Abstract
Prader–Willi syndrome (PWS) is a rare genetic condition characterized by hypotonia, intellectual disability, and hypothalamic dysfunction, causing pituitary hormone deficiencies and hyperphagia, ultimately leading to obesity. PWS is most often caused by the loss of expression of a cluster of genes on chromosome 15q11.2-13. Patients with Prader–Willi-like syndrome (PWLS) display features of the PWS phenotype without a classical PWS genetic defect. We describe a 46-year-old patient with PWLS, including hypotonia, intellectual disability, hyperphagia, and pituitary hormone deficiencies. Routine genetic tests for PWS were normal, but a homozygous missense variant NM_003097.3(SNRPN):c.193C>T, p.(Arg65Trp) was identified. Single nucleotide polymorphism array showed several large regions of homozygosity, caused by high-grade consanguinity between the parents. Our functional analysis, the ‘Pipeline for Rapid in silico, in vivo, in vitro Screening of Mutations’ (PRiSM) screen, showed that overexpression of SNRPN-p.Arg65Trp had a dominant negative effect, strongly suggesting pathogenicity. However, it could not be confirmed that the variant was responsible for the phenotype of the patient. In conclusion, we present a unique homozygous missense variant in SNURF-SNRPN in a patient with PWLS. We describe the diagnostic trajectory of this patient and the possible contributors to her phenotype in light of the current literature on the genotype–phenotype relationship in PWS.
1. Introduction
Prader–Willi syndrome (PWS) is a rare genetic condition (estimated prevalence of 1:10,000–1:30,000), affecting multiple organ systems [1]. In the neonatal period, PWS is characterized by muscular hypotonia and feeding difficulties, which usually require tube feeding. Later in infancy, patients switch to hyperphagia (overeating) due to abnormal satiety response to food intake [1,2,3]. Endocrine features include growth hormone deficiency, hypothyroidism, hypogonadism, and, rarely, central adrenal insufficiency [1,3,4]. Both hyperphagia and pituitary dysfunction contribute to abnormal weight gain, which can ultimately lead to obesity. Additionally, temperature regulation and pain registration are often disturbed [1,2,5]. The majority of these features can be explained by hypothalamic dysfunction [6]. Moreover, patients with PWS often display developmental delay, autism spectrum disorders (ASD), ASD-like behavior [1,7], and challenging behavior [3]. The physical appearance is characterized by short stature, obesity, almond-shaped eyes, strabismus, small bitemporal diameter, a thin upper lip, small hands and feet, and tapering fingers [1,3]. Commonly used consensus diagnostic criteria are those proposed by Holm et al. [8] in 1993 (Table 1).

Table 1.
Consensus criteria Prader–Willi syndrome and the score of the index case.
PWS is usually caused by the absence of expression of a cluster of paternally expressed genes on chromosome 15q11.2-q13. The maternal allele is imprinted, and therefore genomic and epigenetic changes lead to PWS only if they occur in the paternally expressed genes. PWS is most commonly caused by a paternal deletion (70–75%), a uniparental maternal disomy 15 (mUPD, 25–30%), an imprinting center defect (ICD, 1–3%), or a paternal chromosomal translocation (rare) [9,10]. The so called ‘Prader–Willi syndrome critical region’ on chromosome 15q11.2-q13 encompasses several genes, including MKRN3, MAGEL2, NDN, NPAP1, SNURF-SNRPN, and numerous non-coding RNAs (ncRNAs) including small nucleolar RNAs (snoRNAs) [10]. The exact function of most of these genes and their exact relation to the PWS phenotype still remains unclear.
In patients with Prader–Willi-like syndrome (PWLS), PWS features are present without the classical PWS genotype (paternal deletion, mUPD, ICD, or translocation of chromosome 15q11.2-13). To provide adequate treatment and genetic counselling to the patients with PWLS and their relatives, it is important to understand the underlying genetic defects and pathways [10]. Cases with PWLS provide novel insight into the complex genotype–phenotype relationship of PWS. Many chromosomal alterations involving different chromosomes have been described in relation to PWLS [10,11,12]. Several cases of PWLS with a small deletion in the PWS critical region have been described. Deletions ranged from 80 to 236 kb and most include SNORD116 and the ‘imprinted in Prader–Willi syndrome’ (IPW) gene [13,14,15,16,17,18,19]. These case reports suggest that the region encoding SNORD116 and IPW is responsible for the key characteristics of PWS, while other genes in the PWS critical region may have smaller phenotypic contributions.
We now present a patient with PWLS, with a normal result on genetic diagnostic testing for PWS, without abnormalities in SNORD116 or IPW. We describe the fascinating diagnostic trajectory and discuss possible explanations for her phenotype in light of the current knowledge on the genotype–phenotype relationship in PWS.
2. Materials and Methods
The patient and her mother, who is legal representative, gave permission for the use of the clinical information and pictures in the current paper.
2.1. Methylation-Specific Multiplex-Ligation Probe Amplification (MS-MPLA)
For the MS-MLPA, we used the Salsa MLPA ME028-B2 PWS/AS probemix (MRC-Holland©), as described by Beygo et al. [20].
2.2. Single Nucleotide Polymorphism (SNP) Array
Genomic DNA was extracted from peripheral blood and hybridized to a Human CytoSNP-12 array (Illumina, San Diego, CA, USA) as reported before [21]. The array was scanned with the Illumina iScan Control. Data were processed using Genome Studio v2.1 software and analyzed with Nexus Copy Number software v5.0 (Biodiscovery, El Segundo, CA, USA).
2.3. Next Generation Sequencing (NSG)
Obesity gene panel sequencing was performed at the genetic diagnostics laboratory of the UMC Utrecht to search for pathogenic abnormalities in the exons of 57 protein coding obesity-associated genes, as described previously [22]. Identified variants were classified according to the American College of Medical Genetics and Genomics guidelines for variant classification.
2.4. RNA Sequencing
A punch biopsy of the skin was taken from the index case and the mother. RNA sequencing was performed at the Institute of Human Genetics, University Hospital Essen. In order to understand whether, and how, the new variant affects gene expression, we compared RNA sequencing data from the patient and her mother to healthy controls and patients with genetically confirmed classical PWS.
2.5. Effect Predictor Programs
To predict whether the variant could affect protein function, we used Alamut VISUAL PLUS TM v.2.15 to access multiple protein prediction programs: Align-GVGD [23], MutationTaster (Build NCBI37/Ensembl 69) [24], sorting intolerant from tolerant (SIFT) [25,26], and Polymorphism Phenotyping v2 (Polyphen-2) [27,28].
2.6. PRiSM Screen
We assessed the effect of the SNRPN variant on protein function and its pathogenicity using our in-house developed functional genomics screen “Pipeline for Rapid in silico, in vivo, in vitro Screening of Mutations (PRiSM)” (for other studies where this has been used see [29,30,31,32]).
2.6.1. Constructs
To obtain the cDNA sequence from human SNRPN (NM_003097.6) a PCR (Phusion high fidelity, Thermo Fisher) was done on the human brain cDNA library, using the following primers: Fw 5′ GGCGCGCCACCATGACTGTTGGCAAGAGTAG 3′ and Rev 5′ TTAATTAACTAAGGTCTTGGTGGACG 3′. The gene was then cloned into our dual promoter expression vector [31,32,33]. Using PCR (Phusion high fidelity, Thermo Fisher) we then introduced the single nucleotide variant using the following primers: SNRPN– c.193c>t (p.Arg65Trp), Fw 5′ CCAGAGCGTGAAGAAAAGTGGGTTTTGGGTCTGGTGT 3′ and Rev 5′ ACAC-CAGACCCAAAACCCACTTTTCTTCACGCTCTGG 3′. The same expression vector without an inserted gene was used as control for all the in vivo and in vitro experiments (control vector).
2.6.2. Mice
FvB/NHsD females were crossed with FvB/NHsD males (ordered at 8–10 weeks old from Envigo) for the neuronal cultures, whereas for the in utero electroporation female, FvB/NHsD (Envigo) were crossed with male C57Bl6/J (ordered at 8–10 weeks old from Charles River). All mice were kept group-housed in IVC cages (Sealsafe 1145T, Tecniplast) with bedding material (Lignocel BK 8/15 from Rettenmayer) on a 12/12 h light/dark cycle in 21 °C (±1 °C), humidity at 40–70%. Food pellets (801727CRM(P) from Special Dietary Service) and water were available ad libitum. All animal experiments were conducted in accordance with the European Commission Council Directive 2010/63/EU (CCD approval AVD101002017893).
2.6.3. HEK-293T Cell Transfections
We cultured HEK-293T cells (not authenticated) in 6-well plates in DMEM/10% Fetal Calf Serum (FCS)/1% penicillin/streptomycin and transfected them when they were 60% confluent with the empty vector control, SNRPN-WT or SNRPN-p.Arg65Trp (3 μg per 6-well dish), using polyethylenimine (PEI) according to the manufacturer instructions (Sigma). Then, 4–6 h after transfection, we changed the medium to reduce toxicity.
2.6.4. Western Blot
Two to three days after the HEK-293T cells were transfected, they were harvested and homogenized in lysis buffer (10 mM Tris-HCl 6.8, 2.5% SDS, 2 mM EDTA with added protease inhibitor cocktail (#P8340, Sigma), phosphatase inhibitor cocktail 2 (#P5726, Sigma), and phosphatase inhibitor cocktail 3 (#P0044, Sigma)). The BCA protein assay kit (Pierce) was used to determine the protein concentration. The lysate concentrations were adjusted to 1 mg/mL. Primary antibodies used: SNRPN (#11070-1-AP, 1:1000, ProteinTech) and RFP (#600401379, 1:2000, Rockland; used to detect tdTomato); secondary antibody: goat anti-rabbit (#926-68021, 1:15,000, LI-COR). LI-COR Odyssey Scanner and Odyssey 3.0 software were used to quantify the blots. The intensity of the SNRPN protein band in the different conditions was normalized against tdTomato (RFP signal). For the analysis, 4 replicates were used.
