996, 273, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Driska, S.P.; Aksoy, M.O.; Murphy, R.A. Myosin light chain phosphorylation associated with contraction in arterial smooth muscle. Am. J. Physiol. 1981, 240, C222–C233. [Google Scholar] [CrossRef]
- Alcala, D.B.; Haldeman, B.D.; Brizendine, R.K.; Krenc, A.K.; Baker, J.E.; Rock, R.S.; Cremo, C.R. Myosin light chain kinase steady-state kinetics: Comparison of smooth muscle myosin II and nonmuscle myosin IIB as substrates. Cell Biochem. Funct. 2016, 34, 469–474. [Google Scholar] [CrossRef]
- Totsukawa, G.; Wu, Y.; Sasaki, Y.; Hartshorne, D.J.; Yamakita, Y.; Yamashiro, S.; Matsumura, F. Distinct roles of MLCK and ROCK in the regulation of membrane protrusions and focal adhesion dynamics during cell migration of fibroblasts. J. Cell Biol. 2004, 164, 427–439. [Google Scholar] [CrossRef] [Green Version]
- Totsukawa, G.; Yamakita, Y.; Yamashiro, S.; Hartshorne, D.J.; Sasaki, Y.; Matsumura, F. Distinct roles of ROCK (Rho-kinase) and MLCK in spatial regulation of MLC phosphorylation for assembly of stress fibers and focal adhesions in 3T3 fibroblasts. J. Cell Biol. 2000, 150, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Leung, T.; Chen, X.Q.; Tan, I.; Manser, E.; Lim, L. Myotonic dystrophy kinase-related Cdc42-binding kinase acts as a Cdc42 effector in promoting cytoskeletal reorganization. Mol. Cell. Biol. 1998, 18, 130–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, I.; Yong, J.; Dong, J.M.; Lim, L.; Leung, T. A tripartite complex containing MRCK modulates lamellar actomyosin retrograde flow. Cell 2008, 135, 123–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shani, G.; Marash, L.; Gozuacik, D.; Bialik, S.; Teitelbaum, L.; Shohat, G.; Kimchi, A. Death-associated protein kinase phosphorylates ZIP kinase, forming a unique kinase hierarchy to activate its cell death functions. Mol. Cell. Biol. 2004, 24, 8611–8626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nehru, V.; Almeida, F.N.; Aspenström, P. Interaction of RhoD and ZIP kinase modulates actin filament assembly and focal adhesion dynamics. Biochem. Biophys. Res. Commun. 2013, 433, 163–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamao, K.; Ono, T.; Matsushita, M.; Hosoya, H. ZIP kinase phosphorylated and activated by Rho kinase/ROCK contributes to cytokinesis in mammalian cultured cells. Exp. Cell Res. 2020, 386, 111707. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, M.; Sellers, J.R.; Adelstein, R.S.; Hidaka, H. Protein kinase C modulates in vitro phosphorylation of the smooth muscle heavy meromyosin by myosin light chain kinase. J. Biol. Chem. 1984, 259, 8808–8814. [Google Scholar] [CrossRef]
- Aguilar-Cuenca, R.; Llorente-González, C.; Chapman, J.R.; Talayero, V.C.; Garrido-Casado, M.; Delgado-Arévalo, C.; Millán-Salanova, M.; Shabanowitz, J.; Hunt, D.F.; Sellers, J.R.; et al. Tyrosine Phosphorylation of the Myosin Regulatory Light Chain Controls Non-muscle Myosin II Assembly and Function in Migrating Cells. Curr. Biol. 2020, 30, 2446–2458.e2446. [Google Scholar] [CrossRef]
- Asensio-Juárez, G.; Llorente-Gonzalez, C.; Vicente-Manzanares, M. Linking the landscape of MYH9-related diseases to the molecular mechanisms that control non-muscle myosin II-A function in cells. Cells 2020, 9, 1458. [Google Scholar] [CrossRef]
- Dartsch, P.C.; Ritter, M.; Haussinger, D.; Lang, F. Cytoskeletal reorganization in NIH 3T3 fibroblasts expressing the ras oncogene. Eur. J. Cell Biol. 1994, 63, 316–325. [Google Scholar]
- Khosravi-Far, R.; Chrzanowska-Wodnicka, M.; Solski, P.A.; Eva, A.; Burridge, K.; Der, C.J. Dbl and Vav mediate transformation via mitogen-activated protein kinase pathways that are distinct from those activated by oncogenic Ras. Mol. Cell. Biol. 1994, 14, 6848–6857. [Google Scholar] [CrossRef] [Green Version]
