
The Multiple Facets of Iron Recycling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
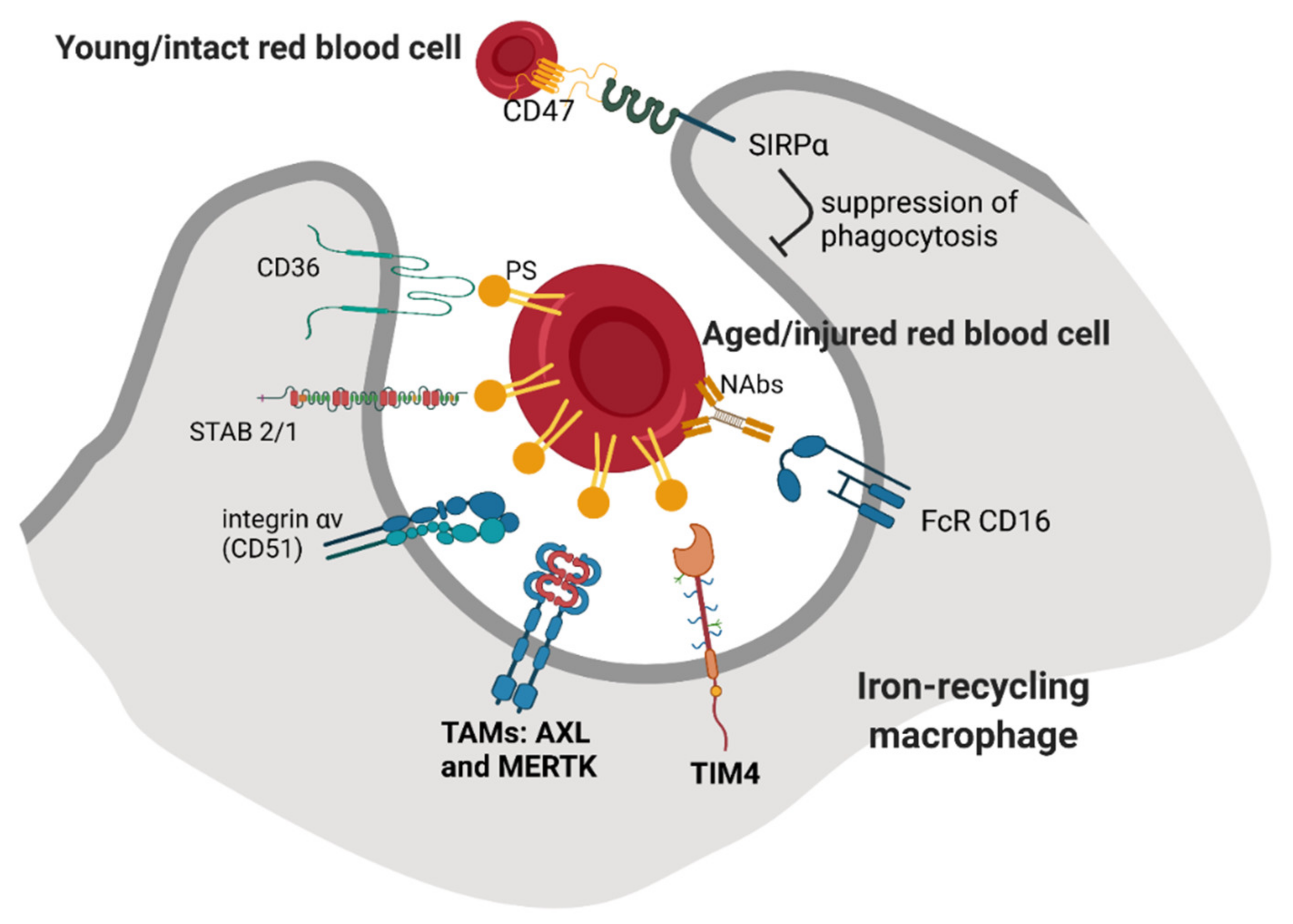
2. Recognition of Aged or Damaged RBCs by Iron-Recycling Macrophages
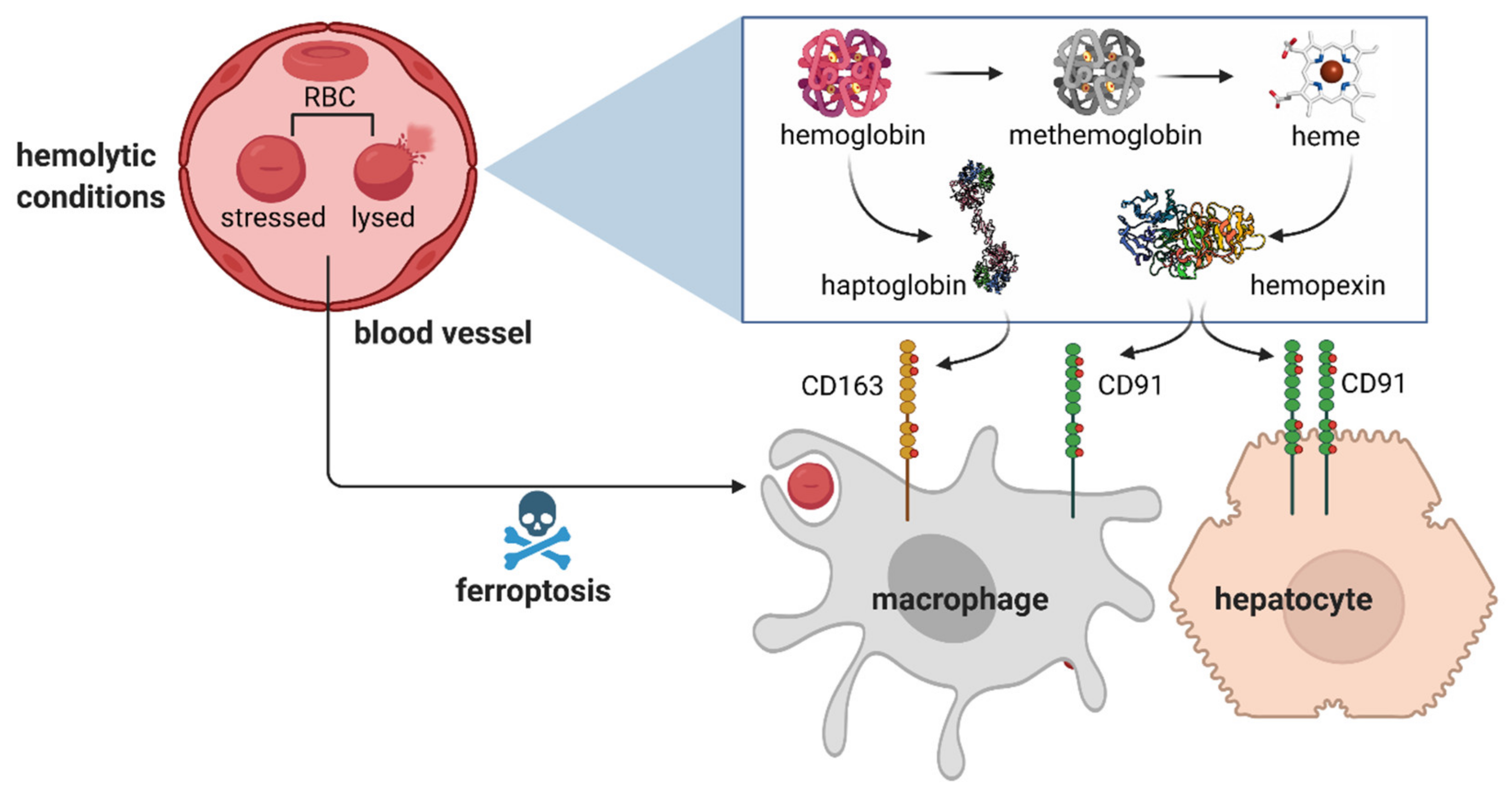
3. Sequestration of Hemolytic Erythrocyte Components
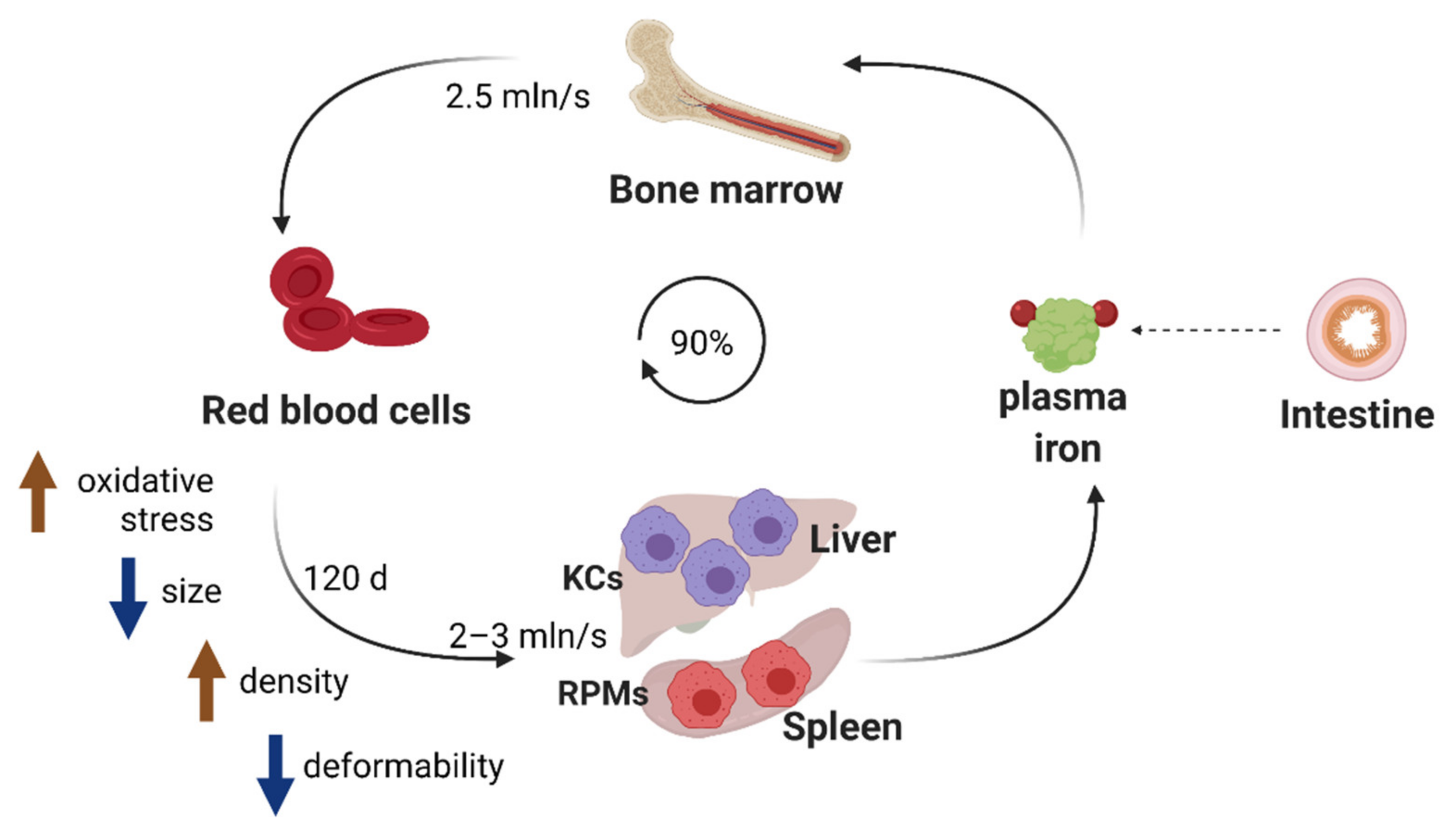
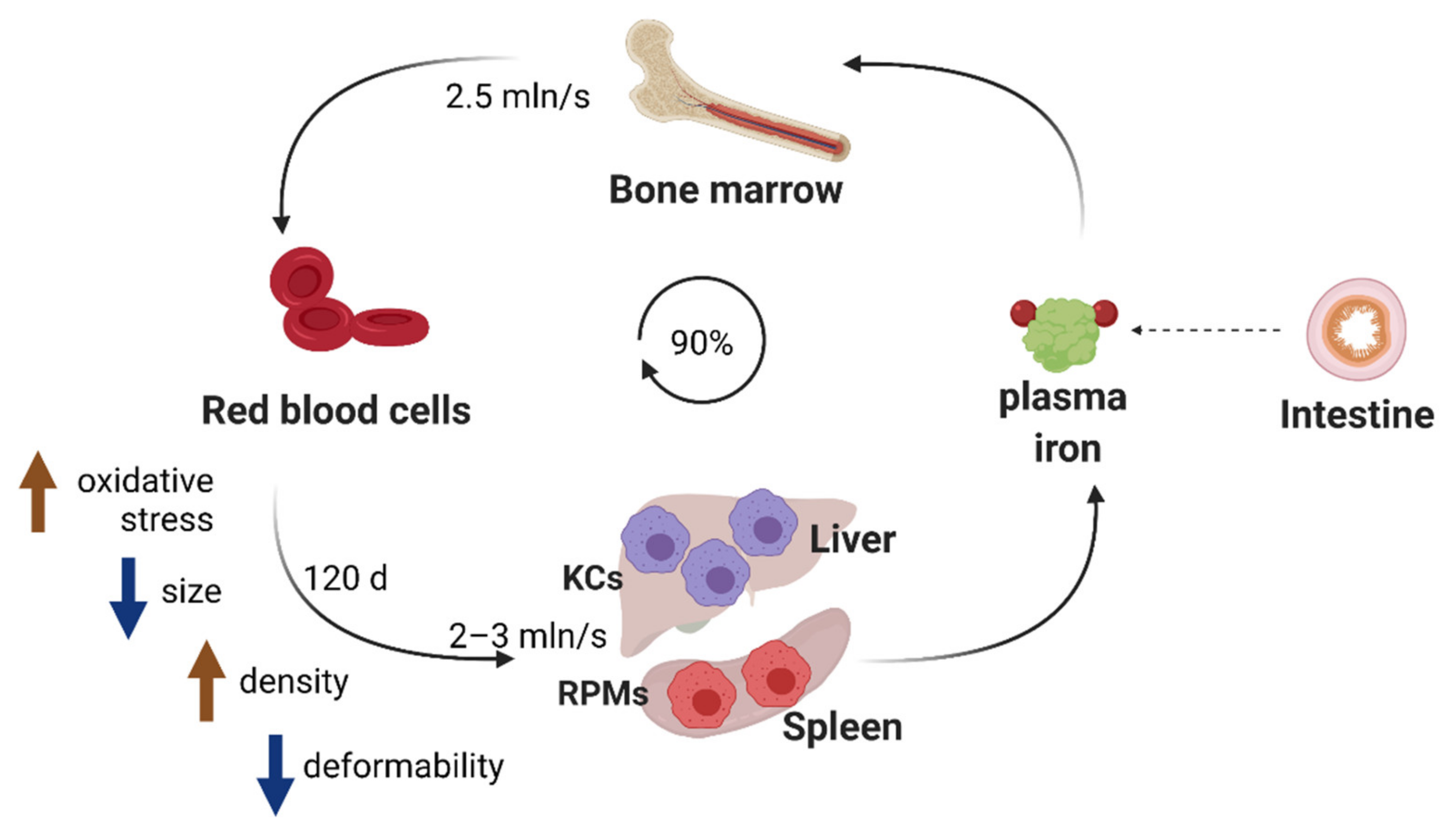
4. Hemolysis-Driven Iron Recycling Model
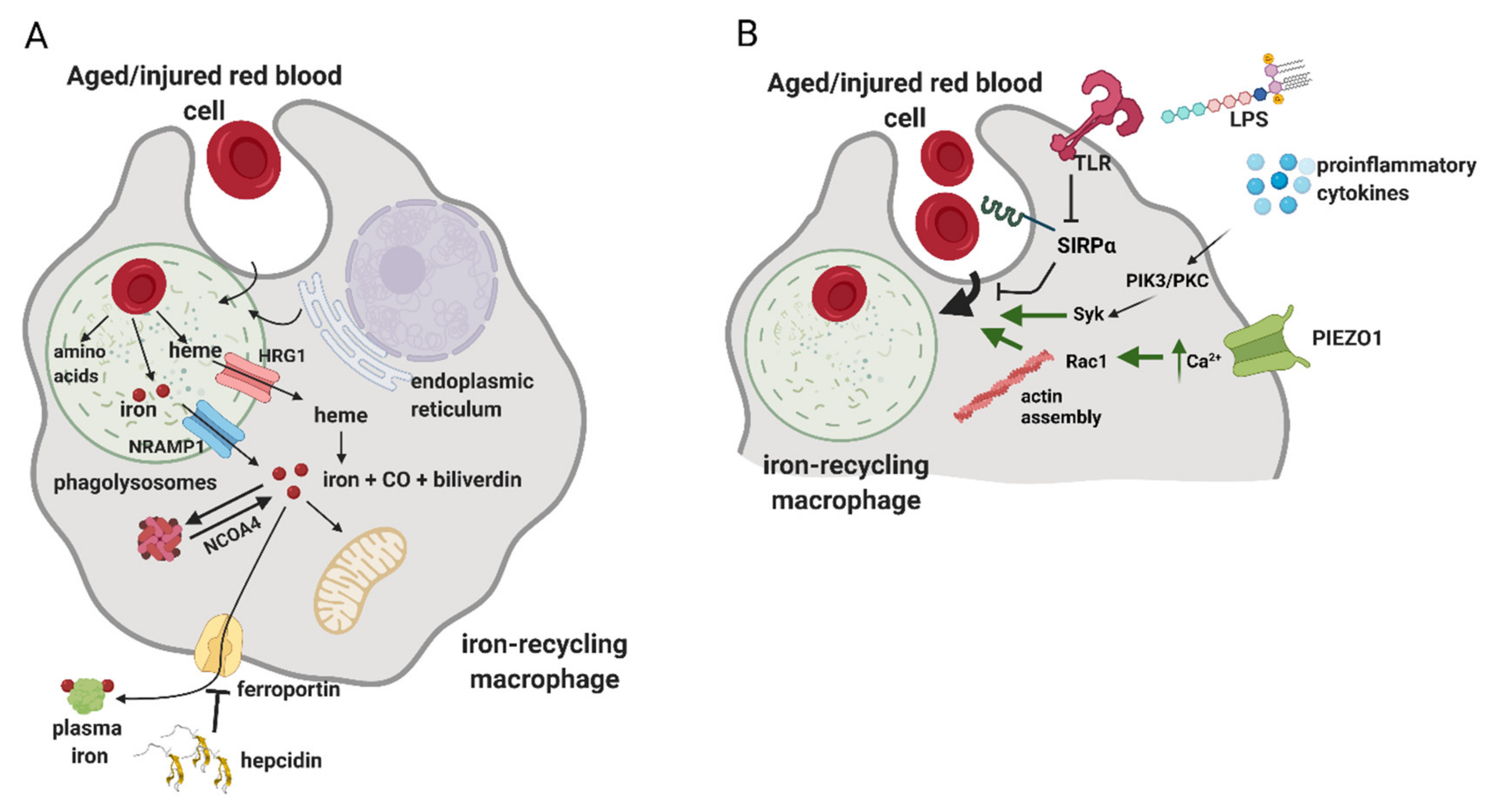
5. Erythrophagocytosis and Intracellular Iron Handling in Iron-Recycling Cells
6. Regulation of the Erythrophagocytosis Rate
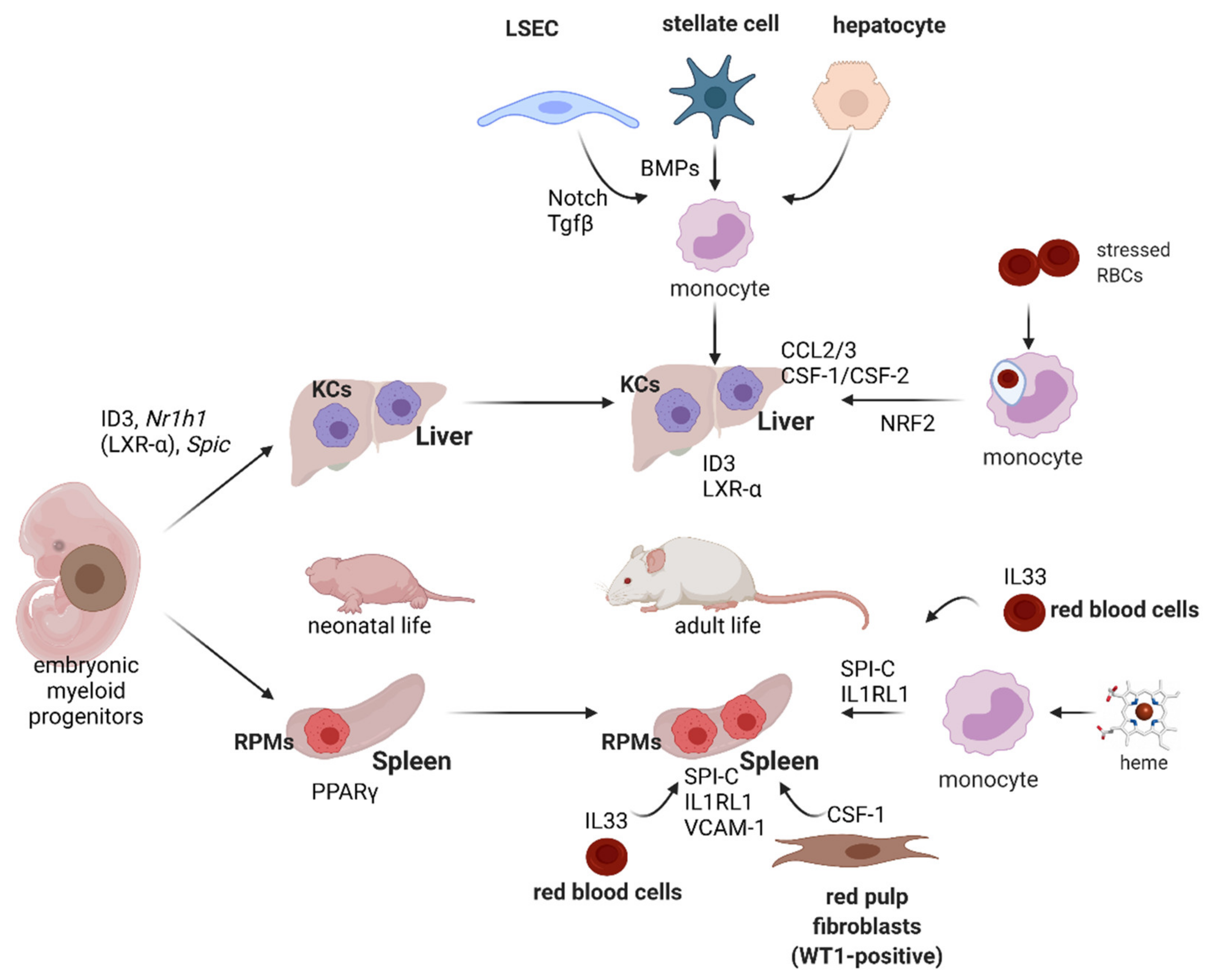
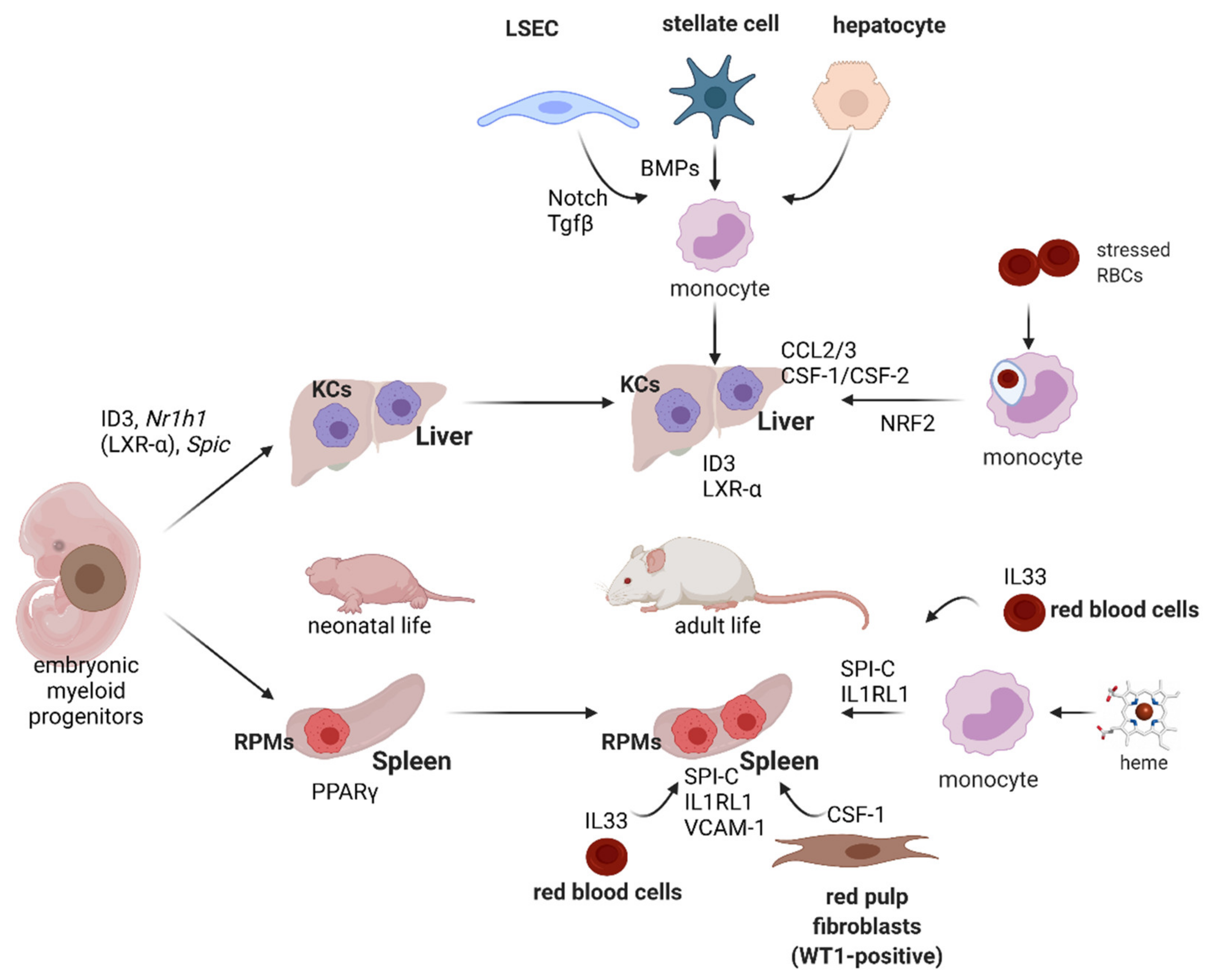
7. Development and Plasticity of Iron-Recycling Macrophages
8. Cross-Talk between Iron Recycling and Macrophage Immune Functions
9. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Palis, J. Primitive and definitive erythropoiesis in mammals. Front. Physiol. 2014, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-J. Regulation of protein synthesis by the heme-regulated eIF2α kinase: Relevance to anemias. Blood 2006, 109, 2693–2699. [Google Scholar] [CrossRef]
- White, C.; Yuan, X.; Schmidt, P.J.; Bresciani, E.; Samuel, T.K.; Campagna, D.; Hall, C.; Bishop, K.; Calicchio, M.L.; Lapierre, A.; et al. HRG1 Is Essential for Heme Transport from the Phagolysosome of Macrophages during Erythrophagocytosis. Cell Metab. 2013, 17, 261–270. [Google Scholar] [CrossRef] [Green Version]
- Moras, M.; Lefevre, S.D.; Ostuni, M.A. From Erythroblasts to Mature Red Blood Cells: Organelle Clearance in Mammals. Front. Physiol. 2017, 8, 1076. [Google Scholar] [CrossRef]
- Bratosin, D.; Mazurier, J.; Tissier, J.; Estaquier, J.; Huart, J.; Ameisen, J.; Aminoff, D.; Montreuil, J. Cellular and molecular mechanisms of senescent erythrocyte phagocytosis by macrophages. A review. Biochimie 1998, 80, 173–195. [Google Scholar] [CrossRef]
- Lutz, H.U. Advances in Experimental Medicine and Biology. In Naturally Occurring Antibodies (NAbs); Springer: Berlin/Heidelberg, Germany, 2012; Volume 750, pp. 0065–2598. [Google Scholar]
- Tyan, M.L. Age-related increase in erythrocyte oxidant sensitivity. Mech. Ageing Dev. 1982, 20, 25–32. [Google Scholar] [CrossRef]
- Zhang, D.-L.; Wu, J.; Shah, B.N.; Greutélaers, K.C.; Ghosh, M.C.; Ollivierre, H.; Su, X.-Z.; Thuma, P.E.; Bedu-Addo, G.; Mockenhaupt, F.P.; et al. Erythrocytic ferroportin reduces intracellular iron accumulation, hemolysis, and malaria risk. Science 2018, 359, 1520–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, R.S. Measurement of Red Cell Lifespan and Aging. Transfus. Med. Hemotherapy 2012, 39, 302–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dholakia, U.; Bandyopadhyay, S.; Hod, E.A.; Prestia, K. Determination of RBC Survival in C57BL/6 and C57BL/6-Tg(UBC–GFP) Mice. Comp. Med. 2015, 65, 196–201. [Google Scholar]
- Muckenthaler, M.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [Green Version]
- Ganz, T. Macrophages and Systemic Iron Homeostasis. J. Innate Immun. 2012, 4, 446–453. [Google Scholar] [CrossRef]
- Higgins, J.M. Red Blood Cell Population Dynamics. Clin. Lab. Med. 2014, 35, 43–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knutson, M.; Wessling-Resnick, M. Iron Metabolism in the Reticuloendothelial System. Crit. Rev. Biochem. Mol. Biol. 2003, 38, 61–88. [Google Scholar] [CrossRef]
- Lasch, J.; Küllertz, G.; Opalka, J.R. Separation of Erythrocytes into Age-Related Fractions by Density or Size? Counterflow Centrifugation. Clin. Chem. Lab. Med. 2000, 38, 629–632. [Google Scholar] [CrossRef]
- Kumar, M.; Coria, A.L.; Cornick, S.; Petri, B.; Mayengbam, S.; Jijon, H.B.; Moreau, F.; Shearer, J.; Chadee, K. Increased intestinal permeability exacerbates sepsis through reduced hepatic SCD-1 activity and dysregulated iron recycling. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Huisjes, R.; Bogdanova, A.; Van Solinge, W.; Schiffelers, R.; Kaestner, L.; Van Wijk, R. Squeezing for Life—Properties of Red Blood Cell Deformability. Front. Physiol. 2018, 9, 656. [Google Scholar] [CrossRef] [PubMed]
- Arashiki, N.; Kimata, N.; Manno, S.; Mohandas, N.; Takakuwa, Y. Membrane Peroxidation and Methemoglobin Formation Are Both Necessary for Band 3 Clustering: Mechanistic Insights into Human Erythrocyte Senescence. Biochemistry 2013, 52, 5760–5769. [Google Scholar] [CrossRef] [Green Version]
- D’Alessandro, A.; Blasi, B. Red blood cell subpopulations in freshly drawn blood: Application of proteomics and metabolomics to a decades-long biological issue. Blood Transfus. 2013, 11, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Joiner, C.H.; Lauf, P.K. Ouabain Binding and Potassium Transport in Young and Old Populations of Human Red Cells. Membr. Biochem. 1978, 1, 187–202. [Google Scholar] [CrossRef] [PubMed]
- Lew, V.L.; Tiffert, T. On the Mechanism of Human Red Blood Cell Longevity: Roles of Calcium, the Sodium Pump, PIEZO1, and Gardos Channels. Front. Physiol. 2017, 8, 977. [Google Scholar] [CrossRef] [Green Version]
