
Genomic Screening to Identify Food Trees Potentially Dispersed by Precolonial Indigenous Peoples


Abstract
:1. Introduction
2. Materials and Methods
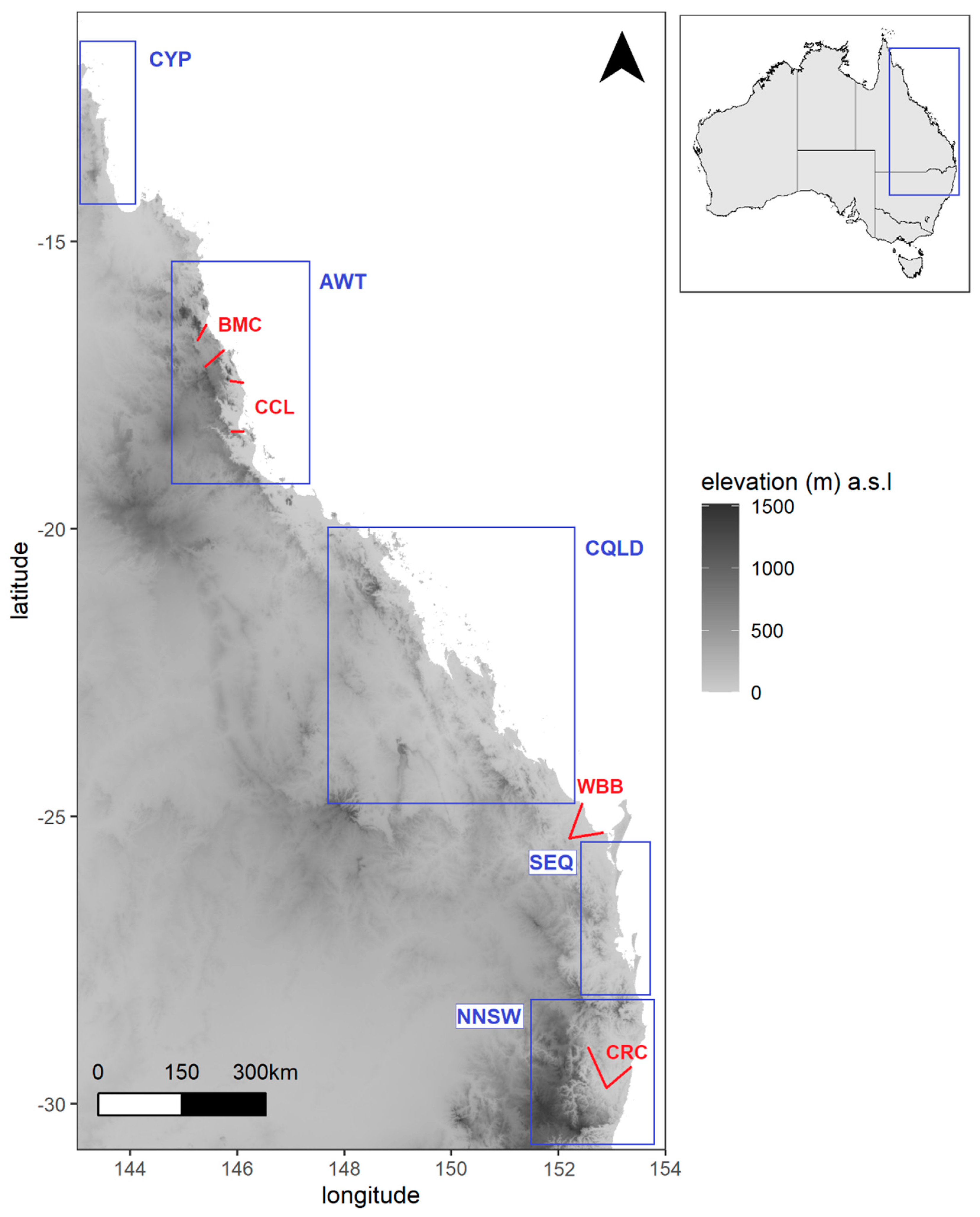
2.1. Study System
2.2. Study Design
2.3. Simulation of Hypothetical Dispersal Scenarios
2.4. Nuclear and Chloroplast Genomic Methods
2.5. Assessment of Fruit Traits and Genetic Connectivity
2.6. Genomic Tests of Dispersal Signals
3. Results
3.1. Fruit Traits and Genetic Connectivity
3.2. Simulation Study
3.3. Genomic Tests of Dispersal Signals
3.3.1. Large Fruit with Known History of Indigenous Use
3.3.2. Large Fruit with Unknown Indigenous Use
3.3.3. Small Fruit with Known History of Indigenous Use
3.3.4. Small Fruit with Unknown Indigenous Use
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Simulation Study to Compare the Genomic Signals of Hypothetical Indigenous versus Faunal-Mediated Dispersal Scenarios
Appendix A.1. Materials & Methods

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Migration Matrix according to Dispersal Scenarios | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Years | gen20 | gen40 | Source | Sink | m | Size | Growth Rate | fd | fd + exp | nd | hd1 | hd2 | hd3 | hd4 | hd5 | hd6 |
| 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 200 | 10 | 5 | 0 | 0 | 0 | 1 | −0.02 | - | - | - | 6 | 6 | - | - | 7 | - |
| 3999 | 199 | 99 | 0 | 0 | 0 | 1 | −0.02 | - | - | - | - | - | 6 | - | - | - |
| 4000 | 200 | 100 | 0 | 0 | 0 | 1 | −0.02 | - | - | - | - | - | - | - | - | 7 |
| 4999 | 249 | 124 | 0 | 1 | 1 | 1 | −0.02 | - | - | - | - | 6 | 2 | 2 | - | - |
| 5000 | 250 | 125 | 0 | 0 | 0 | 1 | −0.02 | 1 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 6000 | 300 | 150 | 0 | 1 | 1 | 1 | −0.02 | - | 1 | - | - | - | - | - | - | - |
| 9000 | 450 | 225 | 0 | 0 | 0 | 1 | −0.005 | 0 | 0 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 18,000 | 900 | 450 | 0 | 0 | 0 | 0.5 | 0.02 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 25,000 | 1250 | 625 | 0 | 0 | 0 | 1 | 0.005 | 3 | 3 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 40,000 | 2000 | 1000 | 0 | 0 | 0 | 1 | −0.005 | 0 | 0 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| 60,000 | 3000 | 1500 | 0 | 0 | 0 | 0.5 | 0.02 | 2 | 2 | - | 2 | 2 | 2 | 2 | 2 | 2 |
| 70,000 | 3500 | 1750 | 0 | 0 | 0 | 1 | −0.005 | 0 | 0 | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| 110,000 | 5500 | 2750 | 0 | 0 | 0 | 1 | −0.02 | 1 | 1 | 5 | 5 | 5 | 5 | 5 | 5 | 5 |
| Matrix | Migration | Nm | Dispersal Vector |
|---|---|---|---|
| 0 | Symmetric distance-weighted migration with barrier between deme2 and deme3 | 0.0005, 0.0002, 0.0000 | Volant fauna |
| 1 | High symmetric distance-weighted migration with no barrier | 0.0200, 0.0100, 0.0050, 0.0025, 0.0012 | Volant fauna |
| 2 | No migration | 0.0000 | NA |
| 3 | Low symmetric distance-weighted migration with barrier between deme2 and deme3 | 0.0025, 0.0012, 0.0000 | Volant fauna |
| 4 | Symmetric stepping-stone with barrier between deme2 and deme3 | 0.0050, 0.0000 | Megafauna |
| 5 | High symmetric stepping-stone migration with no barrier | 0.0200, 0.0000 | Megafauna |
| 6 | Low island migration model | 0.0025 | Human |
| 7 | Low asymmetric stepping-stone migration | 0.0025 | Human |
Appendix A.2. Results









References
