
Tumor Microenvironment Characterization in Breast Cancer Identifies Prognostic Pathway Signatures

Abstract
:1. Introduction
2. Materials and Methods
2.1. Breast Cancer Datasets and Preprocessing
2.2. KEGG Pathway Information
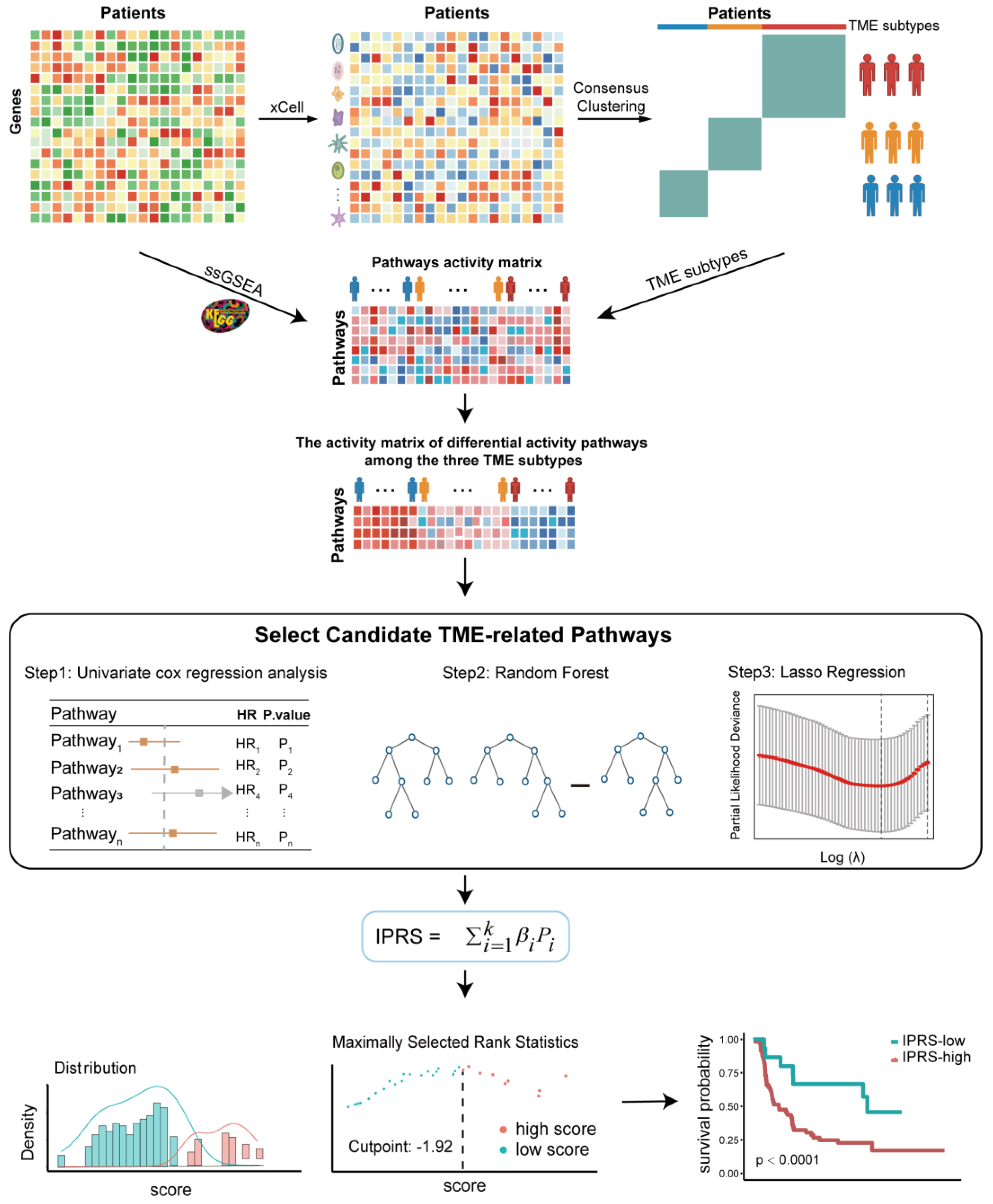
2.3. Identification of Breast Cancer TME Phenotype Based on the Infiltration of Immune Cells
2.4. Construction of the Pathway-Activity-Based Classification for Breast Cancer Patients
2.5. The Correlation between the IPRS and Immune-Related Features
3. Results
3.1. The Landscape of TME in Breast Cancer and Clinicopathological Characteristics of TME Phenotypes
3.2. Construction of the Pathway-Activity-Based Classification for BRCA Patients
3.3. The Correlation between the IPRS and Immune-Related Features
3.4. Validation of the Prognostic Value of the IPRS Model
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hance, K.W.; Anderson, W.F.; Devesa, S.S.; Young, H.A.; Levine, P.H. Trends in inflammatory breast carcinoma incidence and survival: The surveillance, epidemiology, and end results program at the National Cancer Institute. J. Natl. Cancer Inst. 2005, 97, 966–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiser, M.R. AJCC 8th Edition: Colorectal Cancer. Ann. Surg. Oncol. 2018, 25, 1454–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, S.; Nor, J.E. Head and neck cancer stem cells. J. Dent. Res. 2012, 91, 334–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bense, R.D.; Sotiriou, C.; Piccart-Gebhart, M.J.; Haanen, J.; van Vugt, M.; de Vries, E.G.E.; Schroder, C.P.; Fehrmann, R.S.N. Relevance of Tumor-Infiltrating Immune Cell Composition and Functionality for Disease Outcome in Breast Cancer. J. Natl. Cancer Inst. 2017, 109, djw192. [Google Scholar] [CrossRef] [Green Version]
- Barnes, T.A.; Amir, E. HYPE or HOPE: The prognostic value of infiltrating immune cells in cancer. Br. J. Cancer 2018, 118, e5. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Balkwill, F.; Charles, K.A.; Mantovani, A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 2005, 7, 211–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quail, D.F.; Bowman, R.L.; Akkari, L.; Quick, M.L.; Schuhmacher, A.J.; Huse, J.T.; Holland, E.C.; Sutton, J.C.; Joyce, J.A. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 2016, 352, aad3018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAuliffe, P.F.; Meric-Bernstam, F.; Mills, G.B.; Gonzalez-Angulo, A.M. Deciphering the role of PI3K/Akt/mTOR pathway in breast cancer biology and pathogenesis. Clin. Breast Cancer 2010, 10 (Suppl. S3), S59–S65. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J. Targeting the phosphoinositide-3 (PI3) kinase pathway in breast cancer. Oncologist 2011, 16 (Suppl. S1), 12–19. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, M.; Xu, F.; Jiang, S. Wnt signaling in breast cancer: Biological mechanisms, challenges and opportunities. Mol. Cancer 2020, 19, 165. [Google Scholar] [CrossRef]