- Andolfo, I.; Alper, S.L.; De Franceschi, L.; Auriemma, C.; Russo, R.; De Falco, L.; Vallefuoco, F.; Esposito, M.R.; Vandorpe, D.H.; Shmukler, B.E.; et al. Multiple clinical forms of dehydrated hereditary stomatocytosis arise from mutations in PIEZO1. Blood 2013, 121, 3925–3935. [Google Scholar] [CrossRef] [Green Version]
- Romero, P.J.; Romero, E.A. The Role of Calcium Metabolism in Human Red Blood Cell Ageing: A Proposal. Blood Cells Mol. Dis. 1999, 25, 9–19. [Google Scholar] [CrossRef]
- Willekens, F.L.A.; Roerdinkholder-Stoelwinder, B.; Groenen-Döpp, Y.A.M.; Bos, H.J.; Bosman, G.J.C.G.M.; Bos, A.G.V.D.; Verkleij, A.J.; Werre, J.M. Hemoglobin loss from erythrocytes in vivo results from spleen-facilitated vesiculation. Blood 2003, 101, 747–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duez, J.; Holleran, J.; Ndour, P.A.; Pionneau, C.; Diakite, S.A.S.; Roussel, C.; Dussiot, M.; Amireault, P.; Avery, V.; Buffet, P. Mechanical clearance of red blood cells by the human spleen: Potential therapeutic applications of a biomimetic RBC filtration method. Transfus. Clin. Biol. 2015, 22, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Klei, T.R.L.; Meinderts, S.M.; Berg, T.K.V.D.; Van Bruggen, R. From the Cradle to the Grave: The Role of Macrophages in Erythropoiesis and Erythrophagocytosis. Front. Immunol. 2017, 8, 73. [Google Scholar] [CrossRef] [Green Version]
- Theurl, I.; Hilgendorf, I.; Nairz, M.; Tymoszuk, P.; Haschka, D.; Asshoff, M.; He, S.; Gerhardt, L.M.S.; Holderried, T.; Seifert, M.; et al. On-demand erythrocyte disposal and iron recycling requires transient macrophages in the liver. Nat. Med. 2016, 22, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Mebius, R.E.; Kraal, G. Structure and function of the spleen. Nat. Rev. Immunol. 2005, 5, 606–616. [Google Scholar] [CrossRef]
- Lee, S.-J.; Park, S.-Y.; Jung, M.-Y.; Bae, S.M.; Kim, I.-S. Mechanism for phosphatidylserine-dependent erythrophagocytosis in mouse liver. Blood 2011, 117, 5215–5223. [Google Scholar] [CrossRef] [Green Version]
- Bonnardel, J.; T’Jonck, W.; Gaublomme, D.; Browaeys, R.; Scott, C.L.; Martens, L.; Vanneste, B.; De Prijck, S.; Nedospasov, S.A.; Kremer, A.; et al. Stellate Cells, Hepatocytes, and Endothelial Cells Imprint the Kupffer Cell Identity on Monocytes Colonizing the Liver Macrophage Niche. Immunity 2019, 51, 638–654.e9. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-H.; Starkey, P.M.; Gordon, S. Quantitative analysis of total macrophage content in adult mouse tissues. Immunochemical studies with monoclonal antibody F4/80. J. Exp. Med. 1985, 161, 475–489. [Google Scholar] [CrossRef] [Green Version]
- Pfefferlé, M.; Ingoglia, G.; Schaer, C.A.; Yalamanoglu, A.; Buzzi, R.M.; Dubach, I.L.; Tan, G.; López-Cano, E.Y.; Schulthess, N.; Hansen, K.; et al. Hemolysis transforms liver macrophages into anti-inflammatory erythrophagocytes. J. Clin. Investig. 2020, 130. [Google Scholar] [CrossRef]
- Youssef, L.A.; Rebbaa, A.; Pampou, S.; Weisberg, S.; Stockwell, B.R.; Hod, E.A.; Spitalnik, S.L. Increased erythrophagocytosis induces ferroptosis in red pulp macrophages in a mouse model of transfusion. Blood 2018, 131, 2581–2593. [Google Scholar] [CrossRef]
- Bian, Z.; Shi, L.; Guo, Y.L.; Lv, Z.; Tang, C.; Niu, S.; Tremblay, A.; Venkataramani, M.; Culpepper, C.; Li, L.; et al. Cd47-Sirpalpha interaction and IL-10 constrain inflammation-induced macrophage phagocytosis of healthy self-cells. Proc. Natl. Acad. Sci. USA 2016, 37, E5434–E5443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Looareesuwan, S.; Ho, M.; Wattanagoon, Y.; White, N.J.; Warrell, D.A.; Bunnag, D.; Harinasuta, T.; Wyler, D.J. Dynamic Alteration in Splenic Function during Acute falciparum Malaria. N. Engl. J. Med. 1987, 317, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Schroit, A.J.; Madsen, J.W.; Tanaka, Y. In vivo recognition and clearance of red blood cells containing phosphatidylserine in their plasma membranes. J. Biol. Chem. 1985, 260. [Google Scholar] [CrossRef]
- Connor, J.; Pak, C.C.; Schroit, A.J. Exposure of phosphatidylserine in the outer leaflet of human red blood cells. Relationship to cell density, cell age, and clearance by mononuclear cells. J. Biol. Chem. 1994, 269, 2399–2404. [Google Scholar] [CrossRef]
- Gottlieb, Y.; Topaz, O.; Cohen, L.A.; Yakov, L.D.; Haber, T.; Morgenstern, A.; Weiss, A.; Berman, K.C.; Fibach, E.; Meyron-Holtz, E.G. Physiologically aged red blood cells undergo erythrophagocytosis in vivo but not in vitro. Haematologica 2012, 97, 994–1002. [Google Scholar] [CrossRef] [Green Version]
- Khandelwal, S.; Saxena, R.K. A role of phosphatidylserine externalization in clearance of erythrocytes exposed to stress but not in eliminating aging populations of erythrocyte in mice. Exp. Gerontol. 2008, 43, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Kuypers, F.A.; Yuan, J.; Lewis, R.A.; Snyder, L.M.; Kiefer, C.R.; Bunyaratvej, A.; Fucharoen, S.; Ma, L.; Styles, L.; De Jong, K.; et al. Membrane phospholipid asymmetry in human thalassemia. Blood 1998, 91, 3044–3051. [Google Scholar] [CrossRef] [PubMed]
