1. Introduction
The tightly packed monolayer of endothelial cells, which form the inner cellular lining of the blood and lymphatic vessels, is known as endothelium. Not only does it serve as a physical barrier between blood and the surrounding tissues [
1], the endothelium also regulates blood fluidity, vascular tone, inflammation, coagulation, and immunity [
2,
3]. Damage to the endothelium contributes to cardiovascular disease, pulmonary hypertension, pathological angiogenesis, thrombotic thrombocytopenic purpura, hepatic veno-occlusive disease, and severe sepsis [
4,
5,
6]. Recent studies on Coronavirus disease 2019 (COVID-19) demonstrate that damage to endothelium as a result of viral infection contributes to vascular inflammation, coagulopathy, and mortality [
7,
8]. Considering the significance for regulating normal physiological functions as well as a variety of pathological conditions, it is crucial to advance our knowledge on endothelial cell biology. In this regard, utilization of an ex vivo primary cell culture system is highly beneficial to better understand endothelial biology. Moreover, ex vivo systems facilitate controlled manipulation of cells throughout experimentation and could prove to be a noteworthy alternative to in vivo studies.
Since their isolation in the early 1970s, human umbilical vein endothelial cells (HUVECs) have been extensively used to understand vascular endothelial function and disease [
9]. HUVECs are derived from veins exclusive to the umbilical cord. This unique biological infrastructure connects a fetus to the mother’s blood supply via the placenta, suggesting HUVECs represent endothelial cells during embryonic stage. Importantly, HUVECs share multiple attributes such as: tube formation ability, angiogenesis, wound healing, similar molecular markers, and exhibit the same modal number of chromosomes observed in post-embryonic endothelial cells. As HUVEC cells are utilized for a myriad of research applications, comprehensive cytogenetic characterization of HUVEC provides a pivotal element for appropriate utilization of these cells for research.