- Hui, M.; Cazet, A.; Nair, R.; Watkins, D.N.; O’Toole, S.A.; Swarbrick, A. The Hedgehog signalling pathway in breast development, carcinogenesis and cancer therapy. Breast Cancer Res. 2013, 15, 203. [Google Scholar] [CrossRef] [Green Version]
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J.; et al. The somatic mutation profiles of 2433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef] [Green Version]
- Yau, C.; Esserman, L.; Moore, D.H.; Waldman, F.; Sninsky, J.; Benz, C.C. A multigene predictor of metastatic outcome in early stage hormone receptor-negative and triple-negative breast cancer. Breast Cancer Res. 2010, 12, R85. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Li, W.; Gu, Z.; Xing, R.; Ma, Y.; Zhang, Q.; Liu, Z. Inhibition of HER2-Positive Breast Cancer Growth by Blocking the HER2 Signaling Pathway with HER2-Glycan-Imprinted Nanoparticles. Angew. Chem. Int. Ed. 2019, 58, 10621–10625. [Google Scholar] [CrossRef]
- Huang, S.; Chong, N.; Lewis, N.E.; Jia, W.; Xie, G.; Garmire, L.X. Novel personalized pathway-based metabolomics models reveal key metabolic pathways for breast cancer diagnosis. Genome Med. 2016, 8, 34. [Google Scholar] [CrossRef]
- Kumar, S.; Nandi, A.; Singh, S.; Regulapati, R.; Li, N.; Tobias, J.W.; Siebel, C.W.; Blanco, M.A.; Klein-Szanto, A.J.; Lengner, C.; et al. Dll1(+) quiescent tumor stem cells drive chemoresistance in breast cancer through NF-kappaB survival pathway. Nat. Commun. 2021, 12, 432. [Google Scholar] [CrossRef]
- Wu, Y.; Wei, X.; Feng, H.; Hu, B.; Liu, B.; Luan, Y.; Ruan, Y.; Liu, X.; Liu, Z.; Liu, J.; et al. Integrated Analysis to Identify a Redox-Related Prognostic Signature for Clear Cell Renal Cell Carcinoma. Oxid. Med. Cell. Longev. 2021, 2021, 6648093. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef] [Green Version]
- Senbabaoglu, Y.; Michailidis, G.; Li, J.Z. Critical limitations of consensus clustering in class discovery. Sci. Rep. 2014, 4, 6207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, K.; Shahmoradgoli, M.; Martinez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [Green Version]
- Lauss, M.; Donia, M.; Harbst, K.; Andersen, R.; Mitra, S.; Rosengren, F.; Salim, M.; Vallon-Christersson, J.; Torngren, T.; Kvist, A.; et al. Mutational and putative neoantigen load predict clinical benefit of adoptive T cell therapy in melanoma. Nat. Commun. 2017, 8, 1738. [Google Scholar] [CrossRef] [PubMed]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Henriques, A.; Munoz-Gonzalez, J.I.; Sanchez-Munoz, L.; Matito, A.; Torres-Rivera, L.; Jara-Acevedo, M.; Caldas, C.; Mayado, A.; Perez-Pons, A.; Garcia-Montero, A.C.; et al. Frequency and prognostic impact of blood-circulating tumor mast cells in mastocytosis. Blood 2022, 139, 572–583. [Google Scholar] [CrossRef]