2.6.5. Primary Hippocampal Cultures
We prepared the primary hippocampal neuronal cultures from FvB/NHsD wild-type mice following the previously described procedure [34]. Briefly, hippocampi from brains of E16.5 embryos were collected in 10 mL ice cold neurobasal medium (NB, Gibco). After incubation of the hippocampi in pre-warmed trypsin/EDTA solution (Invitrogen) at 37° for 20 min, they were dissociated in 1.5 mL NB medium supplemented with 2% B27, 1% penicillin/streptomycin, and 1% glutamax (Invitrogen). Neurons were then plated on poly-D-lysine (25 mg/mL, Sigma) coated coverslips (1*106 cells per coverslip) in 12 well plates containing 1 mL of supplemented NB for each coverslip. The plates were stored at 37°/5% CO2.
2.6.6. Neuronal Transfection and Immunocytochemistry
After three days in vitro (DIV), neurons were transfected with an empty vector control (1.8 μg per coverslip), SNRPN-WT, or SNRPN-p.Arg65Trp (2.5 μg per coverslip). For transfection, Lipofectamine was used according to the manufacturer’s instructions (Invitrogen). Five days post-transfection, neurons were fixed with 4% paraformaldehyde (PFA)/10% sucrose, for the neuronal morphology analysis. Antibody staining was done overnight at 4 °C with MAP2 (1:500, #188004, Synaptic System) and SNRPN (#11070-1-AP, 1:100, ProteinTech) in GDB buffer (0.2% BSA, 0.8 M NaCl, 0.5% Triton X-100, 30 mM phosphate buffer, pH 7.4). Secondary antibodies: anti-guinea-pig-Alexa647 (#706-605-148) and anti-rabbit-Alexa488 (#711-545-152) conjugated secondary antibody (1:200, Jackson ImmunoResearch). Mowiol-DABCO (Sigma) mounting medium was used to mount the coverslips. The LSM700 was used to acquire confocal images.
At least 10 confocal images (20× objective, 0.5 zoom, 1024 × 1024 pixels) of different transfected neurons (identified by the red staining from the tdTomato) were obtained from each condition, with at least two independent experimental replications. Total neurite length and arborization (the number of branching of each primary neurite) was analyzed, using the NeuronJ plugin of ImageJ. All values were normalized against the mean values of the empty vector control. Analysis was done by an experimenter blinded for the transfection conditions.
2.6.7. In Utero Electroporation
The in utero electroporation was done as described previously [32]. In short, pregnant FvB/NHsD mice at E14.5 of gestation were anesthetized, and the uterus was exposed. Through the uterus wall, the DNA construct (1.5–3 μg/μL, diluted in fast green (0.05%)) was injected in the lateral ventricle of the embryos, using a glass pipette controlled by a Picospritzer® III device. Using tweezer-type electrodes connected to a pulse generator (ECM 830, BTX Harvard Apparatus), five electrical square pulses of 45 V (50 ms per pulse and 150 ms inter-pulse interval (ipi)) were delivered. The positive pole of the tweezers was placed on top of the developing somatosensory cortex. Plasmids injected: empty vector control, SNRPN-WT, or SNRPN-p.Arg65Trp. After birth, pups (M/F) were sacrificed at P1 for histochemical processing.
2.6.8. Immunohistochemistry
Mice (deeply anesthetized with an overdose of Nembutal) underwent transcardial perfusion with 4% paraformaldehyde (PFA) and the brains were post-fixed in 4% PFA. After embedding the brains in gelatin and cryoprotecting them in 30% sucrose in 0.1 M phosphate buffer (PB) for 2–4 h, they were frozen on dry ice, and sectioned coronally using a freezing microtome (50 μm thick). Free-floating sections were blocked in PBS containing 10% normal horse serum (NHS) and 0.5% Triton X-100 for one hour and then incubated with primary antibody RFP (#600401379, 1:2000, Rockland) in PBS containing 2% NHS, 0.5% Triton X-100, at ambient temperature overnight. Secondary antibody used: Cy3 donkey-anti-rabbit (1:400, Jackson ImmunoResearch) diluted in PBS containing 2% NHS, 0.5% Triton-X 100. 4′,6-diamidino-2-phenylindole solution (DAPI, 1:10,000, Invitrogen) was used as a counterstain and Mowiol was used to mount the sections on glass. Images were acquired using a LSM700 confocal microscope (Zeiss) with a 10× objective.
Confocal images (10× objective, 0.5 zoom, 1024 × 1024 pixels) obtained from 2–3 non-consecutive sections from at least 3 successfully targeted animals per condition were used for the neuronal migration analysis (previously described [31,32,33]).
2.6.9. Statistical Analysis
We assumed normally distributed data. For the in vitro and in vivo overexpression experiments, statistical difference was determined using one-way analysis of variance (ANOVA) followed by Dunnett’s post-hoc test for multiple comparisons. Two-tailed unpaired t-test (dual comparison) was used for the Western blot analysis. We analyzed neuronal migration based on the proportion of electroporated cells that migrated to the cortical plate at P1 (defined as the most proximal 40% of dorsoventral distance between the pia and ventricle (first four of ten equally spaced bins)).
2.7. Literature Review
We performed a search on Embase, Medline, the Web of Science Core Collection, Cochrane Central Register of Controlled Trials, and Google Scholar for case reports of patients with genetic alterations (e.g., translocations, deletions, genetic variants) in the PWS critical region on the paternal chromosome that affected only part of the PWS critical region. We included case reports and case series that provide a description of the genotype and phenotype of each individual case. We included articles about patients with and without features of PWS. Exclusion criteria were articles that were not available online, articles that were not available in English, patients with genetic alterations of the PWS critical region on the maternal chromosome only, case reports where the entire PWS critical region or the PWS-imprinting center (IC) was affected, case reports where the affected region extended beyond the PWS critical region, and case reports with insufficient genotyping or phenotyping. For the full search strategy, see Table S2.
3. Results
3.1. Patient Description
The female patient presented at our hospital at the age of 46 years, after she had been referred for treatment of morbid-obesity. She fulfilled the criteria for the clinical diagnosis of PWS, as is shown in Table 1.
3.1.1. Medical History
There were little fetal movements during pregnancy. The index case was born after a pregnancy of 40 weeks with a birth weight of three kilograms (−0.5 SD). Postnatally, she was cyanotic and hypotonic. Her neonatal feeding difficulties required tube feeding for which she was hospitalized for several months. Her feeding difficulties persisted until she was 1.5 years old, after which she developed hyperphagia, resulting in obesity at the age of four. When she was twelve years old, her weight was 125 kg according to her mother. Secondary to her obesity, she had developed type 2 diabetes mellitus and dyslipidaemia, for which she was treated with metformin and simvastatin, respectively. Psychomotor development was severely delayed; she started walking at the age of eight years and talking at the age of 40 years. She received special education. She had primary amenorrhea.
3.1.2. Anamnesis
The patient had clear hyperphagia: She could easily eat four full plates of food. Her mother, who was the primary caregiver, reported challenging behavior including temper tantrums, stealing food, skin picking, and trichotillomania.
3.1.3. Family History
The parents of the index case were first-degree relatives. The mother of the index case had many (half-) siblings, of which most were illiterate or had great difficulty reading and writing, mainly caused by lack of education during childhood. Although both the mother of the index case and multiple siblings had short stature and were overweight, none of the family members had the same phenotype as the index case. However, the mother had lost contact with most relatives and, therefore, family health history was incomplete. The mother of the patient was 157 cm tall and weighed 91 kg (body mass index (BMI) 37 kg/m2).
3.1.4. Physical Examination
Physical examination revealed a typical PWS appearance of the index case, with obesity (BMI 34 kg/m2), hypotonia, kyphosis, high-arched feet (requiring orthopedic shoes), and small hands with tapering fingers. Facial features included narrow bitemporal diameter, almond-shaped eyes, and strabismus (Figure 1). She had some breast formation, but exact Tanner stage for breast development was hard to assess due to her obesity. She had Tanner stage 3 pubic hair development. Her height was 158 cm (−2 SD, [35]), her weight 85 kg, and her head circumference 56.5 cm (+0.7 SD).
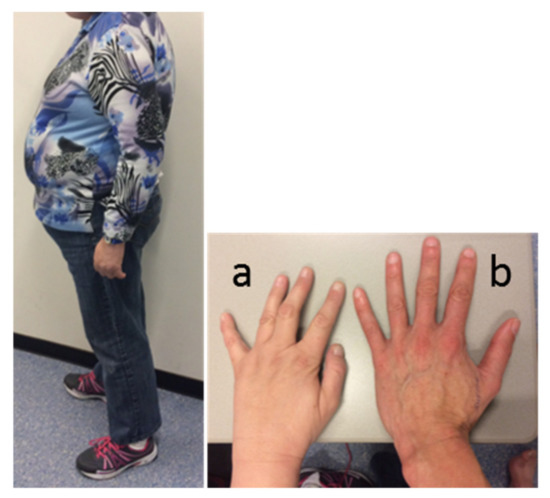
Figure 1.
Appearance of the index case. Picture of the patient’s body, and left hand (a) compared to the hand of a healthy female control (b).