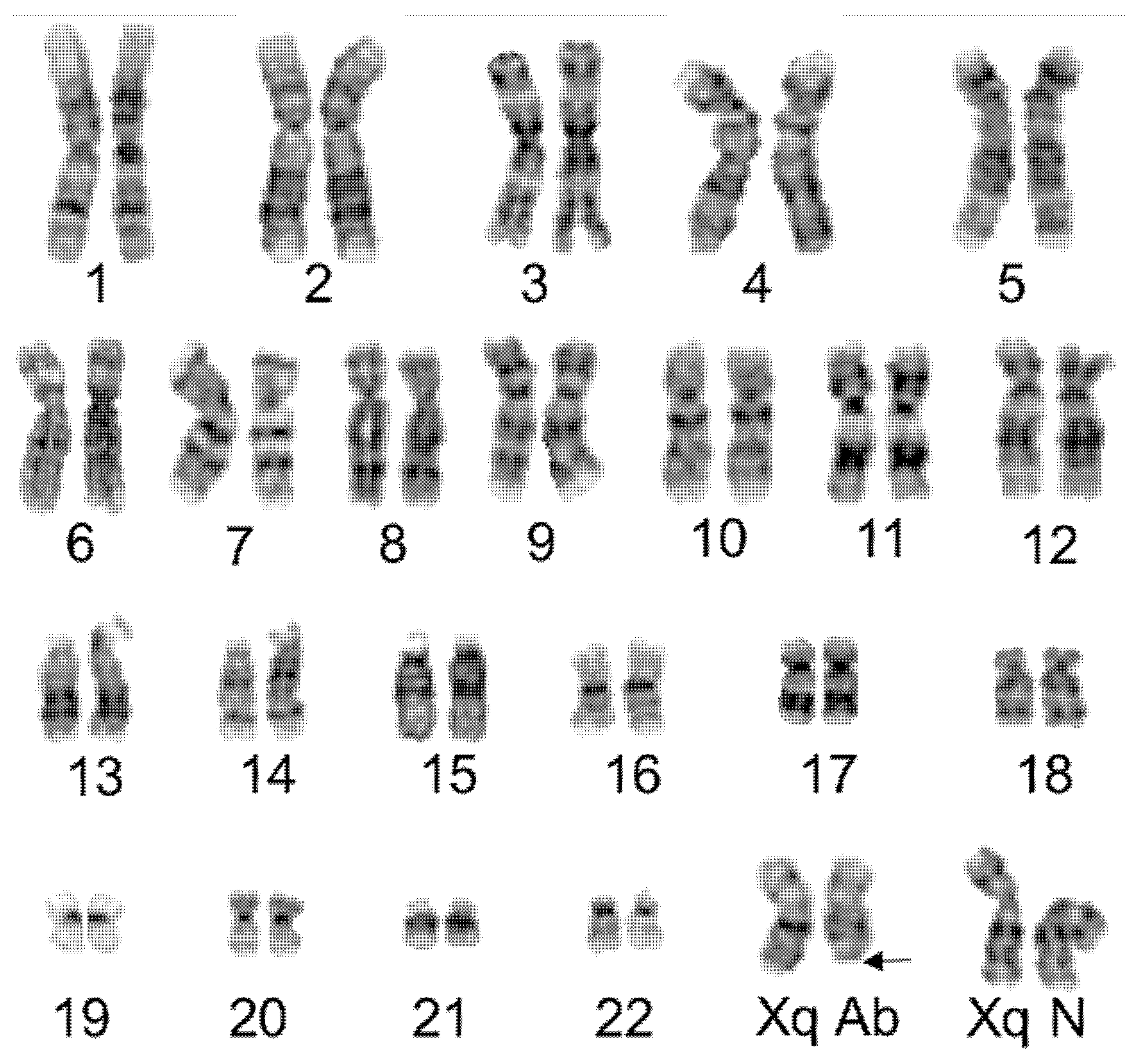
Conventional cytogenetic characterization has shown HUVECs to have a modal number of 46 when chromosomes are harvested during early culture passages [
10]. Previous studies with conventional cytogenetic techniques reveal that long-term culture of HUVECs results in spontaneous loss of chromosome 13 or partial gain of chromosome 11, suggesting that extended culture of HUVECs is prone to induce chromosomal aberrations [
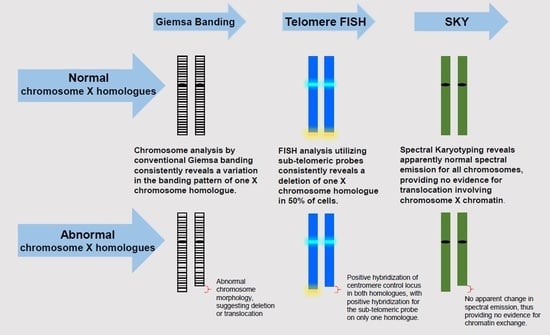
10]. However, characterization of HUVECs utilizing molecular cytogenetic methods during early culture passages has not been examined. In order to potentially identify cryptic chromosomal aberrations in HUVECs during earlier passages, we used conventional G-banding in conjunction with spectral karyotyping (SKY) and fluorescence in situ hybridization (FISH), as each technique, when used alone, poses limitations.
G-banding allows visualization of recognizable bands with alternating light (GC rich) versus dark (AT rich) staining intensities (individual bands are approximately ~5–10 × 106 base pairs of DNA), which are unique to each chromosome. G-banding serves the primary function of identifying aneuploidy and chromosomal rearrangements. While quite useful, this technique is limited by chromosome length and resolution of banding pattern. A low band level of resolution precludes the detection of rearrangements for some preparations, while allowing other types of aberrations to be more recognizable (pericentromeric inversions).
SKY can provide vital information to augment conventional G-band analysis. This molecular technique allows hybridization with various combinations of five fluorochromes, resulting in simultaneous identification of all chromosomes. Advanced analysis assigns pseudo colors to each chromosome based on the unique spectral emission of fluorochromes. Because SKY detects changes in a chromosome-specific manner, it facilitates identification of inter-chromosomal rearrangements, but does not reveal intrachromosomal rearrangements, such as interstitial deletions, duplications, or inversions.