- Wood, B.L.; Gibson, D.F.; Tait, J.F. Increased erythrocyte phosphatidylserine exposure in sickle cell disease: Flow-cytometric measurement and clinical associations. Blood 1996, 88, 1873–1880. [Google Scholar] [CrossRef] [Green Version]
- de Jong, K.; Geldwerth, D.; Kuypers, F.A. Oxidative damage does not alter membrane phospholipid asymmetry in human erythrocytes. Biochemistry 1997, 36, 6768–6776. [Google Scholar] [CrossRef]
- Sakai, M.; Troutman, T.D.; Seidman, J.S.; Ouyang, Z.; Spann, N.J.; Abe, Y.; Ego, K.M.; Bruni, C.M.; Deng, Z.; Schlachetzki, J.C.; et al. Liver-Derived Signals Sequentially Reprogram Myeloid Enhancers to Initiate and Maintain Kupffer Cell Identity. Immunity 2019, 51, 655–670.e8. [Google Scholar] [CrossRef]
- Akilesh, H.M.; Buechler, M.B.; Duggan, J.M.; Hahn, W.O.; Matta, B.; Sun, X.; Gessay, G.; Whalen, E.; Mason, M.; Presnell, S.R.; et al. Chronic TLR7 and TLR9 signaling drives anemia via differentiation of specialized hemophagocytes. Science 2019, 363, eaao5213. [Google Scholar] [CrossRef]
- Oh, M.-H.; Collins, S.L.; Sun, I.-H.; Tam, A.J.; Patel, C.H.; Arwood, M.L.; Chan-Li, Y.; Powell, J.D.; Horton, M.R. mTORC2 Signaling Selectively Regulates the Generation and Function of Tissue-Resident Peritoneal Macrophages. Cell Rep. 2017, 20, 2439–2454. [Google Scholar] [CrossRef] [Green Version]
- Zagórska, A.; Través, P.G.; Jiménez-García, L.; Strickland, J.D.; Oh, J.; Tapia, F.J.; Mayoral, R.; Burrola, P.; Copple, B.L.; Lemke, G. Differential regulation of hepatic physiology and injury by the TAM receptors Axl and Mer. Life Sci. Alliance 2020, 3, e202000694. [Google Scholar] [CrossRef] [PubMed]
- Hanayama, R.; Tanaka, M.; Miwa, K.; Shinohara, A.; Iwamatsu, A.; Nagata, S. Identification of a factor that links apoptotic cells to phagocytes. Nature 2002, 417, 182–187. [Google Scholar] [CrossRef]
- McGilvray, I.D.; Serghides, L.; Kapus, A.; Rotstein, O.D.; Kain, K.C. Nonopsonic monocyte/macrophage phagocytosis of Plasmodium falciparum-parasitized erythrocytes: A role for CD36 in malarial clearance. Blood 2000, 96, 3231–3240. [Google Scholar] [CrossRef]
- Totino, P.R.R.; Daniel-Ribeiro, C.T.; Ferreira-Da-Cruz, M.D.F. Evidencing the Role of Erythrocytic Apoptosis in Malarial Anemia. Front. Cell. Infect. Microbiol. 2016, 6, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pain, A.; Urban, B.C.; Kai, O.; Casals-Pascual, C.; Shafi, J.; Marsh, K.; Roberts, D.J. A non-sense mutation in Cd36 gene is associated with protection from severe malaria. Lancet 2001, 357, 1502–1503. [Google Scholar] [CrossRef]
- Hochreiter-Hufford, A.E.; Lee, C.S.; Kinchen, J.; Sokolowski, J.D.; Arandjelovic, S.; Call, J.; Klibanov, A.L.; Yan, Z.; Mandell, J.W.; Ravichandran, K. Phosphatidylserine receptor BAI1 and apoptotic cells as new promoters of myoblast fusion. Nature 2013, 497, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, N.; Karisola, P.; Peña-Cruz, V.; Dorfman, D.M.; Jinushi, M.; Umetsu, S.E.; Butte, M.; Nagumo, H.; Chernova, I.; Zhu, B.; et al. TIM-1 and TIM-4 Glycoproteins Bind Phosphatidylserine and Mediate Uptake of Apoptotic Cells. Immunity 2007, 27, 927–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, G.J.; Casasnovas, J.M.; Umetsu, D.T.; DeKruyff, R.H. TIM genes: A family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol. Rev. 2010, 235, 172–189. [Google Scholar] [CrossRef] [Green Version]
- Alderman, E.M.; Fudenberg, H.H.; Lovins, R.E. Binding of immunoglobulin classes to subpopulations of human red blood cells separated by density-gradient centrifugation. Blood 1980, 55, 817–822. [Google Scholar] [CrossRef] [Green Version]
- Kay, M.M. Mechanism of removal of senescent cells by human macrophages in situ. Proc. Natl. Acad. Sci. USA 1975, 72, 3521–3525. [Google Scholar] [CrossRef] [Green Version]
- Christian, J.A.; Rebar, A.H.; Boon, G.D.; Low, P. Senescence of canine biotinylated erythrocytes: Increased autologous immunoglobulin binding occurs on erythrocytes aged in vivo for 104 to 110 days. Blood 1993, 82, 3469–3473. [Google Scholar] [CrossRef] [Green Version]
- Singer, J.A.; Jennings, L.K.; Jackson, C.W.; Dockter, M.E.; Morrison, M.; Walker, W.S. Erythrocyte homeostasis: Antibody-mediated recognition of the senescent state by macrophages. Proc. Natl. Acad. Sci. USA 1986, 83, 5498–5501. [Google Scholar] [CrossRef] [Green Version]
- Beppu, M.; Mizukami, A.; Nagoya, M.; Kikugawa, K. Binding of anti-band 3 autoantibody to oxidatively damaged erythrocytes. Formation of senescent antigen on erythrocyte surface by an oxidative mechanism. J. Biol. Chem. 1990, 265, 3226–3233. [Google Scholar] [CrossRef]
- Kannan, R.; Labotka, R.; Low, P.S. Isolation and characterization of the hemichrome-stabilized membrane protein aggregates from sickle erythrocytes. Major site of autologous antibody binding. J. Biol. Chem. 1988, 263, 13766–13773. [Google Scholar] [CrossRef]
- Kannan, R.; Yuan, J.; Low, P.S. Isolation and partial characterization of antibody- and globin-enriched complexes from membranes of dense human erythrocytes. Biochem. J. 1991, 278, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arese, P.; Turrini, F.; Schwarzer, E. Band 3/Complement-mediated Recognition and Removal of Normally Senescent and Pathological Human Erythrocytes. Cell. Physiol. Biochem. 2005, 16, 133–146. [Google Scholar] [CrossRef]