- Sander, E.E.; ten Klooster, J.P.; van Delft, S.; van der Kammen, R.A.; Collard, J.G. Rac downregulates Rho activity: Reciprocal balance between both GTPases determines cellular morphology and migratory behavior. J. Cell Biol. 1999, 147, 1009–1022. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, B.K.; Lou, M.; Zheng, Y.; Lang, R.A. Balanced Rac1 and RhoA activities regulate cell shape and drive invagination morphogenesis in epithelia. Proc. Natl. Acad. Sci. USA 2011, 108, 18289–18294. [Google Scholar] [CrossRef] [Green Version]
- Klemke, R.L.; Cai, S.; Giannini, A.L.; Gallagher, P.J.; de Lanerolle, P.; Cheresh, D.A. Regulation of cell motility by mitogen-activated protein kinase. J. Cell Biol. 1997, 137, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Connell, L.E.; Helfman, D.M. Myosin light chain kinase plays a role in the regulation of epithelial cell survival. J. Cell Sci. 2006, 119, 2269–2281. [Google Scholar] [CrossRef] [Green Version]
- Beningo, K.A.; Hamao, K.; Dembo, M.; Wang, Y.L.; Hosoya, H. Traction forces of fibroblasts are regulated by the Rho-dependent kinase but not by the myosin light chain kinase. Arch. Biochem. Biophys. 2006, 456, 224–231. [Google Scholar] [CrossRef] [Green Version]
- Kwong, L.; Wozniak, M.A.; Collins, A.S.; Wilson, S.D.; Keely, P.J. R-Ras promotes focal adhesion formation through focal adhesion kinase and p130(Cas) by a novel mechanism that differs from integrins. Mol. Cell. Biol. 2003, 23, 933–949. [Google Scholar] [CrossRef] [Green Version]
- Furuhjelm, J.; Peranen, J. The C-terminal end of R-Ras contains a focal adhesion targeting signal. J. Cell Sci. 2003, 116, 3729–3738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Self, A.J.; Caron, E.; Paterson, H.F.; Hall, A. Analysis of R-Ras signalling pathways. J. Cell Sci. 2001, 114, 1357–1366. [Google Scholar] [CrossRef]
- Lagarrigue, F.; Kim, C.; Ginsberg, M.H. The Rap1-RIAM-talin axis of integrin activation and blood cell function. Blood 2016, 128, 479–487. [Google Scholar] [CrossRef] [Green Version]
- Elosegui-Artola, A.; Trepat, X.; Roca-Cusachs, P. Control of Mechanotransduction by Molecular Clutch Dynamics. Trends Cell Biol. 2018, 28, 356–367. [Google Scholar] [CrossRef]
- del Rio, A.; Perez-Jimenez, R.; Liu, R.; Roca-Cusachs, P.; Fernandez, J.M.; Sheetz, M.P. Stretching single talin rod molecules activates vinculin binding. Science 2009, 323, 638–641. [Google Scholar] [CrossRef]
- Wennstrom, S.; Hawkins, P.; Cooke, F.; Hara, K.; Yonezawa, K.; Kasuga, M.; Jackson, T.; Claesson-Welsh, L.; Stephens, L. Activation of phosphoinositide 3-kinase is required for PDGF-stimulated membrane ruffling. Curr. Biol. 1994, 4, 385–393. [Google Scholar] [CrossRef]
- Wennstrom, S.; Siegbahn, A.; Yokote, K.; Arvidsson, A.K.; Heldin, C.H.; Mori, S.; Claesson-Welsh, L. Membrane ruffling and chemotaxis transduced by the PDGF beta-receptor require the binding site for phosphatidylinositol 3’ kinase. Oncogene 1994, 9, 651–660. [Google Scholar] [PubMed]
- Brachmann, S.M.; Yballe, C.M.; Innocenti, M.; Deane, J.A.; Fruman, D.A.; Thomas, S.M.; Cantley, L.C. Role of phosphoinositide 3-kinase regulatory isoforms in development and actin rearrangement. Mol. Cell. Biol. 2005, 25, 2593–2606. [Google Scholar] [CrossRef] [Green Version]
- Ponti, A.; Machacek, M.; Gupton, S.L.; Waterman-Storer, C.M.; Danuser, G. Two distinct actin networks drive the protrusion of migrating cells. Science 2004, 305, 1782–1786. [Google Scholar] [CrossRef] [Green Version]