FISH allows a more fine-tuned analysis by identifying locus- and/or region-specific structural chromosomal aberrations, including the sub-telomeric regions. The Vysis ToTelVysion probes specifically hybridize the p and q sub-telomeres of chromosomes 1 to 12 and 16 to 20; the q sub-telomeres of the acrocentric chromosomes (13, 14, 15, 21, and 22); and the Xp/Yp and Xq/Yq pseudo-autosomal region sub-telomeres (
Table 1). In addition, several of the ToTelVysion probes contain unique sequence locus-specific identifier probes or α satellite centromere enumeration probes (CEP) for identifying specific chromosomes within the mixtures (
Table 1). However, ToTelVysion probes allow analysis of only 64 specific target areas in the entire genome.
In the current study, we used G-banding, SKY, and the Vysis ToTelVysion FISH probe kit. Our studies reveal a novel cytogenetic alteration in primary HUVECs. In addition, this novel aberration is present in an immortal endothelial cell line derived by combining HUVECs with human adenocarcinoma cell line, suggesting this previously unidentified aberration is perhaps a characteristic feature of HUVECs. This novel discovery has potential to have formidable or tremendous impact for endothelial biology research.
2. Materials and Methods
2.1. Cell Culture
HUVEC cells (batch #0000560571) were purchased from Lonza (Walkersville, MD, USA) and cultured in 50/50 vascular cell basal medium (Cat # PCS-100-030, ATCC, Manassas, VA, USA) supplemented with endothelial cell growth kit-BBE (Cat # PCS-100-040, ATCC) and CHANG Medium D (Cat # 99404; FujiFilm Irvine Scientific, Santa Ana, CA, USA). EA.hy926 cells (ATCC) were cultured in Dulbecco’s Modified Eagle’s Medium (ATCC) supplemented with 10% Fetal Bovine Serum (ATCC) and 1% antibiotics. Cells were grown in a humidified incubator with 5% CO2 at 37 °C. Cells were subcultured every 2 to 3 d after detachment with a brief 0.25% trypsin-EDTA (Gibco, MA, USA) treatment. All cytogenetic experiments with HUVECs were performed by passage number 5.
2.2. Metaphase Chromosome Preparation
Metaphase chromosomes were prepared as previously described [
11,
12]. Semi-confluent cells were treated with KaryoMAX colcemid (Gibco, Waltham, MA, USA) at a final concentration of 75 ng/mL for 20 min to inhibit spindle fiber formation, thus arresting the cells in metaphase. The mitogen-containing media was removed, the cells were thoroughly rinsed with pre-warmed PBS with no Ca++ and Mg++ (Gibco) and treated with trypsin for detachment. All contents of the flask were collected into a 15 mL centrifuge tube and then centrifuged at 1000 rpm at room temperature for 5 min. The supernatant was carefully removed, the cell pellet was gently resuspended with a transfer pipet, 2 mL of prewarmed hypotonic solution (75 mM KCl; Gibco) was added in a dropwise manner. The final volume was brought to 6 mL and the tube was incubated in a water bath at 37 °C for 15 min. Cells were then fixed with room temperature fixative (3 to 1 volume of methanol and glacial acetic acid). Cells were washed an additional two times by repeating aspiration, fresh fixative resuspension, and centrifugation. Finally, metaphase spreads were prepared by dropwise application of cell suspension to clean slides at room temperature.
2.3. G-Banding
G-banded chromosomes were prepared as described elsewhere [
11,
12]. Slides were dried overnight at 66 °C and incubated with 0.025% trypsin for 1 min, gently rinsed with Tyrode’s buffer (Sigma, St. Louis, MO, USA), and stained with Giemsa solution (Sigma) for 5 min. Karyotype integrity and band resolution of metaphase chromosomes was determined according to the International System for Human Cytogenetic Nomenclature. Images were captured with Zeiss Imazer.Z2 microscope equipped with GenASIs Case Data Manager system, version 7.2.2.40970 (ASI, Carlsbad, CA, USA) for the analysis of karyotypes. At least 30 G-banded metaphase spreads were photographed and analyzed for each cell type.