- Klei, T.R.L.; De Back, D.Z.; Asif, P.J.; Verkuijlen, P.J.J.H.; Veldthuis, M.; Ligthart, P.C.; Berghuis, J.; Clifford, E.; Beuger, B.M.; Berg, T.K.V.D.; et al. Glycophorin-C sialylation regulates Lu/BCAM adhesive capacity during erythrocyte aging. Blood Adv. 2018, 2, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldenborg, P.-A.; Zheleznyak, A.; Fang, Y.-F.; Lagenaur, C.F.; Gresham, H.D.; Lindberg, F.P. Role of CD47 as a Marker of Self on Red Blood Cells. Science 2000, 288, 2051–2054. [Google Scholar] [CrossRef]
- Olsson, M.; Oldenborg, P.A. CD47 on experimentally senescent murine RBCs inhibits phagocytosis following Fcgamma receptor-mediated but not scavenger receptor-mediated recognition by macrophages. Blood 2008, 112, 4259–4267. [Google Scholar] [CrossRef] [Green Version]
- Sosale, N.G.; Rouhiparkouhi, T.; Bradshaw, A.M.; Dimova, R.; Lipowsky, R.; Discher, D.E. Cell rigidity and shape override CD47′s “self”-signaling in phagocytosis by hyperactivating myosin-II. Blood 2015, 125, 542–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khandelwal, S.; Van Rooijen, N.; Saxena, R.K. Reduced expression of CD47 during murine red blood cell (RBC) senescence and its role in RBC clearance from the circulation. Transfusion 2007, 47, 1725–1732. [Google Scholar] [CrossRef]
- Stewart, A.; Urbaniak, S.; Turner, M.; Bessos, H. The application of a new quantitative assay for the monitoring of integrin-associated protein CD47 on red blood cells during storage and comparison with the expression of CD47 and phosphatidylserine with flow cytometry. Transfusion 2005, 45, 1496–1503. [Google Scholar] [CrossRef]
- Kharitonenkov, A.; Chen, Z.; Sures, I.; Wang, H.; Schilling, J.; Ullrich, A. A family of proteins that inhibit signalling through tyrosine kinase receptors. Nature 1997, 386, 181–186. [Google Scholar] [CrossRef]
- Burger, P.; Hilarius-Stokman, P.; de Korte, D.; Berg, T.K.V.D.; Van Bruggen, R. CD47 functions as a molecular switch for erythrocyte phagocytosis. Blood 2012, 119, 5512–5521. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Huang, Q.; Xiao, W.; Zhao, Y.; Pi, J.; Xu, H.; Zhao, H.; Xu, J.; Evans, C.E.; Jin, H. Advances in Anti-Tumor Treatments Targeting the CD47/SIRPalpha Axis. Front. Immunol. 2020, 11, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayi, K.; Lu, Z.; Serghides, L.; Ho, J.M.; Finney, C.; Wang, J.C.Y.; Liles, W.C.; Kain, K.C. CD47-SIRPalpha Interactions Regulate Macrophage Uptake of Plasmodium falciparum-Infected Erythrocytes and Clearance of Malaria In Vivo. Infect. Immun. 2016, 84, 2002–2011. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, R.; Khandelwal, S.; Kozakai, Y.; Sahu, B.; Kumar, S. CD47 regulates the phagocytic clearance and replication of the Plasmodium yoelii malaria parasite. Proc. Natl. Acad. Sci. USA 2015, 112, 3062–3067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, B.S.W.D.H. The Illustrated Pathology of the Spleen; Cambridge University Press: Cambridge, UK, 2000. [Google Scholar]
- Garby, L.; Noyes, W.D. Studies on hemoglobin metabolism. I. the kinetic properties of the plasma hemoglobin pool in normal man. J. Clin. Investig. 1959, 38, 1479–1483. [Google Scholar] [CrossRef] [Green Version]
- Kato, G.; Steinberg, M.H.; Gladwin, M.T. Intravascular hemolysis and the pathophysiology of sickle cell disease. J. Clin. Investig. 2017, 127, 750–760. [Google Scholar] [CrossRef]
- Schaer, D.J.; Vinchi, F.; Ingoglia, G.; Tolosano, E.; Buehler, P.W. Haptoglobin, hemopexin, and related defense pathways-basic science, clinical perspectives, and drug development. Front. Physiol. 2014, 5, 415. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Kinzie, E.; Berger, F.G.; Lim, S.K.; Baumann, H. Haptoglobin, an inflammation-inducible plasma protein. Redox Rep. 2001, 6, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, M.; Graversen, J.H.; Jacobsen, C.; Sonne, O.; Hoffman, H.-J.; Law, S.A.; Moestrup, S.K. Identification of the haemoglobin scavenger receptor. Nature 2001, 409, 198–201. [Google Scholar] [CrossRef]
- Svendsen, P.; Graversen, J.H.; Etzerodt, A.; Hager, H.; Røge, R.; Grønbæk, H.; Christensen, E.I.; Møller, H.J.; Vilstrup, H.; Moestrup, S.K. Antibody-Directed Glucocorticoid Targeting to CD163 in M2-type Macrophages Attenuates Fructose-Induced Liver Inflammatory Changes. Mol. Ther.-Methods Clin. Dev. 2017, 4, 50–61. [Google Scholar] [CrossRef] [Green Version]
- Klei, T.R.L.; Dalimot, J.; Nota, B.; Veldthuis, M.; Mul, E.; Rademakers, T.; Hoogenboezem, M.; Nagelkerke, S.Q.; Van Ijcken, W.F.; Oole, E.; et al. Hemolysis in the spleen drives erythrocyte turnover. Blood 2020, 136, 1579–1589. [Google Scholar] [CrossRef] [PubMed]
- Fagoonee, S.; Gburek, J.; Hirsch, E.; Marro, S.; Moestrup, S.K.; Laurberg, J.M.; Christensen, E.I.; Silengo, L.; Altruda, F.; Tolosano, E. Plasma Protein Haptoglobin Modulates Renal Iron Loading. Am. J. Pathol. 2005, 166, 973–983. [Google Scholar] [CrossRef] [Green Version]