- Ens, E.; Walsh, F.; Clarke, P. Aboriginal People and Australia’s Vegetation: Past and Current Interactions. In Australian Vegetation; Keith, D., Ed.; Cambridge University Press: Cambridge, UK, 2017. [Google Scholar]
- Anderson, M.; Keeley, J. Native Peoples’ Relationship to the California Chaparral. In Valuing Chaparral; Springer: Cham, Switzerland, 2018; pp. 79–121. ISBN 978-3-319-68302-7. [Google Scholar]
- Anderson, M.K.; Rosenthal, J. An Ethnobiological Approach to Reconstructing Indigenous Fire Regimes in the Foothill Chaparral of the Western Sierra Nevada. J. Ethnobiol. 2015, 35, 4–36. [Google Scholar] [CrossRef]
- Delcourt, P.A.; Delcourt, H.R.; Jones, E.L. Human Ecosystems in Eastern North America Since the Pleistocene; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Denham, T.; Donohue, M.; Booth, S. Horticultural experimentation in Northern Australia reconsidered. Antiquity 2009, 83, 634–648. [Google Scholar] [CrossRef]
- Ross, N.J. Modern tree species composition reflects ancient Maya “forest gardens” in northwest Belize. Ecol. Appl. 2011, 21, 75–84. [Google Scholar] [CrossRef]
- Abrams, M.D.; Nowacki, G.J. Native Americans as active and passive promoters of mast and fruit trees in the eastern USA. Holocene 2008, 18, 1123–1137. [Google Scholar] [CrossRef]
- Gammage, B. The Biggest Estate on Earth: How Aborigines Made Australia; Allen & Unwin: Crows Nest, NSW, Australia, 2011; ISBN 9781742377483. [Google Scholar]
- Denham, T.; Atchison, J.; Austin, J.; Bestel, S.; Bowdery, D.; Crowther, A.; Dolby, N.; Fairbairn, A.; Field, J.; Kennedy, A.; et al. Archaeobotany in Australia and New Guinea: Practice, potential and prospects. Aust. Archaeol. 2009, 68, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Pascoe, B. Dark Emu; Magabala Books: Broome, WA, Australia, 2014; ISBN 9781921248016. [Google Scholar]
- Hynes, R.A.; Chase, A.K. Plants, Sites and Domiculture: Aboriginal Influence upon Plant Communities in Cape York Peninsula. Archaeol. Ocean. 1982, 17, 38–50. [Google Scholar] [CrossRef]
- Warren, R.J. Ghosts of Cultivation Past—Native American Dispersal Legacy Persists in Tree Distribution. PLoS ONE 2016, 11, e0150707. [Google Scholar] [CrossRef]
- Silcock, J.L. Aboriginal Translocations: The Intentional Propagation and Dispersal of Plants in Aboriginal Australia. J. Ethnobiol. 2018, 28, 390–405. [Google Scholar] [CrossRef]
- Loeb, R.E. Evidence of Prehistoric Corn (Zea mays) and Hickory (Carya spp.) Planting in New York City: Vegetation History of Hunter Island, Bronx County, New York. J. Torrey Bot. Soc. 1998, 125, 74–86. [Google Scholar] [CrossRef]
- McLauchlan, K. Plant cultivation and forest clearance by prehistoric North Americans: Pollen evidence from Fort Ancient, Ohio, USA. Holocene 2003, 13, 557–566. [Google Scholar] [CrossRef]
- Anderson, M.K. Tending the Wild: Native American Knowledge and the Management of California’s Natural Resources; University of California Press: Oakland, CA, USA, 2005; ISBN 0520238567. [Google Scholar]
- Hastorf, C.A. The Cultural Life of Early Domestic Plant Use. Antiquity 1998, 72, 773–782. [Google Scholar] [CrossRef]
- Turner, N.J.; Ari, Y.; Berkes, F.; Davidson-Hunt, I.; Ertug, Z.F.; Miller, A. Cultural Management of Living Trees: An International Perspective. J. Ethnobiol. 2009, 29, 237–270. [Google Scholar] [CrossRef]