- Saito, T.; Nishikawa, H.; Wada, H.; Nagano, Y.; Sugiyama, D.; Atarashi, K.; Maeda, Y.; Hamaguchi, M.; Ohkura, N.; Sato, E.; et al. Two FOXP3(+)CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat. Med. 2016, 22, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Peng, L.S.; Zhao, Y.L.; Shi, Y.; Mao, X.H.; Chen, W.; Pang, K.C.; Liu, X.F.; Liu, T.; Zhang, J.Y.; et al. CD8(+) T cells that produce interleukin-17 regulate myeloid-derived suppressor cells and are associated with survival time of patients with gastric cancer. Gastroenterology 2012, 143, 951–962.e8. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Sun, H.; Dong, K.; Hu, L.; Wang, Q.; Zhuang, Q.; Zhu, Y.; Zhang, X.; Shao, Y.; Tang, H.; et al. The thermogenic activity of adjacent adipocytes fuels the progression of ccRCC and compromises anti-tumor therapeutic efficacy. Cell Metab. 2021, 33, 2021–2039.e8. [Google Scholar] [CrossRef]
- Gong, K.; Jiao, J.; Xu, C.; Dong, Y.; Li, D.; He, D.; Zhao, D.; Yu, J.; Sun, Y.; Zhang, W.; et al. The targetable nanoparticle BAF312@cRGD-CaP-NP represses tumor growth and angiogenesis by downregulating the S1PR1/P-STAT3/VEGFA axis in triple-negative breast cancer. J. Nanobiotechnol. 2021, 19, 165. [Google Scholar] [CrossRef] [PubMed]
- Franses, J.W.; Baker, A.B.; Chitalia, V.C.; Edelman, E.R. Stromal endothelial cells directly influence cancer progression. Sci. Transl. Med. 2011, 3, 66ra65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.N.; Yang, K.D.; Chen, C.; He, Z.C.; Wang, Q.H.; Feng, H.; Lv, S.Q.; Wang, Y.; Mao, M.; Liu, Q.; et al. Pericytes augment glioblastoma cell resistance to temozolomide through CCL5-CCR5 paracrine signaling. Cell Res. 2021, 31, 1072–1087. [Google Scholar] [CrossRef] [PubMed]
- Engblom, C.; Pfirschke, C.; Zilionis, R.; Da Silva Martins, J.; Bos, S.A.; Courties, G.; Rickelt, S.; Severe, N.; Baryawno, N.; Faget, J.; et al. Osteoblasts remotely supply lung tumors with cancer-promoting SiglecF(high) neutrophils. Science 2017, 358, eaal5081. [Google Scholar] [CrossRef] [PubMed]
- Heichler, C.; Scheibe, K.; Schmied, A.; Geppert, C.I.; Schmid, B.; Wirtz, S.; Thoma, O.M.; Kramer, V.; Waldner, M.J.; Buttner, C.; et al. STAT3 activation through IL-6/IL-11 in cancer-associated fibroblasts promotes colorectal tumour development and correlates with poor prognosis. Gut 2020, 69, 1269–1282. [Google Scholar] [CrossRef] [PubMed]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Michea, P.; Noel, F.; Zakine, E.; Czerwinska, U.; Sirven, P.; Abouzid, O.; Goudot, C.; Scholer-Dahirel, A.; Vincent-Salomon, A.; Reyal, F.; et al. Adjustment of dendritic cells to the breast-cancer microenvironment is subset specific. Nat. Immunol. 2018, 19, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef]
- Wong, R.S. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef] [Green Version]
- Loveless, R.; Bloomquist, R.; Teng, Y. Pyroptosis at the forefront of anticancer immunity. J. Exp. Clin. Cancer Res. 2021, 40, 264. [Google Scholar] [CrossRef]
- New, M.I. Nonclassical congenital adrenal hyperplasia and the polycystic ovarian syndrome. Ann. N. Y. Acad. Sci. 1993, 687, 193–205. [Google Scholar] [CrossRef]