- Marro, S.; Barisani, D.; Chiabrando, D.; Fagoonee, S.; Muckenthaler, M.U.; Stolte, J.; Meneveri, R.; Haile, D.; Silengo, L.; Altruda, F.; et al. Lack of Haptoglobin Affects Iron Transport Across Duodenum by Modulating Ferroportin Expression. Gastroenterology 2007, 133, 1261–1271.e3. [Google Scholar] [CrossRef] [PubMed]
- Boretti, F.S.; Baek, J.H.; Palmer, A.F.; Schaer, D.J.; Buehler, P.W. Modeling hemoglobin and hemoglobin:haptoglobin complex clearance in a non-rodent species-pharmacokinetic and therapeutic implications. Front. Physiol. 2014, 5, 385. [Google Scholar] [CrossRef] [Green Version]
- Schaer, D.J.; Schaer, C.; Buehler, P.W.; Boykins, R.A.; Schoedon, G.; Alayash, A.I.; Schaffner, A. CD163 is the macrophage scavenger receptor for native and chemically modified hemoglobins in the absence of haptoglobin. Blood 2006, 107, 373–380. [Google Scholar] [CrossRef] [Green Version]
- Etzerodt, A.; Kjolby, M.; Nielsen, M.J.; Maniecki, M.; Svendsen, P.; Moestrup, S.K. Plasma Clearance of Hemoglobin and Haptoglobin in Mice and Effect of CD163 Gene Targeting Disruption. Antioxid. Redox Signal. 2013, 18, 2254–2263. [Google Scholar] [CrossRef]
- Van Avondt, K.; Nur, E.; Zeerleder, S. Mechanisms of haemolysis-induced kidney injury. Nat. Rev. Nephrol. 2019, 15, 671–692. [Google Scholar] [CrossRef]
- Vinchi, F.; De Franceschi, L.; Ghigo, A.; Townes, T.; Cimino, J.; Silengo, L.; Hirsch, E.; Altruda, F.; Tolosano, E. Hemopexin Therapy Improves Cardiovascular Function by Preventing Heme-Induced Endothelial Toxicity in Mouse Models of Hemolytic Diseases. Circulation 2013, 127, 1317–1329. [Google Scholar] [CrossRef] [Green Version]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Milbauer, L.; Abdulla, F.; Alayash, A.I.; Smith, A.; Nath, K.A.; Hebbel, R.P.; Vercellotti, G.M. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 2014, 123, 377–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hvidberg, V.; Maniecki, M.B.; Jacobsen, C.; Højrup, P.; Møller, H.J.; Moestrup, S.K. Identification of the receptor scavenging hemopexin-heme complexes. Blood 2005, 106, 2572–2579. [Google Scholar] [CrossRef]
- Kovtunovych, G.; Eckhaus, M.A.; Ghosh, M.C.; Ollivierre-Wilson, H.; Rouault, T.A. Dysfunction of the heme recycling system in heme oxygenase 1–deficient mice: Effects on macrophage viability and tissue iron distribution. Blood 2010, 116, 6054–6062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haldar, M.; Kohyama, M.; So, A.Y.-L.; Kc, W.; Wu, X.; Briseno, C.; Satpathy, A.; Kretzer, N.M.; Arase, H.; Rajasekaran, N.S.; et al. Heme-Mediated SPI-C Induction Promotes Monocyte Differentiation into Iron-Recycling Macrophages. Cell 2014, 156, 1223–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Basatemur, G.; Scott, I.C.; Chiarugi, D.; Clement, M.; Harrison, J.; Jugdaohsingh, R.; Yu, X.; Newland, S.; Jolin, H.E.; et al. Interleukin-33 Signaling Controls the Development of Iron-Recycling Macrophages. Immunity 2020, 52, 782–793.e5. [Google Scholar] [CrossRef]
- Bennett, L.F.; Liao, C.; Quickel, M.D.; Yeoh, B.S.; Vijay-Kumar, M.; Hankey-Giblin, P.; Prabhu, K.S.; Paulson, R.F. Inflammation induces stress erythropoiesis through heme-dependent activation of SPI-C. Sci. Signal. 2019, 12, eaap7336. [Google Scholar] [CrossRef]
- Pek, R.H.; Yuan, X.; Rietzschel, N.; Zhang, J.; Jackson, L.; Nishibori, E.; Ribeiro, A.; Simmons, W.; Jagadeesh, J.; Sugimoto, H.; et al. Hemozoin produced by mammals confers heme tolerance. eLife 2019, 8, e49503. [Google Scholar] [CrossRef]
- Santarino, I.; Viegas, M.S.; Domingues, N.; Ribeiro, A.M.; Soares, M.P.; Vieira, O.V. Involvement of the p62/NRF2 signal transduction pathway on erythrophagocytosis. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Asare, P.F.; Roscioli, E.; Hurtado, P.R.; Tran, H.B.; Mah, C.Y.; Hodge, S. LC3-Associated Phagocytosis (LAP): A Potentially Influential Mediator of Efferocytosis-Related Tumor Progression and Aggressiveness. Front. Oncol. 2020, 10, 1298. [Google Scholar] [CrossRef] [PubMed]
- Delaby, C.; Rondeau, C.; Pouzet, C.; Willemetz, A.; Pilard, N.; Desjardins, M.; Canonne-Hergaux, F. Subcellular localization of iron and heme metabolism related proteins at early stages of erythrophagocytosis. PLoS ONE 2012, 7, e42199. [Google Scholar] [CrossRef]
- Gagnon, E.; Duclos, S.; Rondeau, C.; Chevet, E.; Cameron, P.H.; Steele-Mortimer, O.; Paiement, J.; Bergeron, J.J.; Desjardins, M. Endoplasmic Reticulum-Mediated Phagocytosis Is a Mechanism of Entry into Macrophages. Cell 2002, 110, 119–131. [Google Scholar] [CrossRef] [Green Version]
- Delaby, C.; Pilard, N.; Hetet, G.; Driss, F.; Grandchamp, B.; Beaumont, C.; Canonne-Hergaux, F. A physiological model to study iron recycling in macrophages. Exp. Cell Res. 2005, 310, 43–53. [Google Scholar] [CrossRef]
- Delaby, C.; Pilard, N.; Puy, H.; Canonne-Hergaux, F. Sequential regulation of ferroportin expression after erythrophagocytosis in murine macrophages: Early mRNA induction by haem, followed by iron-dependent protein expression. Biochem. J. 2008, 411, 123–131. [Google Scholar] [CrossRef] [Green Version]