3.1.5. Biochemistry and Imaging
Laboratory results revealed central hypogonadism and a lowered Insulin-like growth factor (IGF)1 level (Table S1). Peak growth hormone value during growth hormone releasing hormone (GHRH) and arginine stimulation test was 2.9 µg/L. This is lower than the cut-off of 4.2 µg/L in obese subjects [36], confirming the diagnosis growth hormone deficiency. Cortisol and thyroid hormone levels were normal. Dual-energy X-ray absorptiometry (DEXA) scan showed osteopenia of the lumbar vertebrae and osteoporosis of the femoral neck.
3.1.6. Polysomnography
Polysomnography showed mild sleep apnea with an apnea–hypopnea index of 6.2 per hour [37].
3.1.7. Features Not Corresponding to PWS
Apart from the typical PWS features, the patient also had features not corresponding to PWS, like celiac disease, tinnitus, hearing loss (requiring a hearing aid), cataract (requiring lens implantation), and arthralgia of the hands and feet. Atypical findings during physical examination were a remarkable overbite, diastemata, multiple naevi, and a small palpebral fissure width. Her relatively tall stature (158 cm, while most PWS females have a height below 150 cm) was also atypical for PWS. Additionally, MRI of the brain also revealed abnormalities not specific for PWS, including septo-optic dysplasia (SOD), with several midline defects, including partial agenesis of the corpus callosum and the septum pellucidum and an atrophic left optic nerve, but the pituitary gland had a normal size [38]. In addition, there was partial agenesis of the sagittal sinus and most likely agenesis of the falx cerebri. Lastly, there was a small lesion in the left-posterior part of the pituitary gland with a maximum diameter of 5 mm, most likely a pituitary incidentaloma. Her clinical, behavioral, and dysmorphic features are shown in Table 2.
Table 2.
Clinical features, dysmorphic features, and challenging behavior associated with PWS and presence in the index case.
As the patient fulfilled the consensus criteria for Prader–Willi syndrome (Table 1), we performed genetic diagnostic testing for PWS. After routine PWS methylation tests, using methylation-specific multiplex-ligation probe amplification (MS-MLPA), ruled out the presence of a deletion, mUPD, or ICD of chromosome 15q11.2-13, we performed additional genetic testing. Obesity gene panel analysis revealed a homozygous variant NM_003097.3(SNRPN):c.193C>T, p.(Arg65Trp) in SNURF-SNRPN (Figure 2), which was classified as a variant of uncertain clinical significance (class 3 according to the American College of Medical Genetics and Genomics guidelines). This variant was mentioned in the supplementary table (individual 111) of the article by Kleinendorst et al. [22]. The mother was heterozygous for the same variant. The father was not available for DNA analysis. Hemizygosity in the index patient was excluded by MS-MLPA analysis. Single nucleotide polymorphism array (SNP) array showed several large regions of homozygosity (ROH), caused by high-grade consanguinity between the parents (Figure 3). The exact coordinates of the breakpoints can be found in Table S3.
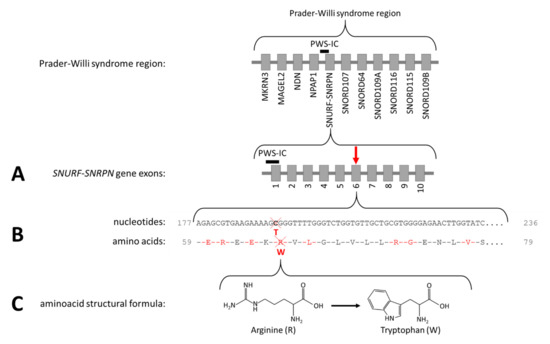
Figure 2.
The Prader–Willi syndrome region and NM_003097.3(SNRPN):c.193C>T, p.(Arg65Trp). (A) The arrow indicates the location of the variant in the SNURF-SNRPN gene. (B) The cross indicates the location of the affected nucleotide and the amino acid. (C) The structural formula of arginine and tryptophan.
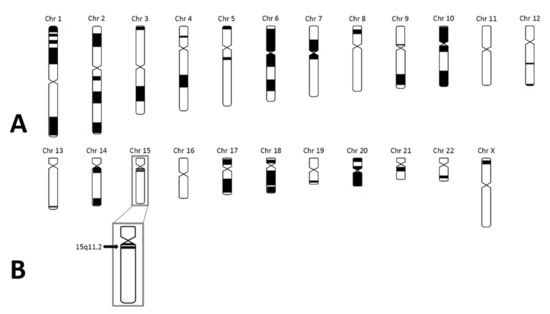
Figure 3.
SNP array results in the index case. (A) Black parts represent homozygous regions in the index case. The number of homozygous regions is suggestive for high-grade consanguinity. (B) The black arrow represents 15q11.2 in which SNURF-SNRPN is located. As shown, SNURF-SNRPN lies within a homozygous region.
SNURF-SNRPN was located in one of the ROH. In silico analysis using variant effect predictor programs Align-GVGD [23], MutationTaster (Build NCBI37/Ensembl 69) [24], sorting intolerant from tolerant (SIFT) [25,26], and Polymorphism Phenotyping v2 (Polyphen-2) [27,28] predicted that NM_003097.3(SNRPN):c.193C>T, p.(Arg65Trp) is deleterious. The variant was not present in the 1000 Genomes Project (Phase 3 release) [42] or in the genome aggregation database (gnomAD v2.1.1) [43]. The protein structure of SNRPN and the location of the changed amino acid is depicted in Figure 4. RNA sequencing analysis showed equal expression of SNRPN and all other genes in the PWS critical region for the index case, the mother, and four healthy, Caucasian volunteers (one male and three females) that served as controls. RNA sequencing confirmed that the mother of the patient had the variant on her paternal chromosome 15.
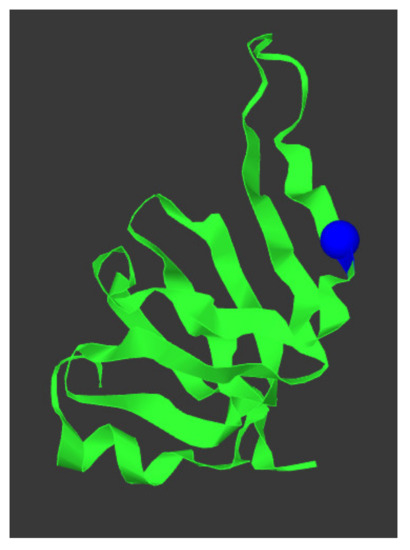
Figure 4.
SNRPN protein structure and location p.(Arg65Trp) variant. The protein structure of SNPRN is given in green, the p.(Arg65Trp) variant is depicted as a blue sphere. This figure was generated using mutation3D [44].
Functional analysis showed that the NM_003097.3(SNRPN):c.193C>T, p.(Arg65Trp) variant did not cause instability of the protein, showing similar expression levels to SNRPN-WT (p = 0.16, two-tailed unpaired Student’s t-test) (Figure 5a). Additionally, overexpression of SNRPN-WT or SNRPN-p.Arg65Trp in a small subset of neurons during embryological development did not alter the migration pattern of the neurons transfected (One-Way ANOVA: F[2,29] = 0.97, p = 0.39) (Figure 5b). However, whereas overexpression of SNRPN-WT showed a small trend in reducing neuronal maturation (Neurite length: One-Way ANOVA: F[2,45] = 6.29, p = 0.003; SNRPN-WT versus empty vector control: p = 0.0627, Tukey’s multiple comparison test; Arborization: One-Way ANOVA: F[2,45] = 5.66, p = 0.006; SNRPN-WT versus empty vector control: p = 0.44, Tukey’s multiple comparison test), overexpression of the SNRPN-p.Arg65Trp variant significantly affected neuronal maturation in vitro, resulting in a decreased neurite length and arborization (Neurite length: SNRPN-p.Arg65Trp versus empty vector control: p = 0.003, Tukey’s multiple comparison test; Arborization: SNRPN-p.Arg65Trp versus empty vector control: p = 0.004, Tukey’s multiple comparison test) (Figure 5c). Taken together, these results suggest that the SNRPN-p.Arg65Trp variant does affect the function of SNRPN, suggesting pathogenicity.
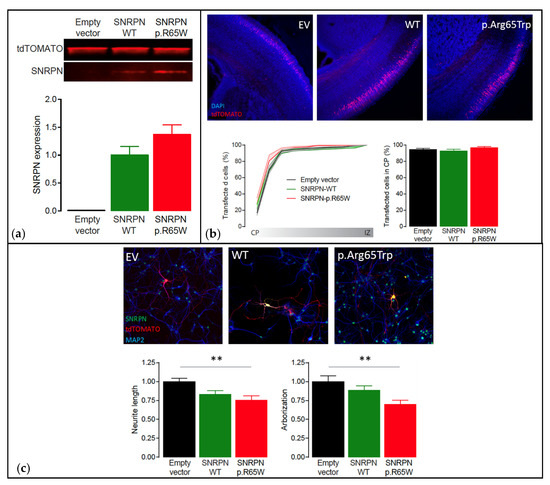
Figure 5.