- Jezkova, T.; Jaeger, J.R.; Marshall, Z.L.; Riddle, B.R. Pleistocene impacts on the phylogeography of the Desert Pocket Mouse (Chaetodipus penicillatus). J. Mammal. 2009, 90, 306–320. [Google Scholar] [CrossRef] [Green Version]
- Keener, C.; Kuhns, E. The Impact of Iroquoian Populations on the Northern Distribution of Pawpaws in the Northeast. N. Am. Archaeol. 2005, 18, 327–342. [Google Scholar] [CrossRef]
- Bean, A.R. A new system for determining which plant species are indigenous in Australia. Aust. Syst. Bot. 2007, 20, 1–43. [Google Scholar] [CrossRef]
- Ross, N.J.; Stevens, M.H.H.; Rupiper, A.W.; Harkreader, I.; Leben, L.A. The ecological side of an ethnobotanical coin: Legacies in historically managed trees. Am. J. Bot. 2014, 101, 1618–1630. [Google Scholar] [CrossRef] [Green Version]
- Levis, C.; Costa, F.R.C.; Bongers, F.; Peña-Claros, M.; Clement, C.R.; Junqueira, A.B.; Neves, E.G.; Tamanaha, E.K.; Figueiredo, F.O.G.; Salomão, R.P.; et al. Persistent effects of pre-Columbian plant domestication on Amazonian forest composition. Science 2017, 355, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Clement, C.R.; de Cristo-Araújo, M.; d’Eeckenbrugge, G.C.; Pereira, A.A.; Picanço-Rodrigues, D. Origin and domestication of native Amazonian crops. Diversity 2010, 2, 72–106. [Google Scholar] [CrossRef] [Green Version]
- Moreira, P.A.; Aguirre-Dugua, X.; Mariac, C.; Zekraoui, L.; Couderc, M.; Rodrigues, D.P.; Casas, A.; Clement, C.R.; Vigouroux, Y. Diversity of Treegourd (Crescentia cujete) Suggests Introduction and Prehistoric Dispersal Routes into Amazonia. Front. Ecol. Evol. 2017, 5, 150. [Google Scholar] [CrossRef] [Green Version]
- Chaïr, H.; Traore, R.E.; Duval, M.F.; Rivallan, R.; Mukherjee, A.; Aboagye, L.M.; Van Rensburg, W.J.; Andrianavalona, V.; de Carvalho, M.A.A.P.; Saborio, F.; et al. Genetic Diversification and Dispersal of Taro (Colocasia esculenta (L.) Schott). PLoS ONE 2016, 11, e0157712. [Google Scholar] [CrossRef] [Green Version]
- Hughes, C.E.; Govindarajulu, R.; Robertson, A.; Filer, D.L.; Harris, S.A.; Bailey, C.D. Serendipitous backyard hybridization and the origin of crops. Proc. Natl. Acad. Sci. USA 2007, 104, 14389–14394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, J.; Sikora, M.; Garud, N.; Flowers, J.M.; Rubinstein, S.; Reynolds, A.; Huang, P.; Jackson, S.; Schaal, B.A.; Bustamante, C.D. Molecular evidence for a single evolutionary origin of domesticated rice. Proc. Natl. Acad. Sci. USA 2011, 108, 8351–8356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankin, A.; Altmüller, J.; Becker, C.; von Korff, M. Targeted resequencing reveals genomic signatures of barley domestication. New Phytol. 2018, 218, 1247–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrier, X.; De Langhe, E.; Donohue, M.; Lentfer, C.; Vrydaghs, L.; Bakry, F.; Carreel, F.; Hippolyte, I.; Horry, J.-P.; Jenny, C.; et al. Multidisciplinary perspectives on banana (Musa spp.) domestication. Proc. Natl. Acad. Sci. USA 2011, 108, 11311–11318. [Google Scholar] [CrossRef] [Green Version]
- Lauterjung, M.B.; Bernardi, A.P.; Montagna, T.; Candido-Ribeiro, R.; da Costa, N.C.F.; Mantovani, A.; dos Reis, M.S. Phylogeography of Brazilian pine (Araucaria angustifolia): Integrative evidence for pre-Columbian anthropogenic dispersal. Tree Genet. Genomes 2018, 14, 1–12. [Google Scholar] [CrossRef]