- Choi, C.K.; Vicente-Manzanares, M.; Zareno, J.; Whitmore, L.A.; Mogilner, A.; Horwitz, A.R. Actin and alpha-actinin orchestrate the assembly and maturation of nascent adhesions in a myosin II motor-independent manner. Nat. Cell Biol. 2008, 10, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Pramanik, M.K.; Iijima, M.; Iwadate, Y.; Yumura, S. PTEN is a mechanosensing signal transducer for myosin II localization in Dictyostelium cells. Genes Cells 2009, 14, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Wessels, D.; Lusche, D.F.; Kuhl, S.; Heid, P.; Soll, D.R. PTEN plays a role in the suppression of lateral pseudopod formation during Dictyostelium motility and chemotaxis. J. Cell Sci. 2007, 120, 2517–2531. [Google Scholar] [CrossRef] [Green Version]
- Signorello, M.G.; Leoncini, G. Effect of 2-arachidonoylglycerol on myosin light chain phosphorylation and platelet activation: The role of phosphatidylinositol 3 kinase/AKT pathway. Biochimie 2014, 105, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Arora, P.D.; Di Gregorio, M.; He, P.; McCulloch, C.A. TRPV4 mediates the Ca(2+) influx required for the interaction between flightless-1 and non-muscle myosin, and collagen remodeling. J. Cell Sci. 2017, 130, 2196–2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koziol-White, C.J.; Yoo, E.J.; Cao, G.; Zhang, J.; Papanikolaou, E.; Pushkarsky, I.; Andrews, A.; Himes, B.E.; Damoiseaux, R.D.; Liggett, S.B.; et al. Inhibition of PI3K promotes dilation of human small airways in a rho kinase-dependent manner. Br. J. Pharmacol. 2016, 173, 2726–2738. [Google Scholar] [CrossRef]
- Shoval, Y.; Berissi, H.; Kimchi, A.; Pietrokovski, S. New modularity of DAP-kinases: Alternative splicing of the DRP-1 gene produces a ZIPk-like isoform. PLoS ONE 2011, 6, e17344. [Google Scholar] [CrossRef]
- Chen, A.S.; Wardwell-Ozgo, J.; Shah, N.N.; Wright, D.; Appin, C.L.; Vigneswaran, K.; Brat, D.J.; Kornblum, H.I.; Read, R.D. Drak/STK17A Drives Neoplastic Glial Proliferation through Modulation of MRLC Signaling. Cancer Res. 2019, 79, 1085–1097. [Google Scholar] [CrossRef]
- Angulo-Urarte, A.; Casado, P.; Castillo, S.D.; Kobialka, P.; Kotini, M.P.; Figueiredo, A.M.; Castel, P.; Rajeeve, V.; Mila-Guasch, M.; Millan, J.; et al. Endothelial cell rearrangements during vascular patterning require PI3-kinase-mediated inhibition of actomyosin contractility. Nat. Commun. 2018, 9, 4826. [Google Scholar] [CrossRef] [Green Version]
- Asokan, S.B.; Johnson, H.E.; Rahman, A.; King, S.J.; Rotty, J.D.; Lebedeva, I.P.; Haugh, J.M.; Bear, J.E. Mesenchymal chemotaxis requires selective inactivation of myosin II at the leading edge via a noncanonical PLCgamma/PKCalpha pathway. Dev. Cell 2014, 31, 747–760. [Google Scholar] [CrossRef] [Green Version]
- Sipeki, S.; Bander, E.; Parker, P.J.; Farago, A. PKCalpha reduces the lipid kinase activity of the p110alpha/p85alpha PI3K through the phosphorylation of the catalytic subunit. Biochem. Biophys. Res. Commun. 2006, 339, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Yoshioka, K.; Aki, S.; Ishimaru, K.; Yamada, H.; Takuwa, N.; Takuwa, Y. Class II phosphatidylinositol 3-kinase alpha and beta isoforms are required for vascular smooth muscle Rho activation, contraction and blood pressure regulation in mice. J. Physiol. Sci. 2020, 70, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunduz, D.; Troidl, C.; Tanislav, C.; Rohrbach, S.; Hamm, C.; Aslam, M. Role of PI3K/Akt and MEK/ERK Signalling in cAMP/Epac-Mediated Endothelial Barrier Stabilisation. Front. Physiol. 2019, 10, 1387. [Google Scholar] [CrossRef] [Green Version]
- Sarker, M.A.K.; Aki, S.; Yoshioka, K.; Kuno, K.; Okamoto, Y.; Ishimaru, K.; Takuwa, N.; Takuwa, Y. Class II PI3Ks alpha and beta Are Required for Rho-Dependent Uterine Smooth Muscle Contraction and Parturition in Mice. Endocrinology 2019, 160, 235–248. [Google Scholar] [CrossRef]
- Franco, M.; Carmena, A. Eph signaling controls mitotic spindle orientation and cell proliferation in neuroepithelial cells. J. Cell Biol. 2019, 218, 1200–1217. [Google Scholar] [CrossRef] [Green Version]