Functional analysis using the PRiSM screen. Abbreviations: Empty vector (EV), wild type (WT). (a) Western blot analysis of transfected HEK293T cells showing that SNRPN expression is normal. Top: Western blot of transfected HEK293T cells showing clear overexpression of SNRPN-WT and SNRPN-p.R65W. Bottom: Quantification of the Western blots showing that the SNRPN-p.R65W missense variant does not alter the expression levels of SNRPN (N = 4 per condition). (b) In utero electroporation of the different SNRPN constructs does not affect neuronal migration in vivo. Top: Representative images from in utero electroporated postnatal brains on day 1. These images show that the far majority of the transfected cells (tdTOMATO+) migrated out to the cortical plate (CP; the outer layer of the cortex). Bottom left: Cumulative distribution of the transfected neurons at P1 from the cortical plate (CP; the outer layer of the cortex) to the intermediate zone (IZ; inner layer of the cortex). Bottom right: Percentage of neurons that migrated out to the superficial layers of the cortex (sum of bins 1 through 4). Number of pictures/animals for each condition = 9/3. (c) Expression of the different SNRPN constructs in mouse primary hippocampal neurons reveal that overexpression of the p.Arg65Trp variant significantly affects neuronal maturation in vitro. Top: Representative images of primary hippocampal neurons transfected with empty vector control, SNRPN-WT or SNRPN-p.Arg65Trp (tdTOMATO+). Neurons are stained for SNRPN (green) and MAP2 (blue). Bottom: Quantitative analysis of the total neurite length and arborization in the different conditions. Number of neurons/number of batches for each condition = 20/2; ** p < 0.01.
3.2. Literature Review
To interpret our findings in the light of the available literature, we performed a thorough literature search for case reports or case series of patients with genetic alterations (e.g., translocations, deletions, genetic variants) in the PWS critical region on the paternal chromosome that affected only part of the PWS critical region. After deduplication, we found 4693 articles, of which the titles and abstracts were reviewed. After full-text screening, we included 22 cases with PWLS. Additionally, we found 29 case reports that described abnormalities in only MAGEL2 (associated with Schaaf–Yang syndrome) and 42 case reports with abnormalities in only MKRN3 (associated with central precocious puberty), which are not included in the table. Most of the 22 cases with PWLS had a translocation or a deletion, leading to a loss of function of one or more genes of the PWS critical region. Additionally, we found one case report of a patient with a small duplication in exon 1 of SNURF, which introduced a premature stop codon in SNURF. The results of this literature review are summarized in Table 3 and Figure 6.
Table 3.
Literature review.
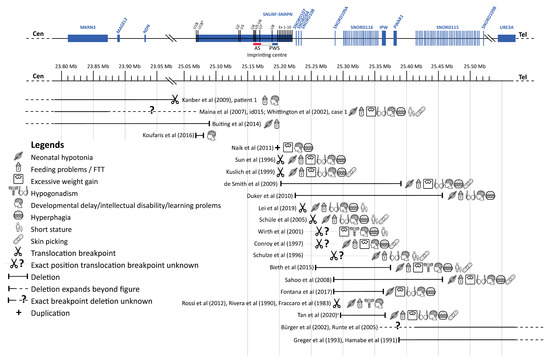
Figure 6.
Overview cases literature review. Abbreviations: Angelman syndrome (AS), failure to thrive (FFT), Prader–Willi syndrome (PWS). a Exact breakpoints for this case are not available, depicted is the minimal deleted region. This figure shows an overview of the cases we found during our literature review. For each case, the affected region is depicted, followed by symbols representing the clinical phenotype of the case. Genes are represented at scale with physical distance in Mb. SnoRNAs are depicted as vertical lines and other genes as boxes. Deletions are depicted according to the genomic deletion coordinates for build hg19. Excessive weight gain was only scored as present if it occurred when the case was 1 to 6 years old. Figure adapted from Fontana et al. [19] and Tan et al. [13]. This figure has been designed using resources from Flaticon.com (from surang, freepik, smashicons, dinosoftlabs and monkik), accessed on 1 March 2021.
4. Discussion
We here present the puzzling diagnostic trajectory of a 46-year-old female patient with PWLS. After genetic testing excluded the classical PWS genetic defects (deletion, mUPD, ICD, or translocation of chromosome 15q11.2-13), we performed additional genetic analyses. Obesity gene panel analysis of 57 obesity-associated genes revealed a homozygous variant NM_003097.3(SNRPN):c.193C>T, p.(Arg65Trp) in exon 6 of the SNURF-SNRPN gene, changing CGG into TGG (Figure 2). This variant was not present in the 1000 Genomes Project (Phase 3 release) [42] or in the genome aggregation database (gnomAD v2.1.1) [43]. The variant changes the non-aromatic and positively charged arginine into the aromatic, non-polar tryptophan (Arg65Trp), which is deleterious according to various variant effect predictor programs. The fact that this homozygous missense variant is located in the PWS critical region suggests a relation with the PWS phenotype of the patient.
While the patient’s phenotype included PWS features, she also had atypical features, including brain lesions at MRI. Although morphological abnormalities in the brain of patients with PWS have been described [67,68,69,70,71], the brain lesions found in our patient were atypical for PWS. SNP array analysis showed multiple large ROH spread across different chromosomes, suggesting that (multiple) autosomal recessive diagnoses could contribute to the complete phenotype in this patient. Additionally, the fact that the mother of the patient had the same variant on her paternal chromosome 15 without having PWLS (she had obesity but did not fulfil any other diagnostic criteria for PWS), suggests that this variant is probably not the only explanation for the phenotype of the index case.
As she had dysgenesis of midline brain structures and optic nerve hypoplasia at MRI with hypothalamic-pituitary dysfunction [38], she fulfilled the criteria for SOD. SOD has a wide variability of clinical features, including some features that were present in our patient, like developmental delay [72], dysmorphic features, and autism-like behavior [73]. However, our patient also had many symptoms not associated with SOD, including hyperphagia, hypotonia, and kyphosis. Although the etiology remains unclear in most cases, SOD has been associated with pathogenic variants in HESX1, OTX2, SOX2, and SOX3 [38,74,75]. One of the ROH of the index patient included SOX3, but none of the ROH included HESX1, OTX2, or SOX2.
The index case has a remarkable family history with a large pedigree. Most family members were short, overweight, and/or illiterate. The latter is remarkable in the context of 10% inadequate literacy in the total Dutch 16–65 year old population [76]. The high prevalence of short stature, obesity and/or illiteracy in this family is suggestive of a genetic component. Unfortunately, other family members were unavailable for genetic analysis.
Interestingly, SNURF-SNRPN is a bi-cistronic gene, which is transcribed exclusively from the paternally inherited chromosome. SNURF-SNRPN is expressed in several tissues and shows the highest RNA expression in the brain and heart, followed by endocrine tissues, male reproductive tissues, and blood [77]. SNURF-SNRPN encodes two proteins: SNURF and SNRPN (also called SmN) [78]. The function of SNURF is unknown [78]. SNRPN is involved in mRNA splicing and predominantly expressed in neurons, especially in central neurons [79,80,81]. SNRPN influences neurite outgrowth, neuron migration, and distribution of dendritic spines in mice [82]. However, the precise role of SNRPN in the development of the PWS phenotype remains largely unknown. Apart from encoding SNURF and SNRPN proteins, SNURF-SNRPN transcription is necessary for production of downstream ncRNAs implicated in many PWS traits [83,84].
Imprinting at 15q11.2-13 is regulated by a bipartite imprinting center defined as the smaller regions of overlap (SRO) found in patients with imprinting defects [85,86,87,88,89,90]. The Angelman syndrome-SRO (AS-SRO) functions to silence paternally expressed genes on the maternal allele. The PWS-SRO functions somatically to activate paternally expressed genes on the paternal allele [91,92,93]. The PWS-SRO spans SNURF-SNRPN exon 1 [94,95]. The missense variant in this patient is in exon 6, about 20 kb downstream of the PWS-SRO and is therefore very unlikely to generate an ICD. Additionally, in our patient, imprinting and expression of the genes in the PWS critical region was normal, excluding an ICD.
The PWS critical region encompasses several genes, including MKRN3, MAGEL2, NDN, SNURF-SNRPN, and ncRNAs including snoRNAs like SNORD115 and SNORD116 [10]. Two individuals with deletions of MKRN3, MAGEL2, and NDN have been described [60,64]. These patients lacked many typical PWS features, suggesting that deletions of these genes cannot cause the full PWS phenotype on their own. However, patients with a heterozygous truncating pathogenic variant in the paternally derived allele of the MAGEL2 gene have Schaaf–Yang syndrome, which has clinical overlap with PWS, including intellectual disability, autism spectrum disorder, neonatal hypotonia, and infantile feeding problems [96,97]. Patients with Schaaf–Yang syndrome also display non-PWS features, for example distal joint contractures. SNORD115 is unlikely to play a role in the PWS phenotype, as individuals with a paternally inherited deletion of SNORD115 do not have PWS features [47,48,56,57]. SNORD116 is suspected to be a strong contributor to the PWS phenotype [98]. Several cases with PWS features and microdeletions or translocations resulting in lack of expression of SNORD116 and IPW indicate an important role of SNORD116 and IPW [13,14,15,16,17,18,19,49,50,51,52,53,58,66], Table 3. However, IPW is a non-protein coding RNA, which is minimally conserved between human and mouse genomes [99,100]. Mice with a targeted deletion of SNORD116 show cognitive deficits, abnormal growth and feeding [101,102], while mice with paternal deletions of portions of SNURF or SNRPN seem to have a normal phenotype [103,104]. It has been suggested that individuals with translocations have a milder phenotype when only SNORD116 is affected, compared to individuals in whom SNURF-SNRPN expression is also abolished. SNORD116 deletions seem to cause most of the major features of PWS, but they may be less prominent [58]. It should be noted that this is hard to investigate as all patients are assessed by different physicians, making it hard to objectively assess patients [84]. Lastly, SNURF could contribute to the PWS phenotype, as is demonstrated by Naik et al. [61], who presented a case with a small duplication in exon 1 of SNURF-SNRPN, which is predicted to only affect SNURF expression, although it cannot be ruled out that SNRPN is also affected. This patient had developmental delay, challenging behavior, and increased appetite, but also tall stature (170 cm (+3.26 SD)) and a large head circumference (57 cm (+2.69 SD)), which are not PWS features and were also not present in the index case that we present.