- Knutson, M.D.; Vafa, M.; Haile, D.J.; Wessling-Resnick, M. Iron loading and erythrophagocytosis increase ferroportin 1 (FPN1) expression in J774 macrophages. Blood 2003, 102, 4191–4197. [Google Scholar] [CrossRef] [PubMed]
- Soe-Lin, S.; Apte, S.S.; Andriopoulos, B.; Andrews, M.C.; Schranzhofer, M.; Kahawita, T.; Garcia-Santos, D.; Ponka, P. Nramp1 promotes efficient macrophage recycling of iron following erythrophagocytosis in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 5960–5965. [Google Scholar] [CrossRef] [Green Version]
- Rajagopal, A.; Rao, A.U.; Amigo, J.; Tian, M.; Upadhyay, S.K.; Hall, C.; Uhm, S.; Mathew, M.K.; Fleming, M.; Paw, B.; et al. Haem homeostasis is regulated by the conserved and concerted functions of HRG-1 proteins. Nature 2008, 453, 1127–1131. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chambers, I.; Yun, S.; Phillips, J.; Krause, M.; Hamza, I. Hrg1 promotes heme-iron recycling during hemolysis in the zebrafish kidney. PLoS Genet. 2018, 14, e1007665. [Google Scholar] [CrossRef] [PubMed]
- Poss, K.D.; Tonegawa, S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc. Natl. Acad. Sci. USA 1997, 94, 10919–10924. [Google Scholar] [CrossRef] [Green Version]
- Nai, A.; Lidonnici, M.R.; Federico, G.; Pettinato, M.; Olivari, V.; Carrillo, F.; Crich, S.G.; Ferrari, G.; Camaschella, C.; Silvestri, L.; et al. NCOA4-mediated ferritinophagy in macrophages is crucial to sustain erythropoiesis in mice. Haematologica 2020, 106, 795–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knutson, M.D.; Oukka, M.; Koss, L.M.; Aydemir, F.; Wessling-Resnick, M. Iron release from macrophages after erythrophagocytosis is up-regulated by ferroportin 1 overexpression and down-regulated by hepcidin. Proc. Natl. Acad. Sci. USA 2005, 102, 1324–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Zhang, F.; An, P.; Guo, X.; Shen, Y.; Tao, Y.; Wu, Q.; Zhang, Y.; Yu, Y.; Ning, B.; et al. Ferroportin1 deficiency in mouse macrophages impairs iron homeostasis and inflammatory responses. Blood 2011, 118, 1912–1922. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin Regulates Cellular Iron Efflux by Binding to Ferroportin and Inducing Its Internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [Green Version]
- Aschemeyer, S.; Qiao, B.; Stefanova, D.; Valore, E.V.; Sek, A.; Ruwe, T.; Vieth, K.R.; Jung, G.; Casu, C.; Rivella, S.; et al. Structure-function analysis of ferroportin defines the binding site and an alternative mechanism of action of hepcidin. Blood 2018, 131, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Fung, E.; Parikh, S.G.; Valore, E.V.; Gabayan, V.; Nemeth, E.; Ganz, T. A mouse model of anemia of inflammation: Complex pathogenesis with partial dependence on hepcidin. Blood 2014, 123, 1129–1136. [Google Scholar] [CrossRef] [Green Version]
- Guida, C.; Altamura, S.; Klein, F.A.; Galy, B.; Boutros, M.; Ulmer, A.J.; Hentze, M.W.; Muckenthaler, M.U. A novel inflammatory pathway mediating rapid hepcidin-independent hypoferremia. Blood 2015, 125, 2265–2275. [Google Scholar] [CrossRef] [Green Version]
- Camaschella, C. Iron deficiency. Blood 2019, 133, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Kautz, L.; Jung, G.; Valore, E.V.; Rivella, S.; Nemeth, E.; Ganz, T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat. Genet. 2014, 46, 678–684. [Google Scholar] [CrossRef] [Green Version]
- Schaer, C.; Vallelian, F.; Imhof, A.; Schoedon, G.; Schaer, D.J. Heme carrier protein (HCP-1) spatially interacts with the CD163 hemoglobin uptake pathway and is a target of inflammatory macrophage activation. J. Leukoc. Biol. 2007, 83, 325–333. [Google Scholar] [CrossRef]
- Ma, S.; Dubin, A.E.; Zhang, Y.; Mousavi, S.A.R.; Wang, Y.; Coombs, A.M.; Loud, M.; Andolfo, I.; Patapoutian, A. A role of PIEZO1 in iron metabolism in mice and humans. Cell 2021, 184, 969–982.e13. [Google Scholar] [CrossRef] [PubMed]
- Schlam, D.; Bagshaw, R.D.; Freeman, S.A.; Collins, R.F.; Pawson, T.; Fairn, G.D.; Grinstein, S. Phosphoinositide 3-kinase enables phagocytosis of large particles by terminating actin assembly through Rac/Cdc42 GTPase-activating proteins. Nat. Commun. 2015, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Beesetty, P.; Rockwood, J.; Kaitsuka, T.; Zhelay, T.; Hourani, S.; Matsushita, M.; Kozak, J.A. Phagocytic activity of splenic macrophages is enhanced and accompanied by cytosolic alkalinization in TRPM7 kinase-dead mice. FEBS J. 2020, 288, 3585–3601. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Martinez-Pomares, L. Physiological roles of macrophages. Pflugers Archiv 2017, 469, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Epelman, S.; LaVine, K.J.; Randolph, G.J. Origin and Functions of Tissue Macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Yona, S.; Kim, K.-W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate Mapping Reveals Origins and Dynamics of Monocytes and Tissue Macrophages under Homeostasis. Immunity 2012, 38, 79–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautier, E.; Shay, T.; Miller, J.; Greter, M.; Jakubzick, C.; Ivanov, S.; Helft, J.; Chow, A.; Elpek, K.G.; the Immunological Genome Consortium; et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 2012, 13, 1118–1128. [Google Scholar] [CrossRef] [Green Version]