- Rossetto, M.; Ens, E.J.; Honings, T.; Wilson, P.D.; Yap, J.Y.S.; Costello, O.; Round, E.R.; Bowern, C. From Songlines to genomes: Prehistoric assisted migration of a rain forest tree by Australian Aboriginal people. PLoS ONE 2017, 12, e0186663. [Google Scholar] [CrossRef] [Green Version]
- Tomimatsu, H.; Kephart, S.R.; Vellend, M. Phylogeography of Camassia quamash in western North America: Postglacial colonization and transport by indigenous peoples. Mol. Ecol. 2009, 18, 3918–3928. [Google Scholar] [CrossRef]
- Lullfitz, A.; Byrne, M.; Knapp, L.; Hopper, S.D. Platysace (Apiaceae) of south-western Australia: Silent story tellers of an ancient human landscape. Biol. J. Linn. Soc. 2020, 130, 61–78. [Google Scholar] [CrossRef]
- Rangan, H.; Bell, K.L.; Baum, D.A.; Fowler, R.; McConvell, P.; Saunders, T.; Spronck, S.; Kull, C.A.; Murphy, D.J. New genetic and linguistic analyses show ancient human influence on baobab evolution and distribution in Australia. PLoS ONE 2015, 10, e0119758. [Google Scholar] [CrossRef] [Green Version]
- IUCN. Guidelines for Reintroductions and Other Conservation Translocations; IUCN: Gland, Switzerland, 2013. [Google Scholar]
- Kerkhove, R. The Great Bunya Gathering: Early Accounts; Ray Kerkhove: Sydney, QLS, Australia, 2012. [Google Scholar]
- Swan, D.K.; Deakin, U. Bunya Tukka Tracks: Investigating Traditional Travelling Routes of Eastern Australia. Master Thesis, Deakin University, Melbourne, VIC, Australia, 2017. [Google Scholar]
- Cosgrove, R.; Field, J.; Ferrier, Å. The archaeology of Australia’s tropical rainforests. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2007, 251, 150–173. [Google Scholar] [CrossRef]
- Jones, R.; Meehan, B. Plant Foods of the Gidjingali: Ethnographic and Archaeological Perspectives from Northern Australia on Tuber and Seed Exploitation. In Foraging and Farming: The Evolution of Plant Exploitation; Taylor & Francis Group: Milton Park, UK, 1989; ISBN 9781317598299. [Google Scholar]
- Rossetto, M.; Mcpherson, H.; Siow, J.; Kooyman, R.; van der Merwe, M.; Wilson, P.D. Where did all the trees come from? A novel multispecies approach reveals the impacts of biogeographical history and functional diversity on rain forest assembly. J. Biogeogr. 2015, 42, 2172–2186. [Google Scholar] [CrossRef]
- Rossetto, M.; Kooyman, R.; Yap, J.Y.S.; Laffan, S.W. From ratites to rats: The size of fleshy fruits shapes species’ distributions and continental rainforest assembly. Proc. R. Soc. B Biol. Sci. 2015, 282, 20151998. [Google Scholar] [CrossRef]
- David, B.; Arnold, L.J.; Delannoy, J.J.; Fresløv, J.; Urwin, C.; Petchey, F.; McDowell, M.C.; Mullett, R.; Mialanes, J.; Wood, R.; et al. Late survival of megafauna refuted for Cloggs Cave, SE Australia: Implications for the Australian Late Pleistocene megafauna extinction debate. Quat. Sci. Rev. 2021, 253, 106781. [Google Scholar] [CrossRef]
- Graham, C.H.; Moritz, C.; Williams, S.E. Habitat history improves prediction of biodiversity in rainforest fauna. Proc. Natl. Acad. Sci. USA 2006, 103, 632–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kooyman, R.; Rossetto, M.; Cornwell, W.; Westoby, M. Phylogenetic tests of community assembly across regional to continental scales in tropical and subtropical rain forests. Glob. Ecol. Biogeogr. 2011, 20, 707–716. [Google Scholar] [CrossRef]