SNORD116 is produced from the same primary transcript as SNURF-SNRPN [83,84] and therefore variants in SNURF-SNRPN could cause a PWS phenotype through a change of expression of SNORD116. However, the RNA sequencing in our patient showed a normal expression of SNORD116. This rules out that the PWLS was caused by lack of SNORD116 expression.
PWS is generally thought to result from loss of expression of the paternal genes on the PWS critical region, as the maternal PWS critical region is imprinted and therefore not expressed. The mother of the index case had the same variant as the index case on her paternal chromosome 15. She did not have a documented intellectual disability or specific PWS features, but she was illiterate and had obesity. This suggests that the SNRPN-p.Arg65Trp variant might not always be pathogenic (incomplete penetrance) or might result in a mild phenotype. There is some evidence from mice that expression of the maternal allele of chromosome 15q11.2-13 can be detected when the paternal allele is not active [105,106]. However, findings in mice are poor predictors of the human situation and therefore the loss of silencing found by Rieusset et al. [105] cannot be extrapolated to humans.
A previous report by Iourov et al. [107] suggested that homozygosity (without mUPD) of part of 15q11.2 could be related to a mild PWLS phenotype, mainly characterized by developmental delay and/or intellectual disability. According to SNP array, homozygosity of 15q11.2 was also present in our patient and could have influenced her phenotype. However, the phenotype of our index case was specific to PWS, whereas the five cases described by Iourov et al. lacked most major phenotypic features of PWS. They did not present hypothalamic dysfunction leading to hyperphagia, pituitary hormone deficiencies, abnormal temperature regulation, or inadequate pain registration.
In our patient, RNA sequencing showed that expression of SNURF-SNRPN and the other genes in the PWS critical region were not affected. However, the change from arginine to tryptophan caused by the homozygous SNRPN variant was predicted to severely affect protein function. In order to understand whether the variant found in this patient indeed is pathogenic and whether it causes a loss of function or a gain of function, we made use of the unbiased functional genomics screen PRiSM. This showed that the variant does not cause protein instability, nor behaves differently from SNRPN-WT in the migration assay. However, whereas overexpression of SNRPN-WT resulted in a small but non-significant trend in reduced neural development in vitro, SNRPN-p.Arg65Trp significantly reduced neural development upon overexpression primary hippocampal neurons. This suggests that the missense variant might alter the function of SNRPN, causing a dominant negative effect on neuronal maturation when overexpressed. These results show that this variant does not cause a loss of function, rather it might suggest that it causes a gain of function. However, future studies are needed to confirm its functional impact.
The SNRPB’/B genes encode the proteins small nuclear ribonucleoprotein-associated polypeptide B’ and B (SmB’/B). They are closely related to SNRPN, which encodes the protein SNRPN, also called small nuclear ribonucleoprotein-associated polypeptide N (SmN). Normally, SNRPN replaces SmB’/B in the brain. It has been demonstrated that the loss of SNRPN in PWS brain tissue causes a compensatory feedback loop that drastically upregulates the levels of SmB’/B. It has been suggested that upregulation of SmB’/B in PWS reduces the severity of the syndrome [108]. However, in the presence of a non-functional SNRPN protein, this compensation mechanism might not be induced, leading to more severe phenotypic features compared to cases where SNRPN is completely absent. RNA sequencing in this patient showed that SNRPB’/B expression in fibroblasts was normal.
5. Conclusions
In conclusion, the finding of this homozygous missense SNURF-SNRPN variant in a patient with virtually all clinical features of PWS suggests that this variant might have caused her PWLS with combined pituitary hormone deficiency (CPHD). Additionally, functional analysis suggested that the variant might affect the function of SNRPN. However, the large ROHs that were found throughout the genome suggests other autosomal recessive conditions that could contribute to her complete phenotype. The fascinating trajectory of genetic and functional analyses performed in this patient shows that the finding of a unique variant in the PWS critical region, in a patient with PWLS, does not necessarily prove a causal relationship between the two.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/genes12060875/s1, Table S1: Endocrine evaluations of the index case, Table S2: Full search strategy (Embase), Table S3: Regions of homozygosity detected by SNP array.
Author Contributions
Conceptualization, L.C.G.d.G., M.M.v.H. and G.M.v.W.; methodology, L.C.G.d.G., M.M.v.H. and G.M.v.W.; formal analysis, B.H., C.G. and G.M.v.W.; investigation, G.M.v.W.; resources, L.C.G.d.G. and G.M.v.W.; writing—original draft preparation, K.P.; writing—review and editing, K.P., G.M.v.W., L.K., A.G.W.R., B.H., C.G., L.J.C.M.v.Z., E.F.C.v.R., A.J.v.d.L., J.L.R., H.T.B., M.M.v.H. and L.C.G.d.G.; visualization, K.P., G.M.v.W. and L.C.G.d.G.; supervision, L.C.G.d.G. and A.J.v.d.L.; project administration, L.C.G.d.G.; funding acquisition, L.C.G.d.G. and G.M.v.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki. All animal experiments were conducted in accordance with the European Commission Council Directive 2010/63/EU (CCD approval AVD101002017893).
Informed Consent Statement
Written informed consent has been obtained from the patient to publish this paper.
Data Availability Statement
Not applicable.
Acknowledgments
We would like to thank the patient and her mother, for their willingness to provide fibroblasts and detailed family information. The authors wish to thank Elise Krabbendam from the Erasmus MC Medical Library for developing the search strategies, and Bas Munting for his technical assistance for the functional assays.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cassidy, S.B.; Schwartz, S.; Miller, J.L.; Driscoll, D.J. Prader-willi syndrome. Genet. Med. 2012, 14, 10–26. [Google Scholar] [CrossRef]
- Angulo, M.A.; Butler, M.G.; Cataletto, M.E. Prader-willi syndrome: A review of clinical, genetic, and endocrine findings. J. Endocrinol. Investig. 2015, 38, 1249–1263. [Google Scholar] [CrossRef]
- Goldstone, A.P.; Holland, A.J.; Hauffa, B.P.; Hokken-Koelega, A.C.; Tauber, M. Recommendations for the diagnosis and management of prader-willi syndrome. J. Clin. Endocrinol. Metab. 2008, 93, 4183–4197. [Google Scholar] [CrossRef]
- Pellikaan, K.; Rosenberg, A.G.W.; Kattentidt-Mouravieva, A.A.; Kersseboom, R.; Bos-Roubos, A.G.; Veen-Roelofs, J.M.C.; van Wieringen, N.; Hoekstra, F.M.E.; van den Berg, S.A.A.; van der Lely, A.J.; et al. Missed diagnoses and health problems in adults with prader-willi syndrome: Recommendations for screening and treatment. J. Clin. Endocrinol. Metab. 2020, 105, e4671–e4687. [Google Scholar] [CrossRef]
- Williams, M.S.; Rooney, B.L.; Williams, J.; Josephson, K.; Pauli, R. Investigation of thermoregulatory characteristics in patients with prader-willi syndrome. Am. J. Med. Genet. 1994, 49, 302–307. [Google Scholar] [CrossRef]
- Wattendorf, D.J.; Muenke, M. Prader-willi syndrome. Am. Fam. Physician 2005, 72, 827–830. [Google Scholar] [PubMed]
- Bennett, J.A.; Germani, T.; Haqq, A.M.; Zwaigenbaum, L. Autism spectrum disorder in prader-willi syndrome: A systematic review. Am. J. Med. Genet. A 2015, 167A, 2936–2944. [Google Scholar] [CrossRef] [PubMed]
- Holm, V.A.; Cassidy, S.B.; Butler, M.G.; Hanchett, J.M.; Greenswag, L.R.; Whitman, B.Y.; Greenberg, F. Prader-willi syndrome: Consensus diagnostic criteria. Pediatrics 1993, 91, 398–402. [Google Scholar]