- Lavin, Y.; Winter, D.; Blecher-Gonen, R.; David, E.; Keren-Shaul, H.; Merad, M.; Jung, S.; Amit, I. Tissue-Resident Macrophage Enhancer Landscapes Are Shaped by the Local Microenvironment. Cell 2014, 159, 1312–1326. [Google Scholar] [CrossRef] [Green Version]
- Gosselin, D.; Link, V.M.; Romanoski, C.E.; Fonseca, G.J.; Eichenfield, D.Z.; Spann, N.J.; Stender, J.D.; Chun, H.B.; Garner, H.; Geissmann, F.; et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 2014, 159, 1327–1340. [Google Scholar] [CrossRef]
- Okreglicka, K.; Iten, I.; Pohlmeier, L.; Onder, L.; Feng, Q.; Kurrer, M.; Ludewig, B.; Nielsen, N.P.; Schneider, C.; Kopf, M. PPARgamma is essential for the development of bone marrow erythroblastic island macrophages and splenic red pulp macrophages. J. Exp. Med. 2021, 218, e20191314. [Google Scholar] [CrossRef] [PubMed]
- Kohyama, M.; Ise, W.; Edelson, B.T.; Wilker, P.R.; Hildner, K.; Mejia, C.; Frazier, W.A.; Murphy, T.L.; Murphy, K.M. Role for Spi-C in the development of red pulp macrophages and splenic iron homeostasis. Nature 2008, 457, 318–321. [Google Scholar] [CrossRef] [PubMed]
- Ulyanova, T.; Phelps, S.R.; Papayannopoulou, T. The macrophage contribution to stress erythropoiesis: When less is enough. Blood 2016, 128, 1756–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karsten, E.; Breen, E.; Herbert, B. Red blood cells are dynamic reservoirs of cytokines. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Bellomo, A.; Mondor, I.; Spinelli, L.; Lagueyrie, M.; Stewart, B.J.; Brouilly, N.; Malissen, B.; Clatworthy, M.R.; Bajénoff, M. Reticular Fibroblasts Expressing the Transcription Factor WT1 Define a Stromal Niche that Maintains and Replenishes Splenic Red Pulp Macrophages. Immunity 2020, 53, 127–142.e7. [Google Scholar] [CrossRef] [PubMed]
- Mass, E.; Ballesteros, I.; Farlik, M.; Halbritter, F.; Günther, P.; Crozet, L.; Jacome-Galarza, C.E.; Händler, K.; Klughammer, J.; Kobayashi, Y.; et al. Specification of tissue-resident macrophages during organogenesis. Science 2016, 353, aaf4238. [Google Scholar] [CrossRef] [Green Version]
- Scott, C.L.; T’Jonck, W.; Martens, L.; Todorov, H.; Sichien, D.; Soen, B.; Bonnardel, J.; De Prijck, S.; Vandamme, N.; Cannoodt, R.; et al. The Transcription Factor ZEB2 Is Required to Maintain the Tissue-Specific Identities of Macrophages. Immunity 2018, 49, 312–325.e5. [Google Scholar] [CrossRef] [Green Version]
- Scott, C.; Zheng, F.; De Baetselier, P.; Martens, L.; Saeys, Y.; De Prijck, S.; Lippens, S.; Abels, C.; Schoonooghe, S.; Raes, G.; et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat. Commun. 2016, 7, 10321. [Google Scholar] [CrossRef]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-Resident Macrophages Self-Maintain Locally throughout Adult Life with Minimal Contribution from Circulating Monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Gu, Y.; Chakarov, S.; Bleriot, C.; Kwok, I.; Chen, X.; Shin, A.; Huang, W.; Dress, R.J.; Dutertre, C.-A.; et al. Fate Mapping via Ms4a3-Expression History Traces Monocyte-Derived Cells. Cell 2019, 178, 1509–1525.e19. [Google Scholar] [CrossRef]
- Kondo, H.; Saito, K.; Grasso, J.P.; Aisen, P. Iron metabolism in the erythrophagocytosing Kupffer cell. Hepatology 1988, 8, 32–38. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Tassa, C.; Shaw, S.Y.; Weissleder, R. Dextran-Coated Iron Oxide Nanoparticles: A Versatile Platform for Targeted Molecular Imaging, Molecular Diagnostics, and Therapy. Acc. Chem. Res. 2011, 44, 842–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirschbaum, K.; Sonner, J.K.; Zeller, M.W.; Deumelandt, K.; Bode, J.; Sharma, R.; Krüwel, T.; Fischer, M.; Hoffmann, A.; da Silva, M.C.; et al. In vivo nanoparticle imaging of innate immune cells can serve as a marker of disease severity in a model of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2016, 113, 13227–13232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, U.; Zia, K.; Iqbal, R.; Saeed, M.; Ashraf, N. Efficacy of a Novel Food Supplement (Ferfer(R)) Containing Microencapsulated Iron in Liposomal Form in Female Iron Deficiency Anemia. Cureus 2019, 11, e4603. [Google Scholar] [CrossRef] [Green Version]
- Chow, A.; Huggins, M.; Ahmed, J.; Hashimoto, D.; Lucas, D.; Kunisaki, Y.; Pinho, S.; Leboeuf, M.; Noizat, C.; Van Rooijen, N.; et al. CD169+ macrophages provide a niche promoting erythropoiesis under homeostasis and stress. Nat. Med. 2013, 19, 429–436. [Google Scholar] [CrossRef] [Green Version]
- Ramos, P.; Casu, C.; Gardenghi, S.; Breda, L.; Crielaard, B.J.; Guy, E.; Marongiu, M.F.; Gupta, R.; Levine, R.L.; Abdel-Wahab, O.; et al. Macrophages support pathological erythropoiesis in polycythemia vera and β-thalassemia. Nat. Med. 2013, 19, 437–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, C.; Prabhu, K.S.; Paulson, R.F. Monocyte-derived macrophages expand the murine stress erythropoietic niche during the recovery from anemia. Blood 2018, 132, 2580–2593. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Xiang, J.; Qian, F.; Diwakar, B.T.; Ruan, B.; Hao, S.; Prabhu, K.S.; Paulson, R.F. Epo receptor signaling in macrophages alters the splenic niche to promote erythroid differentiation. Blood 2020, 136, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Kidder, K.; Bian, Z.; Shi, L.; Liu, Y. Inflammation Unrestrained by SIRPalpha Induces Secondary Hemophagocytic Lymphohistiocytosis Independent of IFN-γ. J. Immunol. 2020, 205, 2821–2833. [Google Scholar] [CrossRef] [PubMed]