2.4. FISH Hybridization
The ToTelVysion kit (Vysis Inc./Abbott Molecular Laboratories, Abbott Park, IL, USA) consists of 15-probe cocktails, to demark the sub-telomere regions of all human chromosomes.
Table 1 describes the probe cocktails. Briefly, before FISH hybridization, slides were dipped in 2× SSC (Sigma) at room temperature for 5 min, then dehydrated in 70%, 80%, and 100% ethanol, respectively, for 2 min each. Slides were then air dried, placed in pre-warmed denature solution (70% formamide (Millipore, Temecula, CA, USA) in 2× SSC) at 71 °C for 1 min, then immediately dehydrated in a cold ethanol series (70%, 85%, 100%) for 2 min each. The appropriate measurement for each probe mixture was individually transferred to clean microcentrifuge tubes. All probes were prewarmed to 37 °C for 5 min, then placed in a water bath preheated to 75–77 °C for 5 min for denaturation. Each denatured probe mixture was applied to respective target area of cells, protected with a coverslip and sealed with rubber cement. Slides were protected from light in a humidified chamber and allowed to hybridize overnight at 37 °C. The following day, hybridized slides were subject to the following wash series pre-warmed to 45 °C: Formamide wash (50% formamide in 2× SSC) series of three—10 min each, followed by a series of two Coplin jars containing 2× SSC—5 min each. Slides were dipped in distilled water, immediately air dried, counterstained with DAPI, and protected with a coverslip. At least 4 cells from each hybridization area were photographed and analyzed for a total of 60 cells (GenASIs software, ASI, Carlsbad, CA, USA).
2.5. Spectral Karyotyping
The SKY kit from Applied Spectral Imaging (ASI) was used for this study. Slides were pre-soaked in 2× SSC solution and dehydrated in 70%, 85%, and 100% ethanol for 10 min each. Following air dry the slides were placed in pre-warmed denature solution (70% formamide (Millipore, Temecula, CA, USA) in 2× SSC) at 71 °C for 1 min, and then instantly dehydrated in a cold ethanol series (70%, 85%, and 100%) for 2 min each. Meanwhile, SKY probe was pre-warmed to 37 °C for 5 min and then denatured in a water bath at 80–81 °C for 7 min. Then, 10 μL of denatured SKY probe was added to the target area of the slide, covered with a 22 × 22-mm coverslip, and carefully sealed with rubber cement. Slides were incubated in a humidified chamber for 48 h at 37 °C for probe hybridization. Hybridized slides were washed with a series pre-warmed to 45 °C: Formamide wash (50% formamide in 2× SSC) series of three—5 min each, followed by a series of two Coplin jars containing 1× SSC—5 min each. Once removed from the 1× SSC solution, the slide was allowed to drain and 100 μL of blocking reagent was applied to the target area and protected with a plastic coverslip. Meanwhile, Cy5 antibody solution (ASI) was reconstituted with filtered 4× SSC. Following a 30 min incubation at 37 °C, the coverslip was carefully removed, Cy5 antibody solution was applied to the target area, protected with a fresh plastic coverslip, and returned to 37 °C incubator. Following 1 h of hybridization, the slide was subject to wash a series (prewarmed at 45 °C) consisting of 3 jars containing 4× SSC with 0.1% Tween-20, 5 min each. Meanwhile, Cy5.5 antibody solution (ASI) was reconstituted with filtered 4× SSC. Upon completion of the wash series, the slide was briefly dipped into distilled water to remove detergent residue, and Cy5.5 antibody solution was applied to the target area and protected with a fresh plastic coverslip. The incubation and 4× SSC with 0.1% Tween-20 wash steps were repeated, as was the brief dip in distilled water. Slides were counterstained with DAPI and covered with a clean glass coverslip. SKY images were captured using an SD200 Spectracube (ASI) mounted on a Zeiss Imager.Z2 microscope. DAPI images were captured and then inverted and enhanced with SKY View software (ASI) to produce G-band–like patterns on the axis of each chromosome. At least 30 SKY images were captured under 63× magnification for spectral analysis. Spectral imaging combines fluorescence microscopy, CCD-imaging and Fourier spectroscopy to enable simultaneous visualization for the entire spectrum at all image points, thereby assigning a unique color to each chromosome.