- Buiting, K.; Cassidy, S.B.; Driscoll, D.J.; Gillessen-Kaesbach, G.; Kanber, D.; Tauber, M.; Schwinger, E.; Horsthemke, B. Clinical utility gene card for: Prader-willi syndrome. Eur. J. Hum. Genet. 2014, 22, 1153. [Google Scholar] [CrossRef]
- Cheon, C.K. Genetics of prader-willi syndrome and Prader-Will-like syndrome. Ann. Pediatr. Endocrinol. Metab. 2016, 21, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.F.; Paiva, C.L. Prader-Willi-like phenotypes: A systematic review of their chromosomal abnormalities. Genet. Mol. Res. 2014, 13, 2290–2298. [Google Scholar] [CrossRef] [PubMed]
- Desch, L.; Marle, N.; Mosca-Boidron, A.L.; Faivre, L.; Eliade, M.; Payet, M.; Ragon, C.; Thevenon, J.; Aral, B.; Ragot, S.; et al. 6q16.3q23.3 duplication associated with Prader-Willi-like syndrome. Mol. Cytogenet. 2015, 8, 42. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Potter, K.J.; Burnett, L.C.; Orsso, C.E.; Inman, M.; Ryman, D.C.; Haqq, A.M. Prader–Willi-like phenotype caused by an atypical 15q11.2 microdeletion. Genes 2020, 11, 128. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, T.; Del Gaudio, D.; German, J.R.; Shinawi, M.; Peters, S.U.; Person, R.E.; Garnica, A.; Cheung, S.W.; Beaudet, A.L. Prader-Willi phenotype caused by paternal deficiency for the hbii-85 c/d box small nucleolar rna cluster. Nat. Genet. 2008, 40, 719–721. [Google Scholar] [CrossRef]
- De Smith, A.J.; Purmann, C.; Walters, R.G.; Ellis, R.J.; Holder, S.E.; Van Haelst, M.M.; Brady, A.F.; Fairbrother, U.L.; Dattani, M.; Keogh, J.M.; et al. A deletion of the hbii-85 class of small nucleolar rnas (snornas) is associated with hyperphagia, obesity and hypogonadism. Hum. Mol. Genet. 2009, 18, 3257–3265. [Google Scholar] [CrossRef] [PubMed]
- Duker, A.L.; Ballif, B.C.; Bawle, E.V.; Person, R.E.; Mahadevan, S.; Alliman, S.; Thompson, R.; Traylor, R.; Bejjani, B.A.; Shaffer, L.G.; et al. Paternally inherited microdeletion at 15q11.2 confirms a significant role for the snord116 c/d box snorna cluster in prader-willi syndrome. Eur. J. Hum. Genet. 2010, 18, 1196–1201. [Google Scholar] [CrossRef]
- Bieth, E.; Eddiry, S.; Gaston, V.; Lorenzini, F.; Buffet, A.; Conte Auriol, F.; Molinas, C.; Cailley, D.; Rooryck, C.; Arveiler, B.; et al. Highly restricted deletion of the snord116 region is implicated in prader-willi syndrome. Eur. J. Hum. Genet. 2015, 23, 252–255. [Google Scholar] [CrossRef]
- Hassan, M.; Butler, M.G. Prader-Willi syndrome and atypical submicroscopic 15q11-q13 deletions with or without imprinting defects. Eur. J. Med. Genet. 2016, 59, 584–589. [Google Scholar] [CrossRef]
- Fontana, P.; Grasso, M.; Acquaviva, F.; Gennaro, E.; Galli, M.L.; Falco, M.; Scarano, F.; Scarano, G.; Lonardo, F. Snord116 deletions cause prader-willi syndrome with a mild phenotype and macrocephaly. Clin. Genet. 2017, 92, 440–443. [Google Scholar] [CrossRef]
- Beygo, J.; Buiting, K.; Ramsden, S.C.; Ellis, R.; Clayton-Smith, J.; Kanber, D. Update of the emqn/acgs best practice guidelines for molecular analysis of prader-willi and angelman syndromes. Eur. J. Hum. Genet. 2019, 27, 1326–1340. [Google Scholar] [CrossRef]
- Srebniak, M.; Boter, M.; Oudesluijs, G.; Joosten, M.; Govaerts, L.; Van Opstal, D.; Galjaard, R.J. Application of snp array for rapid prenatal diagnosis: Implementation, genetic counselling and diagnostic flow. Eur. J. Hum. Genet. 2011, 19, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Kleinendorst, L.; Massink, M.P.G.; Cooiman, M.I.; Savas, M.; van der Baan-Slootweg, O.H.; Roelants, R.J.; Janssen, I.C.M.; Meijers-Heijboer, H.J.; Knoers, N.; Ploos van Amstel, H.K.; et al. Genetic obesity: Next-generation sequencing results of 1230 patients with obesity. J. Med. Genet. 2018, 55, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Tavtigian, S.V.; Deffenbaugh, A.M.; Yin, L.; Judkins, T.; Scholl, T.; Samollow, P.B.; de Silva, D.; Zharkikh, A.; Thomas, A. Comprehensive statistical study of 452 brca1 missense substitutions with classification of eight recurrent substitutions as neutral. J. Med. Genet. 2006, 43, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. Mutationtaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. Sift missense predictions for genomes. Nat. Protoc. 2016, 11, 1–9. [Google Scholar] [CrossRef]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. Sift web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using polyphen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7.20.1–7.20.41. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Van Woerden, G.M.; Bos, M.; de Konink, C.; Distel, B.; Avagliano Trezza, R.; Shur, N.E.; Barañano, K.; Mahida, S.; Chassevent, A.; Schreiber, A.; et al. Taok1 is associated with neurodevelopmental disorder and essential for neuronal maturation and cortical development. Hum. Mutat. 2021, 42, 445–459. [Google Scholar] [CrossRef]
- Heimer, G.; van Woerden, G.M.; Barel, O.; Marek-Yagel, D.; Kol, N.; Munting, J.B.; Borghei, M.; Atawneh, O.M.; Nissenkorn, A.; Rechavi, G.; et al. Netrin-g2 dysfunction causes a rett-like phenotype with areflexia. Hum. Mutat. 2020, 41, 476–486. [Google Scholar] [CrossRef]
- Reijnders, M.R.F.; Kousi, M.; van Woerden, G.M.; Klein, M.; Bralten, J.; Mancini, G.M.S.; van Essen, T.; Proietti-Onori, M.; Smeets, E.E.J.; van Gastel, M.; et al. Variation in a range of mtor-related genes associates with intracranial volume and intellectual disability. Nat. Commun. 2017, 8, 1052. [Google Scholar] [CrossRef]
- Küry, S.; van Woerden, G.M.; Besnard, T.; Proietti Onori, M.; Latypova, X.; Towne, M.C.; Cho, M.T.; Prescott, T.E.; Ploeg, M.A.; Sanders, S.; et al. De novo mutations in protein kinase genes camk2a and camk2b cause intellectual disability. Am. J. Hum. Genet. 2017, 101, 768–788. [Google Scholar] [CrossRef]
- Proietti Onori, M.; Koopal, B.; Everman, D.B.; Worthington, J.D.; Jones, J.R.; Ploeg, M.A.; Mientjes, E.; van Bon, B.W.; Kleefstra, T.; Schulman, H.; et al. The intellectual disability-associated camk2g p.Arg292pro mutation acts as a pathogenic gain-of-function. Hum. Mutat. 2018, 39, 2008–2024. [Google Scholar] [CrossRef]
- Goslin, K.; Banker, G. Culturing Nerve Cells; MIT Press: Cambridge, MA, USA, 1991. [Google Scholar]
- Fredriks, A.M.; van Buuren, S.; Burgmeijer, R.J.F.; Meulmeester, J.F.; Beuker, R.J.; Brugman, E.; Roede, M.J.; Verloove-Vanhorick, S.P.; Wit, J.-M. Continuing positive secular growth change in the netherlands 1955–1997. Pediatr. Res. 2000, 47, 316–323. [Google Scholar] [CrossRef]
- Corneli, G.; Di Somma, C.; Baldelli, R.; Rovere, S.; Gasco, V.; Croce, C.G.; Grottoli, S.; Maccario, M.; Colao, A.; Lombardi, G.; et al. The cut-off limits of the gh response to gh-releasing hormone-arginine test related to body mass index. Eur. J. Endocrinol. 2005, 153, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Ruehland, W.R.; Rochford, P.D.; O’Donoghue, F.J.; Pierce, R.J.; Singh, P.; Thornton, A.T. The new aasm criteria for scoring hypopneas: Impact on the apnea hypopnea index. Sleep 2009, 32, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Kelberman, D.; Dattani, M.T. Genetics of septo-optic dysplasia. Pituitary 2007, 10, 393–407. [Google Scholar] [CrossRef]
- Fintini, D.; Pedicelli, S.; Bocchini, S.; Bizzarri, C.; Grugni, G.; Cappa, M.; Crinò, A. 25oh vitamin d levels in pediatric patients affected by prader–willi syndrome. J. Endocrinol. Investig. 2018, 41, 739–742. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, L.; Joensson, I.M.; Froekjaer, J.B.; Krogh, K.; Farholt, S. A descriptive study of colorectal function in adults with prader-willi syndrome: High prevalence of constipation. BMC Gastroenterol. 2014, 14, 63. [Google Scholar] [CrossRef] [PubMed]
- Sinnema, M.; Maaskant, M.A.; van Schrojenstein Lantman-de Valk, H.M.; van Nieuwpoort, I.C.; Drent, M.L.; Curfs, L.M.; Schrander-Stumpel, C.T. Physical health problems in adults with prader-willi syndrome. Am. J. Med. Genet. A 2011, 155A, 2112–2124. [Google Scholar] [CrossRef]
- Auton, A.; Abecasis, G.R.; Altshuler, D.M.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.J.; Lapcevic, R.; Romero, A.E.; Yoon, M.; Das, J.; Beltrán, J.F.; Mort, M.; Stenson, P.D.; Cooper, D.N.; Paccanaro, A.; et al. Mutation3d: Cancer gene prediction through atomic clustering of coding variants in the structural proteome. Hum. Mutat. 2016, 37, 447–456. [Google Scholar] [CrossRef]