- Mellick, R.; Lowe, A.; Rossetto, M. Consequences of long- and short-term fragmentation on the genetic diversity and differentiation of a late successional rainforest conifer. Aust. J. Bot. 2011, 59, 351–362. [Google Scholar] [CrossRef]
- Fahey, M.; Rossetto, M.; Wilson, P.D.; Ho, S.Y.W. Habitat preference differentiates the Holocene range dynamics but not barrier effects on two sympatric, congeneric trees (Tristaniopsis, Myrtaceae). Heredity 2019, 123, 532–548. [Google Scholar] [CrossRef]
- Moritz, C.; Hoskin, C.J.; MacKenzie, J.B.; Phillips, B.L.; Tonione, M.; Silva, N.; VanDerWal, J.; Williams, S.E.; Graham, C.H. Identification and dynamics of a cryptic suture zone in tropical rainforest. Proc. R. Soc. B Biol. Sci. 2009, 276, 1235–1244. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.; Moritz, C. Rainforest Refugia and Evolution in Australia’s Wet Tropics. Proc. Biol. Sci. 1999, 266, 191–196. [Google Scholar] [CrossRef] [Green Version]
- Edwards, R.D.; Crisp, M.D.; Cook, D.H.; Cook, L.G. Congruent biogeographical disjunctions at a continent-wide scale: Quantifying and clarifying the role of biogeographic barriers in the Australian tropics. PLoS ONE 2017, 12, e0174812. [Google Scholar] [CrossRef] [Green Version]
- Costion, C.M.; Lowe, A.J.; Rossetto, M.; Kooyman, R.M.; Breed, M.F.; Ford, A.; Crayn, D.M. Building a Plant DNA Barcode Reference Library for a Diverse Tropical Flora: An Example from Queensland, Australia. Diversity 2016, 8, 5. [Google Scholar] [CrossRef]
- Metcalfe, D.J.; Ford, A.J. Floristics and Plant Biodiversity of the Rainforests of the Wet Tropics; Blackwell Publishing: Hoboken, NJ, USA, 2008. [Google Scholar]
- Heslewood, M.M.; Lowe, A.J.; Crayn, D.M.; Rossetto, M. Contrasting levels of connectivity and localised persistence characterise the latitudinal distribution of a wind-dispersed rainforest canopy tree. Genetica 2014, 142, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Elmouttie, D.; Mather, P.B. Can rodents enhance germination rates in rainforest seeds? Ecol. Manag. Restor. 2012, 13, 203–207. [Google Scholar] [CrossRef]
- Hocknull, S.A.; Zhao, J.; Feng, Y.; Webb, G.E. Responses of Quaternary rainforest vertebrates to climate change in Australia. Earth Planet. Sci. Lett. 2007, 264, 317–331. [Google Scholar] [CrossRef]
- Rossetto, M.; Crayn, D.; Ford, A.; Mellick, R.; Sommerville, K. The influence of environment and life-history traits on the distribution of genes and individuals: A comparative study of 11 rainforest trees. Mol. Ecol. 2009, 18, 1422–1438. [Google Scholar] [CrossRef]
- Bradford, M.G.; Dennis, A.J.; Westcott, D.A. Diet and dietary preferences of the southern cassowary (Casuarius casuarius) in North Queensland, Australia. Biotropica 2008, 40, 338–343. [Google Scholar] [CrossRef]
- Dennis, A.J.; Westcott, D.A. Reducing complexity when studying seed dispersal at community scales: A functional classification of vertebrate seed dispersers in tropical forests. Oecologia 2006, 149, 620–634. [Google Scholar] [CrossRef]
- Clarke, P.A. Progress Report on the Anthropological Aspects of the Indigenous Plant Food Database; University of Adelaide: Adelaide, SA, Australia, 2018; Available online: https://theoranafoundation.org/projects/indigenous-food-database/ (accessed on 5 November 2021).