2.6. Successive Rehybridization of Slides
To prepare the slide for repeat hybridization, it was soaked in 4× SSC with 1% tween solution for 10 min, briefly rinsed in distilled water and placed into a clean Coplin jar of freshly prepared 4× SSC with 1% tween solution for an additional 10 min. The slide was gently and thoroughly rinsed in distilled water to remove any traces of detergent. It was then dehydrated in 70%, 85%, and 100% ethanol for 10 min each.
4. Discussion
Freshly isolated primary endothelial cells from a healthy individual exhibit a normal chromosome pattern during early passages as detected by conventional cytogenetic techniques. However, endothelial cells have a tendency to accrue numerical and/or structural cytogenetic abnormalities following long-term ex vivo culture [
13]. Moreover, cytogenetic alterations are common in aged individuals in comparison to their younger counterparts [
14]. An earlier longitudinal study of human age-related chromosomal analysis showed that in vitro aging of adult endothelial cells results in a gain of chromosome 11 (trisomy 11), and that incidence of trisomy 11 proportionately increases over the time [
13]. Similarly, a study by Zhang demonstrated that long-term culture of HUVECs results in a loss of chromosome 13 and 16, and a partial gain of chromosome 11 [
10]. These studies clearly suggest that aging is a major predisposition factor for increased frequency of chromosomal aberrations; however, the nature of chromosomal aberrations is dependent upon the origin of endothelial cells. However, these studies exclusively used the conventional G-banding method, which typically yields 350–550 bands per haploid set with each band representing ~5–10 × 10
6 base pairs of DNA. Identification of aberrations below this resolution is a major limitation of conventional cytogenetic techniques. Further, systematic analysis with relative high-resolution cytogenetic techniques have not been used in identifying chromosomal aberrations in HUVECs during early passages.
Molecular cytogenetic techniques allow identification of chromosomal aberrations at the level of 100 to 400 kilobase pairs. We used conventional and molecular cytogenetic techniques (SKY and FISH) to identify chromosomal aberrations in HUVECs during as early as passage five. We, for the first time, confirmed an Xq terminal deletion in half of the HUVECs. Terminal ends of each chromosome are protected by telomeres, which are comprised of repetitive DNA sequences. Telomeres play a crucial role in maintaining chromosomal integrity. Damage to the telomere or reduction of telomere length occurs gradually as dividing cells lose some part of the telomere with each replicative cycle. Telomere shortening/loss is a major contributing factor for numerical chromosomal aberrations in various cell types, including endothelial cells [
14]. Although we identified the Xq terminal deletion, we did not observe any numerical aberrations in HUVECs, nor did we observe integration of this deleted Xq region into any other chromosome (i.e., translocation). We have not investigated the functional implications of the Xq deleted clone in endothelial cells. Notably, a deletion of Xq in peripheral blood lymphocytes contributes to abnormalities of menstruation and infertility in females [
15]. Others have demonstrated that Xq deletion is associated with ovarian dysfunction [
16,
17]. Thus, our findings have a strong clinical relevance.
As primary cells, HUVECs have a finite lifespan and limited expansion capacity. Moreover, HUVECs, like all other primary cells, require special nutrients for their optimum growth and expansion, which are absent in most markedly available basal media. Various labs have used different growth media for HUVEC culture [
9,
18,
19,
20,
21]. We found mixing of endothelial growth and Chang medium D in equal volume provides optimum culture condition for HUVECs. Considering the limitations of primary endothelial cell culture, an immortal endothelial cell line, called EA.hy926, was generated by fusing HUVECs with A549 lung cancer cells [
22]. Because we observed a terminal deletion of Xq in half of the HUVECs, and considering that HUVECs are one of the parental cell types of EA.hy926, we hypothesized whether EA.hy926 cells could possibly carry the same Xq terminal deletion. Surprisingly, we observed all EA.hy926 cells show a terminal deletion of Xq, suggesting that the HUVEC clone carrying the Xq terminal deletion was involved in generation of EA.hy926.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}