- Motahari Rad, H.; Mowla, S.J.; Ramazanali, F.; Rezazadeh Valojerdi, M. Characterization of altered microRNAs related to different phenotypes of polycystic ovarian syndrome (PCOS) in serum, follicular fluid, and cumulus cells. Taiwan J. Obstet. Gynecol. 2022, 61, 768–779. [Google Scholar] [CrossRef]
- Nunnery, S.E.; Mayer, I.A. Targeting the PI3K/AKT/mTOR Pathway in Hormone-Positive Breast Cancer. Drugs 2020, 80, 1685–1697. [Google Scholar] [CrossRef]
- Keenan, T.E.; Guerriero, J.L.; Barroso-Sousa, R.; Li, T.; O’Meara, T.; Giobbie-Hurder, A.; Tayob, N.; Hu, J.; Severgnini, M.; Agudo, J.; et al. Molecular correlates of response to eribulin and pembrolizumab in hormone receptor-positive metastatic breast cancer. Nat. Commun. 2021, 12, 5563. [Google Scholar] [CrossRef]
- Shehwana, H.; Keskus, A.G.; Ozdemir, S.E.; Acikgoz, A.A.; Biyik-Sit, R.; Cagnan, I.; Gunes, D.; Jahja, E.; Cingir-Koker, S.; Olmezer, G.; et al. CHRNA5 belongs to the secondary estrogen signaling network exhibiting prognostic significance in breast cancer. Cell. Oncol. 2021, 44, 453–472. [Google Scholar] [CrossRef] [PubMed]
- Di, J.; Zheng, B.; Kong, Q.; Jiang, Y.; Liu, S.; Yang, Y.; Han, X.; Sheng, Y.; Zhang, Y.; Cheng, L.; et al. Prioritization of candidate cancer drugs based on a drug functional similarity network constructed by integrating pathway activities and drug activities. Mol. Oncol. 2019, 13, 2259–2277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Han, X.; Kong, Q.; Cheng, L. psSubpathway: A software package for flexible identification of phenotype-specific subpathways in cancer progression. Bioinformatics 2020, 36, 2303–2305. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Li, C.; Yang, H.; Xu, Y.; Zhang, C.; Ma, J.; Shi, X.; Liu, W.; Shang, D.; Yao, Q.; et al. A novel dysregulated pathway-identification analysis based on global influence of within-pathway effects and crosstalk between pathways. J. R. Soc. Interface 2015, 12, 20140937. [Google Scholar] [CrossRef]
- Han, X.; Kong, Q.; Liu, C.; Cheng, L.; Han, J. SubtypeDrug: A software package for prioritization of candidate cancer subtype-specific drugs. Bioinformatics 2021, 37, 2491–2493. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, Y.; Wu, J.; Qiu, J.; Li, J.; Wang, Q.; Jiang, Y.; Han, J. A novel pathway mutation perturbation score predicts the clinical outcomes of immunotherapy. Brief. Bioinform. 2022, 23, bbac360. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.; Jiang, Y.; Yang, Y.; Li, X.; Qiu, J.; Wu, J.; Cheng, L.; Han, J. CNA2Subpathway: Identification of dysregulated subpathway driven by copy number alterations in cancer. Brief. Bioinform. 2021, 22, bbaa413. [Google Scholar] [CrossRef]
- Wu, J.; Li, X.; Wang, Q.; Han, J. DRviaSPCN: A software package for drug repurposing in cancer via a subpathway crosstalk network. Bioinformatics 2022, btac611. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathways | Coef | HR | 95%CI | p Value |
|---|---|---|---|---|
| Progesterone-mediated oocyte maturation | 6.27 | 530.25 | 6.21–45,308.78 | <0.01 |