- Maiden, J.H. The Useful Native Plants of Australia; Trubner: London, UK, 1889. [Google Scholar]
- Roth, W.E. North Queensland Ethnography. In Food: Its Search, Capture, and Preparation; Home Secretary’ Departmant: Brisbane, QLD, Australia, 1901; Volume 3. [Google Scholar]
- Coyyan. The Aboriginals. In North Herreditary (The Tablelander); Columns I-X; National Library of Australia: Canberra, ACT, Australia, 1918. [Google Scholar]
- Dixon, R.M.W. Searching for Aboriginal Languages: Memoirs of a Field Worker; Cambridge University Press: Cambridge, UK, 1983. [Google Scholar]
- Birtles, T.G. Australia’s Ever-Changing Forests III. In Proceedings of the Third National Conference on Australian Forest History, Jervis, NSW, Australia, 24–27 November 1996; Dargavel, J., Ed.; The Australian National University Press: Canberra, ACT, Australia, 1996; pp. 169–187. [Google Scholar]
- Anderson, C. Traditional material culture of the Kuku-Yalanji of Bloomfield River, north Queensland. Rec. South Aust. Mus. 1996, 29, 63–83. [Google Scholar]
- Ferrier, Å.; Cosgrove, R. Aboriginal Exploitation of Toxic Nuts as a Late-Holocene Subsistence Strategy in Australia’s Tropical Rainforests. In Peopled Landscapes: Archaeological and Biogeographic Approaches to Landscapes; ANU Press: Acton, ACT, Australia, 2018. [Google Scholar]
- Maiden, J.H. Native food-plants. Agric. Gaz. N. S. W. 1900, 10, 117–130. [Google Scholar]
- Low, T. Bush Tucker. In Australia’s Wild Food Harvest; Angus & Robertson: Sydney, QLS, Australia, 1989. [Google Scholar]
- Field, J.; Cosgrove, R.; Fullagar, R.; Lance, B. Starch Residues on Grinding Stones in Private Collections: A Study of Morahs from the Tropical Rainforests of NE Queensland. In Archaeological Science under a Microscope: Studies in Residue and Ancient DNA Analysis in Honour of Thomas H. Loy; ANU Press: Sydney, QLS, Australia, 2009; pp. 103–120. [Google Scholar]
- Cribb, A.B.; Cribb, J.W. Wild Food in Australia; Fontana/Collins: Sydney, QLS, Australia, 1982. [Google Scholar]
- Ferrier, Å. Journeys into the Rainforest. Archaeology of Culture Change and Continuity on the Evelyn Tableland, North Queensland, 43rd ed.; Board, T.A.E., Ed.; ANU Press: Sydney, QLS, Australia, 2015; ISBN 9781925022872. [Google Scholar]
- Harris, D.R. The interplay of ethnographic and archaeological knowledge in the study of past human subsistence in the tropics. J. R. Anthropol. Inst. 2006, 12, S63–S78. [Google Scholar] [CrossRef]
- Cherikoff, V.; Isaacs, J. The Bush Food Handbook: How to Gather, Grow, Process and Cook Australian Wild Foods; Ti Tree Press: Sydney, QLS, Australia, 1989. [Google Scholar]
- Australia, G. Episode 28: Booderee Botanic Gardens; Australian Broadcasting Corporation: Sydney, QLS, Australia, 2014. [Google Scholar]
- Sansaloni, C.; Petroli, C.; Jaccoud, D.; Carling, J.; Detering, F.; Grattapaglia, D.; Kilian, A. Diversity Arrays Technology (DArT) and next-generation sequencing combined: Genome-wide, high throughput, highly informative genotyping for molecular breeding of Eucalyptus. BMC Proc. 2011, 5, 54. [Google Scholar] [CrossRef] [Green Version]
- Coissac, E. A de Novo Assembler Dedicated to Organelle Genome Assembling. 2016. Available online: https//pypi.python.org/pypi/ORG.asm/0.2.04 (accessed on 3 March 2021).
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Bradburd, G.S.; Ralph, P.L.; Coop, G.M. Disentangling the effects of geographic and ecological isolation on genetic differentiation. Evolution 2013, 67, 3258–3273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weir, B.S.; Hill, W.G. Estimating F-statistics. Annu. Rev. Genet. 2002, 36, 721–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; Mcglinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Package “vegan” Title Community Ecology Package. Community Ecol. Packag. 2019, 2, 1–295. [Google Scholar]
- Frichot, E.; Mathieu, F.; Trouillon, T.; Bouchard, G.; François, O. Fast and efficient estimation of individual ancestry coefficients. Genetics 2014, 196, 973–983. [Google Scholar] [CrossRef] [Green Version]
- Lawson, D.J.; van Dorp, L.; Falush, D. A tutorial on how not to over-interpret Structure and Admixture bar plots. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Leigh, J.W.; Bryant, D. Popart: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 9, 1110–1116. [Google Scholar] [CrossRef]
- Hopkins, M.S.; Graham, A.W. The viability of seeds of rainforest species after experimental soil burials under tropical wet lowland forest in north-eastern Australia. Aust. J. Ecol. 1987, 12, 97–108. [Google Scholar] [CrossRef]
- Excoffier, L.; Dupanloup, I.; Huerta-Sánchez, E.; Foll, M. Robust demographic inference from genomic and SNP data. PLoS Genet. 2013, 9, e1003905. [Google Scholar] [CrossRef] [Green Version]
- Excoffier, L.; Marchi, N.; Marques, D.A.; Matthey-Doret, R.; Gouy, A.; Sousa, V.C. fastsimcoal2: Demographic inference under complex evolutionary scenarios. Bioinformatics 2021, 37, 4882–4885. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, M.; Beaumont, L.; Das, S.; Yap, S. Bioclimatic Discordance: Combining Molecular and Environmental Data to Identify Floristic Refugia and Corridors. Final Report Prepared for the NSW Office of Environment and Heritage. April 2018, pp. 1–10. Available online: https://climatechange.environment.nsw.gov.au/.