- Rossi, E.; Giorda, R.; Bonaglia, M.C.; Di Candia, S.; Grechi, E.; Franzese, A.; Soli, F.; Rivieri, F.; Patricelli, M.G.; Saccilotto, D.; et al. De novo unbalanced translocations in Prader-Willi and angelman syndrome might be the reciprocal product of inv dup(15)s. PLoS ONE 2012, 7, e39180. [Google Scholar] [CrossRef] [PubMed]
- Gunay-Aygun, M.; Schwartz, S.; Heeger, S.; O’Riordan, M.A.; Cassidy, S.B. The changing purpose of prader-willi syndrome clinical diagnostic criteria and proposed revised criteria. Pediatrics 2001, 108, E92. [Google Scholar] [CrossRef] [PubMed]
- Greger, V.; Woolf, E.; Lalande, M. Cloning of the breakpoints of a submicroscopic deletion in an angelman syndrome patient. Hum. Mol. Genet. 1993, 2, 921–924. [Google Scholar] [CrossRef]
- Hamabe, J.I.; Kuroki, Y.; Imaizumi, K.; Sugimoto, T.; Fukushima, Y.; Yamaguchi, A.; Izumikawa, Y.; Niikawa, N. DNA deletion and its parental origin in angelman syndrome patients. Am. J. Med. Genet. 1991, 41, 64–68. [Google Scholar] [CrossRef]
- Schulze, A.; Hansen, C.; Skakkebaek, N.E.; Brondum-Nielsen, K.; Ledbeter, D.H.; Tommerup, N. Exclusion of snrpn as a major determinant of prader-willi syndrome by a translocation breakpoint. Nat. Genet. 1996, 12, 452–454. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Nicholls, R.D.; Butler, M.G.; Saitoh, S.; Hainline, B.E.; Palmer, C.G. Breakage in the snrpn locus in a balanced 46,xy,t(15;19) prader-willi syndrome patient. Hum. Mol. Genet. 1996, 5, 517–524. [Google Scholar] [CrossRef]
- Conroy, J.M.; Grebe, T.A.; Becker, L.A.; Tsuchiya, K.; Nicholls, R.D.; Buiting, K.; Horsthemke, B.; Cassidy, S.B.; Schwartz, S. Balanced translocation 46,xy,t(2;15)(q37.2;q11.2) associated with atypical prader-willi syndrome. Am. J. Hum. Genet. 1997, 61, 388–394. [Google Scholar] [CrossRef][Green Version]
- Kuslich, C.D.; Kobori, J.A.; Mohapatra, G.; Gregorio-King, C.; Donlon, T.A. Prader-Willi syndrome is caused by disruption of the snrpn gene. Am. J. Hum. Genet. 1999, 64, 70–76. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wirth, J.; Back, E.; Hüttenhofer, A.; Nothwang, H.G.; Lich, C.; Groß, S.; Menzel, C.; Schinzel, A.; Kioschis, P.; Tommerup, N.; et al. A translocation breakpoint cluster disrupts the newly defined 3′ end of the snurf-snrpn transcription unit on chromosome 15. Hum. Mol. Genet. 2001, 10, 201–210. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Maina, E.N.; Webb, T.; Soni, S.; Whittington, J.; Boer, H.; Clarke, D.; Holland, A. Analysis of candidate imprinted genes in pws subjects with atypical genetics: A possible inactivating mutation in the snurf/snrpn minimal promoter. J. Hum. Genet. 2007, 52, 297–307. [Google Scholar] [CrossRef]
- Whittington, J.; Holland, A.; Webb, T.; Butler, J.; Clarke, D.; Boer, H. Relationship between clinical and genetic diagnosis of prader-willi syndrome. J. Med. Genet. 2002, 39, 926–932. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bürger, J.; Horn, D.; Tönnies, H.; Neitzel, H.; Reis, A. Familial interstitial 570 kbp deletion of the ube3a gene region causing angelman syndrome but not prader-willi syndrome. Am. J. Med. Genet. 2002, 111, 233–237. [Google Scholar] [CrossRef]
- Runte, M.; Varon, R.; Horn, D.; Horsthemke, B.; Buiting, K. Exclusion of the c/d box snorna gene cluster hbii-52 from a major role in prader-willi syndrome. Hum. Genet. 2005, 116, 228–230. [Google Scholar] [CrossRef]
- Schüle, B.; Albalwi, M.; Northrop, E.; Francis, D.I.; Rowell, M.; Slater, H.R.; Gardner, R.J.; Francke, U. Molecular breakpoint cloning and gene expression studies of a novel translocation t(4;15)(q27;q11.2) associated with prader-willi syndrome. BMC Med. Genet. 2005, 6, 18. [Google Scholar] [CrossRef]
- Calounova, G.; Hedvicakova, P.; Silhanova, E.; Kreckova, G.; Sedlacek, Z. Molecular and clinical characterization of two patients with prader-willi syndrome and atypical deletions of proximal chromosome 15q. Am. J. Med. Genet. Part A 2008, 146, 1955–1962. [Google Scholar] [CrossRef]
- Kanber, D.; Giltay, J.; Wieczorek, D.; Zogel, C.; Hochstenbach, R.; Caliebe, A.; Kuechler, A.; Horsthemke, B.; Buiting, K. A paternal deletion of mkrn3, magel2 and ndn does not result in prader-willi syndrome. Eur. J. Hum. Genet. 2009, 17, 582–590. [Google Scholar] [CrossRef]
- Naik, S.; Thomas, N.S.; Davies, J.H.; Lever, M.; Raponi, M.; Baralle, D.; Temple, I.K.; Caliebe, A. Novel tandem duplication in exon 1 of the snurf/snrpn gene in a child with transient excessive eating behaviour and weight gain. Mol. Syndr. 2011, 2, 76–80. [Google Scholar] [CrossRef]
- Rivera, H.; Zuffardi, O.; Gargantini, L. Nonreciprocal and jumping translocations of 15q1----qter in prader-willi syndrome. Am J. Med. Genet. 1990, 37, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Fraccaro, M.; Zuffardi, O.; Buhler, E. Deficiency, transposition, and duplication of one 15q region may be alternatively associated with prader-willi (or a similar) syndrome. Analysis of seven cases after varying ascertainment. Hum. Genet. 1983, 64, 388–394. [Google Scholar] [CrossRef]
- Buiting, K.; Di Donato, N.; Beygo, J.; Bens, S.; von der Hagen, M.; Hackmann, K.; Horsthemke, B. Clinical phenotypes of magel2 mutations and deletions. Orphanet J. Rare Dis. 2014, 9, 40. [Google Scholar] [CrossRef]
- Koufaris, C.; Alexandrou, A.; Papaevripidou, I.; Alexandrou, I.; Christophidou-Anastasiadou, V.; Sismani, C. Deletion of snurf/snrpn u1b and u1b* upstream exons in a child with developmental delay and excessive weight. J. Genet. 2016, 95, 621–624. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Mitsuhashi, S.; Miyake, N.; Ohta, T.; Liang, D.; Wu, L.; Matsumoto, N. Translocation breakpoint disrupting the host snhg14 gene but not coding genes or snornas in typical prader-willi syndrome. J. Hum. Genet. 2019, 64, 647–652. [Google Scholar] [CrossRef]
- Yamada, K.; Matsuzawa, H.; Uchiyama, M.; Kwee, I.L.; Nakada, T. Brain developmental abnormalities in prader-willi syndrome detected by diffusion tensor imaging. Pediatrics 2006, 118, e442–e448. [Google Scholar] [CrossRef] [PubMed]
- Azor, A.M.; Cole, J.H.; Holland, A.J.; Dumba, M.; Patel, M.C.; Sadlon, A.; Goldstone, A.P.; Manning, K.E. Increased brain age in adults with prader-willi syndrome. Neuroimage Clin. 2019, 21, 101664. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhang, Y.; von Deneen, K.M.; Zhu, H.; Gao, J.H. Brain structural alterations in obese children with and without prader-willi syndrome. Hum. Brain Mapp. 2017, 38, 4228–4238. [Google Scholar] [CrossRef]
- Lukoshe, A.; White, T.; Schmidt, M.N.; van der Lugt, A.; Hokken-Koelega, A.C. Divergent structural brain abnormalities between different genetic subtypes of children with prader-willi syndrome. J. Neurodev. Disord. 2013, 5, 31. [Google Scholar] [CrossRef]
- Miller, J.L.; Couch, J.A.; Schmalfuss, I.; He, G.; Liu, Y.; Driscoll, D.J. Intracranial abnormalities detected by three-dimensional magnetic resonance imaging in prader–willi syndrome. Am. J. Med. Genet. Part A 2007, 143A, 476–483. [Google Scholar] [CrossRef]
- Miller, S.P.; Shevell, M.I.; Patenaude, Y.; Poulin, C.; O’Gorman, A.M. Septo-optic dysplasia plus: A spectrum of malformations of cortical development. Neurology 2000, 54, 1701–1703. [Google Scholar] [CrossRef]
- Polizzi, A.; Pavone, P.; Iannetti, P.; Manfré, L.; Ruggieri, M. Septo-optic dysplasia complex: A heterogeneous malformation syndrome. Pediatr. Neurol. 2006, 34, 66–71. [Google Scholar] [CrossRef] [PubMed]
- McNay, D.E.; Turton, J.P.; Kelberman, D.; Woods, K.S.; Brauner, R.; Papadimitriou, A.; Keller, E.; Keller, A.; Haufs, N.; Krude, H.; et al. Hesx1 mutations are an uncommon cause of septooptic dysplasia and hypopituitarism. J. Clin. Endocrinol. Metab. 2007, 92, 691–697. [Google Scholar] [CrossRef] [PubMed]