| Acute myeloid leukemia | −3.20 | 0.04 | 0.0003–4.96 | 0.19 |
| Bladder cancer | 5.72 | 304.04 | 9.93–9311.16 | <0.01 |
| Neuroactive ligand-receptor interaction | −1.90 | 0.15 | 0.001–12.03 | 0.40 |
| Apoptosis | −8.67 | 0.0001 | 0.0001–0.03 | <0.01 |
| Longevity regulating pathway—multiple species | −3.15 | 0.043 | 0.0003–6.28 | 0.22 |
| Ovarian steroidogenesis | −1.33 | 0.27 | 0.02–4.34 | 0.35 |
| Estrogen signaling pathway | −2.17 | 0.11 | 0.0005–24.11 | 0.43 |
| Amoebiasis | −0.54 | 0.58 | 0.02–19.24 | 0.76 |
| Training Cohort | Univariable Analysis | Multivariable Analysis | ||||
|---|---|---|---|---|---|---|
| HR | 95%CI | p Value | HR | 95%CI | p Value | |
| IPRS | 2.70 | 2.20–3.40 | <0.01 | 1.67 | 1.178–2.37 | <0.01 |
| Subtype | 0.86 | 0.64–1.20 | 0.31 | |||
| Claudin-low | 0.98 | 0.69–1.40 | 0.91 | |||
| Her2 | 1.32 | 0.95–1.83 | 0.10 | |||
| LumA | 1.18 | 0.82–1.70 | 0.38 | |||
| LumB | 1.31 | 0.92–1.88 | 0.13 | |||
| NC | 1.37 | 0.33–5.71 | 0.67 | |||
| Normal | 1.39 | 0.90–2.16 | 0.14 | |||
| ER status | 0.85 | 0.74–0.97 | 0.021 | 0.95 | 0.71–1.25 | 0.69 |
| HER2 status | 1.50 | 1.20–1.70 | <0.01 | 1.22 | 0.96–1.56 | 0.10 |
| Grade | 1.30 | 1.20–1.40 | <0.01 | 1.04 | 0.91–1.18 | 0.61 |
| PR status | 0.79 | 0.70–0.89 | <0.01 | 0.89 | 0.745–1.06 | 0.17 |
| Tumor size | 1.00 | 1–1 | <0.01 | 1.01 | 1.00–1.01 | <0.01 |
| Tumor stage | 1.80 | 1.60–2.00 | <0.01 | 1.50 | 1.31–1.72 | <0.01 |
| Pathways | NES | p Value | p Adjust |
|---|---|---|---|
| Hallmark Pathways | |||
| Hallmark interferon γ response | 1.75 | 0.0000153 | 0.0001091 |
| Hallmark G2M checkpoint | 1.71 | 0.0000597 | 0.0002987 |
| Hallmark inflammatory response | −1.73 | 0.0000195 | 0.0001217 |
| Hallmark P53 pathway | −1.70 | 0.0000593 | 0.0002987 |
| Hallmark apoptosis | −1.60 | 0.0007173 | 0.0032604 |
| Hallmark IL6 JAK STAT3 signaling | −1.73 | 0.0090204 | 0.0214772 |
| KEGG C2 Pathways | |||
| KEGG ECM receptor interaction | 1.76 | 0.0002955 | 0.0108816 |
| KEGG leukocyte transendothelial migration | −1.77 | 0.0002973 | 0.0108816 |
| KEGG WNT signaling pathway | −1.64 | 0.0003632 | 0.0110781 |
| KEGG B cell receptor signaling Pathway | −1.69 | 0.0016411 | 0.0250264 |
| KEGG JAK STAT signaling pathway | −1.59 | 0.0014223 | 0.0250264 |
| KEGG MAPK signaling pathway | −1.49 | 0.0009607 | 0.0250264 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Qiu, J.; Han, J.; Li, X.; Jiang, Y. Tumor Microenvironment Characterization in Breast Cancer Identifies Prognostic Pathway Signatures. Genes 2022, 13, 1976. https://doi.org/10.3390/genes13111976
Li J, Qiu J, Han J, Li X, Jiang Y. Tumor Microenvironment Characterization in Breast Cancer Identifies Prognostic Pathway Signatures. Genes. 2022; 13(11):1976. https://doi.org/10.3390/genes13111976
Chicago/Turabian StyleLi, Ji, Jiayue Qiu, Junwei Han, Xiangmei Li, and Ying Jiang. 2022. "Tumor Microenvironment Characterization in Breast Cancer Identifies Prognostic Pathway Signatures" Genes 13, no. 11: 1976. https://doi.org/10.3390/genes13111976