| Dispersal Trait | Signal 1 | Signal 2 | Signal 3 | Signal 4 |
|---|---|---|---|---|
| Small fruit faunal dispersed | ✗ Low Fst values & IBD (long-term dispersal) | ✓ Admixture between sites (dispersal following long-term isolation) ✗ Homogeneity among sites (long-term dispersal) | ✓ Within-site outliers (recent LDD) ✗ No outliers (long-term dispersal) | ✗ Range-wide haplotype sharing (long-term dispersal) ✓ Single widespread haplotype (recent rapid dispersal) |
| Small fruit Indigenous dispersed | ✓ Low Fst values & absence of IBD (recent rapid dispersal) | ✓ Admixture between sites (dispersal following long-term isolation) ✗ Homogeneity among sites (long-term dispersal) | ✓ Within-site outliers (dispersal following long-term isolation) | ✗ Range-wide haplotype sharing (long-term dispersal) ✓ Single widespread haplotype (recent rapid dispersal) |
| Large fruit faunal dispersed | ✗ High Fst values with or without IBD (long-term isolation) | ✗ Structure across barriers (long-term isolation) | ✗ Differentiation amongst sites and no outliers (long-term isolation) | ✗ Haplotype divergence (long-term isolation) |
| Large fruit Indigenous dispersed | ✓ Low Fst values & absence of IBD (recent rapid dispersal) | ✓ Admixture between sites (dispersal following long-term isolation) | ✓ Within-site outliers (dispersal following long-term isolation) | ✓ Haplotype sharing between differentiated sites (dispersal following long-term isolation) ✓ Single widespread haplotype (recent rapid dispersal) |
| Family | Species | Common Names | Fruit Trait | Max. Fruit Width (mm) | Seed Number & Traits | nDNA Markers (SNPs) | cpDNA Sequence (bp) | Mantel score (p = 0.05) * Three Sites Only | Reported Indigenous Use |
|---|---|---|---|---|---|---|---|---|---|
| Study Species | |||||||||
| Fabaceae | C. australe | Moreton Bay chestnut, black bean, bean tree | LO | 45 | 3–5 L | 38,124 18,443 (north) 20,705 (south) | 0.67 (p = 0.04) AWT 0.43 (p = 0.18) NNSW | ‘Black bean was a staple food of many northern rainforest Aboriginal people and is still prepared and eaten today.’ (cited [59]). Ethnographic records of consumption by Indigenous peoples (AWT) [60,61,62,63,64]. Seed detoxification described in the AWT [65,66] and in NNSW/SEQ [32,67,68]. | |
| Lauraceae | Bielschmiedia bancroftii | Yellow walnut, yellow nut, Canary ash | LO | 75 × 62 | 1 L | 2080 | 108,132 | 0.36 (p = 0.33) AWT | Seed preparation described in the AWT [61]. Archaeological evidence of seed processing in the AWT [39,69]. |
| Lauraceae | Endiandra insignis | Hairy walnut | LF | 90 × 100 | 1 L | 13,913 | 106,112 | 0.99 (p = 0.17) AWT * | Seed preparation described in the AWT [61]. Bush tucker guide (AWT) [70]. Archaeological evidence of seed processing (AWT) [69]. |
| Sapotaceae | P. australis | Black apple, brush apple, wild plum, native plum | LF | 50 | 1–5 S | 24,873 | 86,899 | 0.63 (p = 0.17) NNSW * | Ethnographic records [67]. Bush tucker guide [70]. |
| Elaeocarpaceae | Elaeocarpus bancroftii | Kuranda quandong, ebony heart, nutwood, Johnstone River almond | LF | 55 × 40 | 1 L | 17,085 | 0.14 (p = 0.32) AWT | Ethnographic records [67,71]. Bush tucker guide [68,70]. Archaeological records of seed preparation [72]. | |
| Lauraceae | Endiandra compressa | LF | 71 × 60 | 1 L | 4025 | 107,869 | 0.91 (p = 0.33) AWT | ||
| Lauraceae | Endiandra globosa | Black walnut | LF | 60 × 60 | 1 L | 24,382 | 107,910 | 0.99 (p = 0.33) AWT | |
| Lauraceae | Endiandra pubens | Hairy walnut | LF | 75 × 75 | 1 L | 23,322 | 107,371 | 0.99 (p = 0.17) NNSW * | |
| Sapotaceae | Niemeyera prunifera | LF | 50 × 50 | 1 L | 22,778 | 84,279 | 0.91 (p = 0.12) AWT | ||
| Sapotaceae | N. whitei | LF | 20–50 | 1 L | 10,669 | 87,841 | 0.61 (p = 0.33) NNSW * | ||