- Lv, Y.; Liu, W.; Ruan, Z.; Xu, Z.; Fu, L. Myosin IIA Regulated Tight Junction in Oxygen Glucose-Deprived Brain Endothelial Cells Via Activation of TLR4/PI3K/Akt/JNK1/2/14-3-3epsilon/NF-kappaB/MMP9 Signal Transduction Pathway. Cell Mol. Neurobiol. 2019, 39, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.S.; Stark, D.J.; Raphael, R.M.; Wen, J.; Su, J.; Zhou, X.; Chang, C.C.; Zu, Y. SDF-1alpha stiffens myeloma bone marrow mesenchymal stromal cells through the activation of RhoA-ROCK-Myosin II. Int. J. Cancer 2015, 136, E219–E229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuel, M.S.; Lopez, J.I.; McGhee, E.J.; Croft, D.R.; Strachan, D.; Timpson, P.; Munro, J.; Schroder, E.; Zhou, J.; Brunton, V.G.; et al. Actomyosin-mediated cellular tension drives increased tissue stiffness and beta-catenin activation to induce epidermal hyperplasia and tumor growth. Cancer Cell 2011, 19, 776–791. [Google Scholar] [CrossRef] [Green Version]
- Miroshnikova, Y.A.; Mouw, J.K.; Barnes, J.M.; Pickup, M.W.; Lakins, J.N.; Kim, Y.; Lobo, K.; Persson, A.I.; Reis, G.F.; McKnight, T.R.; et al. Tissue mechanics promote IDH1-dependent HIF1alpha-tenascin C feedback to regulate glioblastoma aggression. Nat. Cell Biol. 2016, 18, 1336–1345. [Google Scholar] [CrossRef]
- Choi, C.; Kwon, J.; Lim, S.; Helfman, D.M. Integrin beta1, myosin light chain kinase and myosin IIA are required for activation of PI3K-AKT signaling following MEK inhibition in metastatic triple negative breast cancer. Oncotarget 2016, 7, 63466–63487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, E.J.; Cao, G.; Koziol-White, C.J.; Ojiaku, C.A.; Sunder, K.; Jude, J.A.; Michael, J.V.; Lam, H.; Pushkarsky, I.; Damoiseaux, R.; et al. Galpha12 facilitates shortening in human airway smooth muscle by modulating phosphoinositide 3-kinase-mediated activation in a RhoA-dependent manner. Br. J. Pharmacol. 2017, 174, 4383–4395. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhu, J.; Huang, Y.; Li, W.; Cheng, H. MYO18B promotes hepatocellular carcinoma progression by activating PI3K/AKT/mTOR signaling pathway. Diagn. Pathol. 2018, 13, 85. [Google Scholar] [CrossRef] [Green Version]
- Jiu, Y.; Kumari, R.; Fenix, A.M.; Schaible, N.; Liu, X.; Varjosalo, M.; Krishnan, R.; Burnette, D.T.; Lappalainen, P. Myosin-18B Promotes the Assembly of Myosin II Stacks for Maturation of Contractile Actomyosin Bundles. Curr. Biol. 2019, 29, 81–92.e85. [Google Scholar] [CrossRef] [Green Version]
- Zhai, K.; Tang, Y.; Zhang, Y.; Li, F.; Wang, Y.; Cao, Z.; Yu, J.; Kou, J.; Yu, B. NMMHC IIA inhibition impedes tissue factor expression and venous thrombosis via Akt/GSK3beta-NF-kappaB signalling pathways in the endothelium. Thromb. Haemost. 2015, 114, 173–185. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soriano, O.; Alcón-Pérez, M.; Vicente-Manzanares, M.; Castellano, E. The Crossroads between RAS and RHO Signaling Pathways in Cellular Transformation, Motility and Contraction. Genes 2021, 12, 819. https://doi.org/10.3390/genes12060819
Soriano O, Alcón-Pérez M, Vicente-Manzanares M, Castellano E. The Crossroads between RAS and RHO Signaling Pathways in Cellular Transformation, Motility and Contraction. Genes. 2021; 12(6):819. https://doi.org/10.3390/genes12060819
Chicago/Turabian StyleSoriano, Olga, Marta Alcón-Pérez, Miguel Vicente-Manzanares, and Esther Castellano. 2021. "The Crossroads between RAS and RHO Signaling Pathways in Cellular Transformation, Motility and Contraction" Genes 12, no. 6: 819. https://doi.org/10.3390/genes12060819
APA StyleSoriano, O., Alcón-Pérez, M., Vicente-Manzanares, M., & Castellano, E. (2021). The Crossroads between RAS and RHO Signaling Pathways in Cellular Transformation, Motility and Contraction. Genes, 12(6), 819. https://doi.org/10.3390/genes12060819