- McCabe, M.J.; Alatzoglou, K.S.; Dattani, M.T. Septo-optic dysplasia and other midline defects: The role of transcription factors: Hesx1 and beyond. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Houtkoop, W. Basisvaardigheden in Nederland; Max Goote Kenniscentrum: Amsterdam, The Netherlands, 1999; pp. 1–196. [Google Scholar]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Gray, T.A.; Saitoh, S.; Nicholls, R.D. An imprinted, mammalian bicistronic transcript encodes two independent proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 5616–5621. [Google Scholar] [CrossRef]
- Glenn, C.C.; Saitoh, S.; Jong, M.T.; Filbrandt, M.M.; Surti, U.; Driscoll, D.J.; Nicholls, R.D. Gene structure, DNA methylation, and imprinted expression of the human snrpn gene. Am. J. Hum. Genet. 1996, 58, 335–346. [Google Scholar] [PubMed]
- Schmauss, C.; Brines, M.L.; Lerner, M.R. The gene encoding the small nuclear ribonucleoprotein-associated protein n is expressed at high levels in neurons. J. Biol. Chem. 1992, 267, 8521–8529. [Google Scholar] [CrossRef]
- Ozcelik, T.; Leff, S.; Robinson, W.; Donlon, T.; Lalande, M.; Sanjines, E.; Schinzel, A.; Francke, U. Small nuclear ribonucleoprotein polypeptide n (snrpn), an expressed gene in the prader-willi syndrome critical region. Nat. Genet. 1992, 2, 265–269. [Google Scholar] [CrossRef]
- Li, H.; Zhao, P.; Xu, Q.; Shan, S.; Hu, C.; Qiu, Z.; Xu, X. The autism-related gene snrpn regulates cortical and spine development via controlling nuclear receptor nr4a1. Sci. Rep. 2016, 6, 29878. [Google Scholar] [CrossRef]
- Runte, M.; Huttenhofer, A.; Gross, S.; Kiefmann, M.; Horsthemke, B.; Buiting, K. The ic-snurf-snrpn transcript serves as a host for multiple small nucleolar rna species and as an antisense rna for ube3a. Hum. Mol. Genet. 2001, 10, 2687–2700. [Google Scholar] [CrossRef]
- Gallagher, R.C.; Pils, B.; Albalwi, M.; Francke, U. Evidence for the role of pwcr1/hbii-85 c/d box small nucleolar rnas in prader-willi syndrome. Am. J. Hum. Genet. 2002, 71, 669–678. [Google Scholar] [CrossRef]
- Dittrich, B.; Buiting, K.; Korn, B.; Rickard, S.; Buxton, J.; Saitoh, S.; Nicholls, R.D.; Poustka, A.; Winterpacht, A.; Zabel, B.; et al. Imprint switching on human chromosome 15 may involve alternative transcripts of the snrpn gene. Nat. Genet. 1996, 14, 163–170. [Google Scholar] [CrossRef]
- Farber, C.; Dittrich, B.; Buiting, K.; Horsthemke, B. The chromosome 15 imprinting centre (ic) region has undergone multiple duplication events and contains an upstream exon of snrpn that is deleted in all angelman syndrome patients with an ic microdeletion. Hum. Mol. Genet. 1999, 8, 337–343. [Google Scholar] [CrossRef][Green Version]
- Buiting, K.; Saitoh, S.; Gross, S.; Dittrich, B.; Schwartz, S.; Nicholls, R.D.; Horsthemke, B. Inherited microdeletions in the angelman and prader-willi syndromes define an imprinting centre on human chromosome 15. Nat. Genet. 1995, 9, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Blaydes, S.M.; Elmore, M.; Yang, T.; Brannan, C.I. Analysis of murine snrpn and human snrpn gene imprinting in transgenic mice. Mamm. Genome 1999, 10, 549–555. [Google Scholar] [CrossRef]
- Horsthemke, B.; Buiting, K. Imprinting defects on human chromosome 15. Cytogenet. Genome Res. 2006, 113, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T.; Gray, T.A.; Rogan, P.K.; Buiting, K.; Gabriel, J.M.; Saitoh, S.; Muralidhar, B.; Bilienska, B.; Krajewska-Walasek, M.; Driscoll, D.J.; et al. Imprinting-mutation mechanisms in prader-willi syndrome. Am. J. Hum. Genet. 1999, 64, 397–413. [Google Scholar] [CrossRef]
- Rabinovitz, S.; Kaufman, Y.; Ludwig, G.; Razin, A.; Shemer, R. Mechanisms of activation of the paternally expressed genes by the prader-willi imprinting center in the prader-willi/angelman syndromes domains. Proc. Natl. Acad. Sci. USA 2012, 109, 7403–7408. [Google Scholar] [CrossRef] [PubMed]
- Perk, J.; Makedonski, K.; Lande, L.; Cedar, H.; Razin, A.; Shemer, R. The imprinting mechanism of the prader-willi/angelman regional control center. EMBO J. 2002, 21, 5807–5814. [Google Scholar] [CrossRef]
- Horsthemke, B.; Wagstaff, J. Mechanisms of imprinting of the prader-willi/angelman region. Am. J. Med. Genet. A 2008, 146A, 2041–2052. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe, J.S.; Nakao, M.; Christian, S.; Örstavik, K.H.; Tommerup, N.; Ledbetter, D.H.; Beaudet, A.L. Deletions of a differentially methylated cpg island at the snrpn gene define a putative imprinting control region. Nat. Genet. 1994, 8, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, R.D.; Saitoh, S.; Horsthemke, B. Imprinting in prader-willi and angelman syndromes. Trends Genet. 1998, 14, 194–200. [Google Scholar] [CrossRef]
- McCarthy, J.; Lupo, P.J.; Kovar, E.; Rech, M.; Bostwick, B.; Scott, D.; Kraft, K.; Roscioli, T.; Charrow, J.; Schrier Vergano, S.A.; et al. Schaaf-yang syndrome overview: Report of 78 individuals. Am. J. Med. Genet. A 2018, 176, 2564–2574. [Google Scholar] [CrossRef] [PubMed]
- Kleinendorst, L.; Pi Castán, G.; Caro-Llopis, A.; Boon, E.M.J.; van Haelst, M.M. The role of obesity in the fatal outcome of schaaf-yang syndrome: Early onset morbid obesity in a patient with a magel2 mutation. Am. J. Med. Genet. A 2018, 176, 2456–2459. [Google Scholar] [CrossRef]
- Shen, M.; Eyras, E.; Wu, J.; Khanna, A.; Josiah, S.; Rederstorff, M.; Zhang, M.Q.; Stamm, S. Direct cloning of double-stranded rnas from rnase protection analysis reveals processing patterns of c/d box snornas and provides evidence for widespread antisense transcript expression. Nucleic Acids Res. 2011, 39, 9720–9730. [Google Scholar] [CrossRef] [PubMed]
- Wevrick, R.; Francke, U. An imprinted mouse transcript homologous to the human imprinted in prader-willi syndrome (ipw) gene. Hum. Mol. Genet. 1997, 6, 325–332. [Google Scholar] [CrossRef]
- Wevrick, R.; Kerns, J.A.; Francke, U. Identification of a novel paternally expressed gene in the prader-willi syndrome region. Hum. Mol. Genet. 1994, 3, 1877–1882. [Google Scholar] [CrossRef]
- Adhikari, A.; Copping, N.A.; Onaga, B.; Pride, M.C.; Coulson, R.L.; Yang, M.; Yasui, D.H.; LaSalle, J.M.; Silverman, J.L. Cognitive deficits in the snord116 deletion mouse model for prader-willi syndrome. Neurobiol. Learn. Mem. 2019, 165, 106874. [Google Scholar] [CrossRef]
- Ding, F.; Li, H.H.; Zhang, S.; Solomon, N.M.; Camper, S.A.; Cohen, P.; Francke, U. Snorna snord116 (pwcr1/mbii-85) deletion causes growth deficiency and hyperphagia in mice. PLoS ONE 2008, 3, e1709. [Google Scholar] [CrossRef]
- Tsai, T.F.; Jiang, Y.H.; Bressler, J.; Armstrong, D.; Beaudet, A.L. Paternal deletion from snrpn to ube3a in the mouse causes hypotonia, growth retardation and partial lethality and provides evidence for a gene contributing to prader-willi syndrome. Hum. Mol. Genet. 1999, 8, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Adamson, T.E.; Resnick, J.L.; Leff, S.; Wevrick, R.; Francke, U.; Jenkins, N.A.; Copeland, N.G.; Brannan, C.I. A mouse model for prader-willi syndrome imprinting-centre mutations. Nat. Genet. 1998, 19, 25–31. [Google Scholar] [CrossRef]
- Rieusset, A.; Schaller, F.; Unmehopa, U.; Matarazzo, V.; Watrin, F.; Linke, M.; Georges, B.; Bischof, J.; Dijkstra, F.; Bloemsma, M.; et al. Stochastic loss of silencing of the imprinted ndn/ndn allele, in a mouse model and humans with prader-willi syndrome, has functional consequences. PLoS Genet. 2013, 9, e1003752. [Google Scholar] [CrossRef]
- Chamberlain, S.J.; Johnstone, K.A.; DuBose, A.J.; Simon, T.A.; Bartolomei, M.S.; Resnick, J.L.; Brannan, C.I. Evidence for genetic modifiers of postnatal lethality in pws-ic deletion mice. Hum. Mol. Genet. 2004, 13, 2971–2977. [Google Scholar] [CrossRef] [PubMed]
- Iourov, I.Y.; Vorsanova, S.G.; Korostelev, S.A.; Zelenova, M.A.; Yurov, Y.B. Long contiguous stretches of homozygosity spanning shortly the imprinted loci are associated with intellectual disability, autism and/or epilepsy. Mol. Cytogenet. 2015, 8, 77. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gray, T.A.; Smithwick, M.J.; Schaldach, M.A.; Martone, D.L.; Graves, J.A.; McCarrey, J.R.; Nicholls, R.D. Concerted regulation and molecular evolution of the duplicated snrpb’/b and snrpn loci. Nucleic Acids Res. 1999, 27, 4577–4584. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).