- Zoller, E.E.; Lykens, J.E.; Terrell, C.E.; Aliberti, J.; Filipovich, A.H.; Henson, P.M.; Jordan, M.B. Hemophagocytosis causes a consumptive anemia of inflammation. J. Exp. Med. 2011, 208, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, A.; Uchida, K.; Chambers, J.; Sato, K.; Hong, J.; Sanjoba, C.; Matsumoto, Y.; Yamagishi, J.; Goto, Y. Hemophagocytosis induced by Leishmania donovani infection is beneficial to parasite survival within macrophages. PLoS Negl. Trop. Dis. 2019, 13, e0007816. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Zhu, D.; Guo, J.; Wang, C. IL-18 promotes erythrophagocytosis and erythrocyte degradation by M1 macrophages in a calcific microenvironment: IL-18 promotes erythrophagocytosis and degradation. Can. J. Cardiol. 2021. online pre-proof. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef] [PubMed]
- Ju, C.; Tacke, F. Hepatic macrophages in homeostasis and liver diseases: From pathogenesis to novel therapeutic strategies. Cell. Mol. Immunol. 2016, 13, 316–327. [Google Scholar] [CrossRef] [Green Version]
- Kurotaki, D.; Kon, S.; Bae, K.; Ito, K.; Matsui, Y.; Nakayama, Y.; Kanayama, M.; Kimura, C.; Narita, Y.; Nishimura, T.; et al. CSF-1–Dependent Red Pulp Macrophages Regulate CD4 T Cell Responses. J. Immunol. 2011, 186, 2229–2237. [Google Scholar] [CrossRef] [Green Version]
- Enders, M.; Franken, L.; Philipp, M.-S.; Kessler, N.; Baumgart, A.-K.; Eichler, M.; Wiertz, E.J.H.; Garbi, N.; Kurts, C. Splenic Red Pulp Macrophages Cross-Prime Early Effector CTL That Provide Rapid Defense against Viral Infections. J. Immunol. 2019, 204, 87–100. [Google Scholar] [CrossRef]
- Hod, E.A.; Zhang, N.; Sokol, S.A.; Wojczyk, B.S.; Francis, R.O.; Ansaldi, D.; Francis, K.P.; Della-Latta, P.; Whittier, S.; Sheth, S.; et al. Transfusion of red blood cells after prolonged storage produces harmful effects that are mediated by iron and inflammation. Blood 2010, 115, 4284–4292. [Google Scholar] [CrossRef] [Green Version]
- Vinchi, F.; da Silva, M.C.; Ingoglia, G.; Petrillo, S.; Brinkman, N.; Zuercher, A.; Cerwenka, A.; Tolosano, E.; Muckenthaler, M. Hemopexin therapy reverts heme-induced proinflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood 2016, 127, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Olonisakin, T.F.; Suber, T.L.; Gonzalez-Ferrer, S.; Xiong, Z.; Peñaloza, H.F.; van der Geest, R.; Xiong, Y.; Osei-Hwedieh, D.O.; Tejero, J.; Rosengart, M.R.; et al. Stressed erythrophagocytosis induces immunosuppression during sepsis through heme-mediated STAT1 dysregulation. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Costa da Silva, M.; Breckwoldt, M.O.; Vinchi, F.; Correia, M.P.; Stojanovic, A.; Thielmann, C.M.; Meister, M.; Muley, T.; Warth, A.; Platten, M.; et al. Iron Induces Anti-tumor Activity in Tumor-Associated Macrophages. Front. Immunol. 2017, 8, 1479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alaluf, E.; Vokaer, B.; Detavernier, A.; Azouz, A.; Splittgerber, M.; Carrette, A.; Boon, L.; Libert, F.; Soares, M.P.; Le Moine, A.; et al. Heme oxygenase-1 orchestrates the immunosuppressive program of tumor-associated macrophages. JCI Insight 2020, 5, e133929. [Google Scholar] [CrossRef]
- Etzerodt, A.; Moulin, M.; Doktor, T.K.; Delfini, M.; Mossadegh-Keller, N.; Bajenoff, M.; Sieweke, M.; Moestrup, S.K.; Auphan-Anezin, N.; Lawrence, T. Tissue-resident macrophages in omentum promote metastatic spread of ovarian cancer. J. Exp. Med. 2020, 217, e20191869. [Google Scholar] [CrossRef] [PubMed]
- Etzerodt, A.; Tsalkitzi, K.; Maniecki, M.; Damsky, W.; Delfini, M.; Baudoin, E.; Moulin, M.; Bosenberg, M.; Graversen, J.H.; Auphan-Anezin, N.; et al. Specific targeting of CD163+ TAMs mobilizes inflammatory monocytes and promotes T cell–mediated tumor regression. J. Exp. Med. 2019, 216, 2394–2411. [Google Scholar] [CrossRef] [PubMed]
- Etzerodt, A.; Moestrup, S.K. CD163 and Inflammation: Biological, Diagnostic, and Therapeutic Aspects. Antioxid. Redox Signal. 2013, 18, 2352–2363. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Slusarczyk, P.; Mleczko-Sanecka, K. The Multiple Facets of Iron Recycling. Genes 2021, 12, 1364. https://doi.org/10.3390/genes12091364
Slusarczyk P, Mleczko-Sanecka K. The Multiple Facets of Iron Recycling. Genes. 2021; 12(9):1364. https://doi.org/10.3390/genes12091364
Chicago/Turabian StyleSlusarczyk, Patryk, and Katarzyna Mleczko-Sanecka. 2021. "The Multiple Facets of Iron Recycling" Genes 12, no. 9: 1364. https://doi.org/10.3390/genes12091364
APA StyleSlusarczyk, P., & Mleczko-Sanecka, K. (2021). The Multiple Facets of Iron Recycling. Genes, 12(9), 1364. https://doi.org/10.3390/genes12091364