| Elaeocarpaceae | E. johnsonii | Kuranda quandong | LO | 40 × 25 | 1 L | 1274 | 0.99 (p = 0.33) AWT * | Bush tucker guide described the seed as edible [73]. | |
| Elaeocarpaceae | Elaeocarpus grandis | Blue quandong, silver quandong, blue fig | SF | 33 × 33 | 1 L | 10,273 | 0.54 (p = 0.13) NBMC 0.13 (p = 0.35) SBMC 0.99 (p = 0.33) CQLD * | ‘You can eat the thin layer of flesh of the ripe purple-blue fruits when flesh is soft.’ (cited [59]). Bush tucker guide describes edible fruit [70]. | |
| Elaeocarpaceae | Elaeocarpus reticulatus | SF | 12 × 12 | 1 S | 14,731 | 0.56 (p < 0.01) NNSW | B. McLeod describes the fruit as “good bush tucker tea” that can be eaten raw or as a jam [74] (NB: reference is from outside of study area). | ||
| Sapotaceae | Pleioluma queenslandica | SF | 22 × 9 | 1 S | 15,270 | 85,895 | |||
| Lauraceae | Endiandra discolor | SF | 17 × 13 | 1 S | 23,081 | 107,031 | −0.05 (p = 0.42) NNSW | ||
| Fst Only | |||||||||
| Lauraceae | Endiandra introrsa | LF | 50 × 50 | 3461 | |||||
| Lauraceae | Bielschmiedia tooram | Brown walnut, Tooram walnut | LF | 55 × 35 | 3461 | Bush tucker guide describes edible fruit [70]. Bush tucker guide describes edible seed [73]. | |||
| Lauraceae | Bielschmiedia volckii | LF | 67 × 65 | 3461 | |||||
| Sapindaceae | Diploglottis australis | Native tamarind, tamarind tree, orange tamarind | SF | 15 | 4640 | 0.88 (p < 0.01) NNSW | Ethnographic sources [60,67] and bush tucker guide [70] describe the culinary properties of the fruit. | ||
| Lauraceae | Neolitsea dealbata | SF | 11 × 11 | 2881 | 0.91 (p < 0.01) NNSW | ||||
| Lauraceae | Cryptocaria glaucesens | SF | 15 × 18 | 14,970 | 0.89 (p < 0.01) NNSW | ||||
| Elaeocarpaceae | Sloanea australis | SF | 17 × 17 | 7429 | 0.59 (p < 0.01) NNSW | ||||
| Myrtaceae | Tristaniopsis laurina | Water gum, kanooka | W | 10 × 6 | 13,841 | 0.59 (p < 0.01) NNSW | |||
| Myrtaceae | Tristaniopsis collina | Mountain water gum | W | 10 × 6 | 10,721 | 0.82 (p < 0.01) NNSW | |||
| Cunoniaceae | Ceratopetalum apetalum | Coachwood | W | >8 | 659 | 0.75 (p < 0.01) NNSW | |||
| Species | Fruit >30 mm | Verified Indigenous Use | Signal 1 nDNA | Signal 2 nDNA | Signal 3 nDNA | Signal 4 cpDNA |
|---|---|---|---|---|---|---|
| C. australe (CYP/AWT) | ✓ | ✓ | ✗ | ✓ | ✗ | ✗ |
| C. australe (SEQ/NNSW) * | ✓ | ✓ | ✓ | ✓ | ✗ | ✗ |
| E. insignis * | ✓ | ✓ | ✗ | ✗ | ✗ | ✓ |
| B. bancroftii * | ✓ | ✓ | ✓ | ✗ | ✓ | ✓ |
| P. australis | ✓ | ✓ | ✗ | ✗ | ✓ | ✗ |
| E. bancroftii * | ✓ | ✓ | ✓ | ✓ | ✗ | |
| E. globosa (AWT) | ✓ | ✗ | ✗ | ✗ | ✓ | ✗ |
| E. globosa (NNSW) | ✓ | ✗ | ✓ | ✗ | ||
| E. compressa | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ |
| E. pubens | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ |
| N. prunifera * | ✓ | ✗ | ✗ | ✗ | ✗ | ✓ |
| N. whitei | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ |
| E. johnsonii | ✓ | ✗ | ✗ | ✗ | ✓ | |
| E. grandis | ✗ | ✓ | ✓ | ✗ | ✓ | |
| E. discolor | ✗ | ✗ | ✓ | ✓ | ✗ | ✓ |
| P. queenslandica | ✗ | ✗ | ✗ | ✓ | ✗ | ✓ |
| E. reticulatus | ✗ | ✗ | ✗ | ✗ | ✗ |
| Species | Dispersal Hypotheses | Follow Up Studies |
|---|---|---|
| C. australe |
|
|
| E. insignis |
|
|
| B. bancroftii |
|
|
| E. bancroftii |
|
|
| N. prunifera |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fahey, M.; Rossetto, M.; Ens, E.; Ford, A. Genomic Screening to Identify Food Trees Potentially Dispersed by Precolonial Indigenous Peoples. Genes 2022, 13, 476. https://doi.org/10.3390/genes13030476
Fahey M, Rossetto M, Ens E, Ford A. Genomic Screening to Identify Food Trees Potentially Dispersed by Precolonial Indigenous Peoples. Genes. 2022; 13(3):476. https://doi.org/10.3390/genes13030476
Chicago/Turabian StyleFahey, Monica, Maurizio Rossetto, Emilie Ens, and Andrew Ford. 2022. "Genomic Screening to Identify Food Trees Potentially Dispersed by Precolonial Indigenous Peoples" Genes 13, no. 3: 476. https://doi.org/10.3390/genes13030476