
Revealing Genetic Differences in Fiber Elongation between the Offspring of Sea Island Cotton and Upland Cotton Backcross Populations Based on Transcriptome and Weighted Gene Coexpression Networks

 ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Sample Collection
2.2. Determination of Fiber Quality Phenotypes
2.3. RNA Extraction, Library Construction, and Sequencing
2.4. Data and Differential Gene Expression Analysis
2.5. WGCNA Construction and Hub-Gene Screening
2.6. qRT–PCR Validated Transcriptome Sequencing
3. Results
3.1. Fiber Phenotypes of Extreme Offspring of Fiber Length
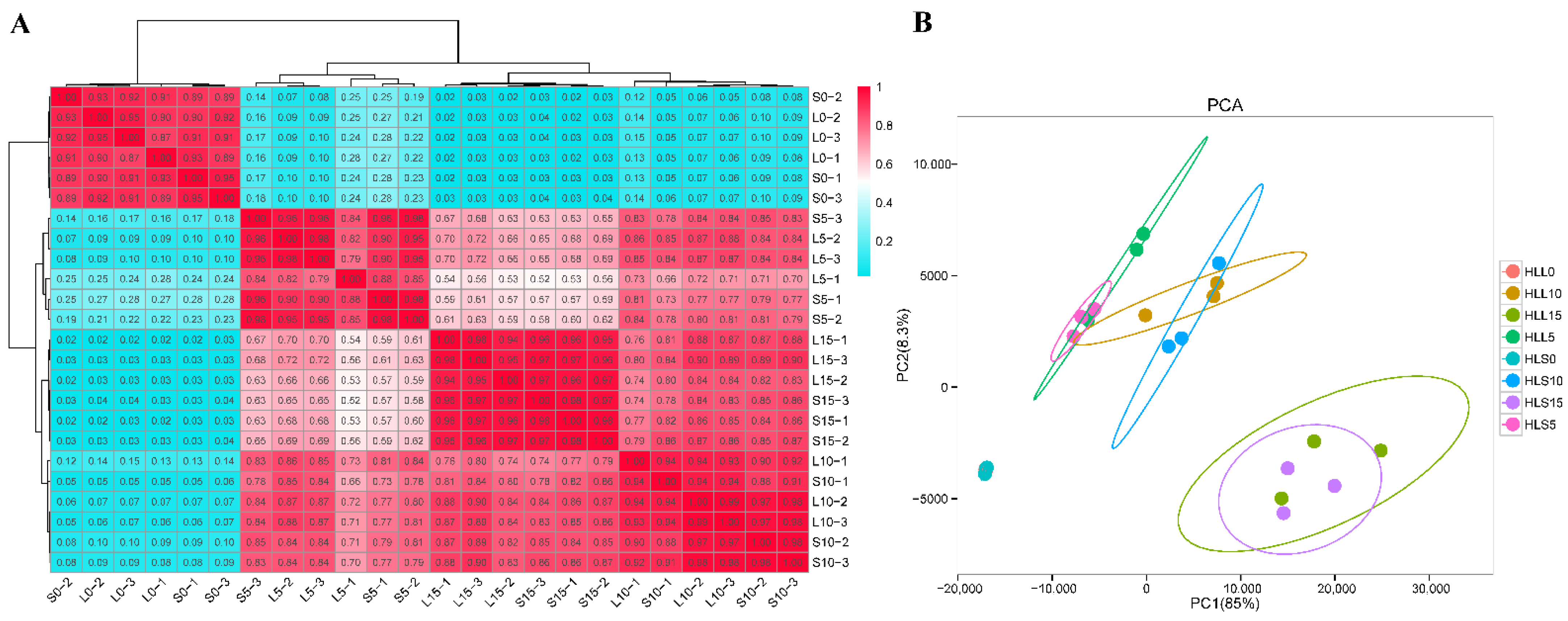
3.2. Overall Analysis of the Transcriptome during Cotton Fiber Development
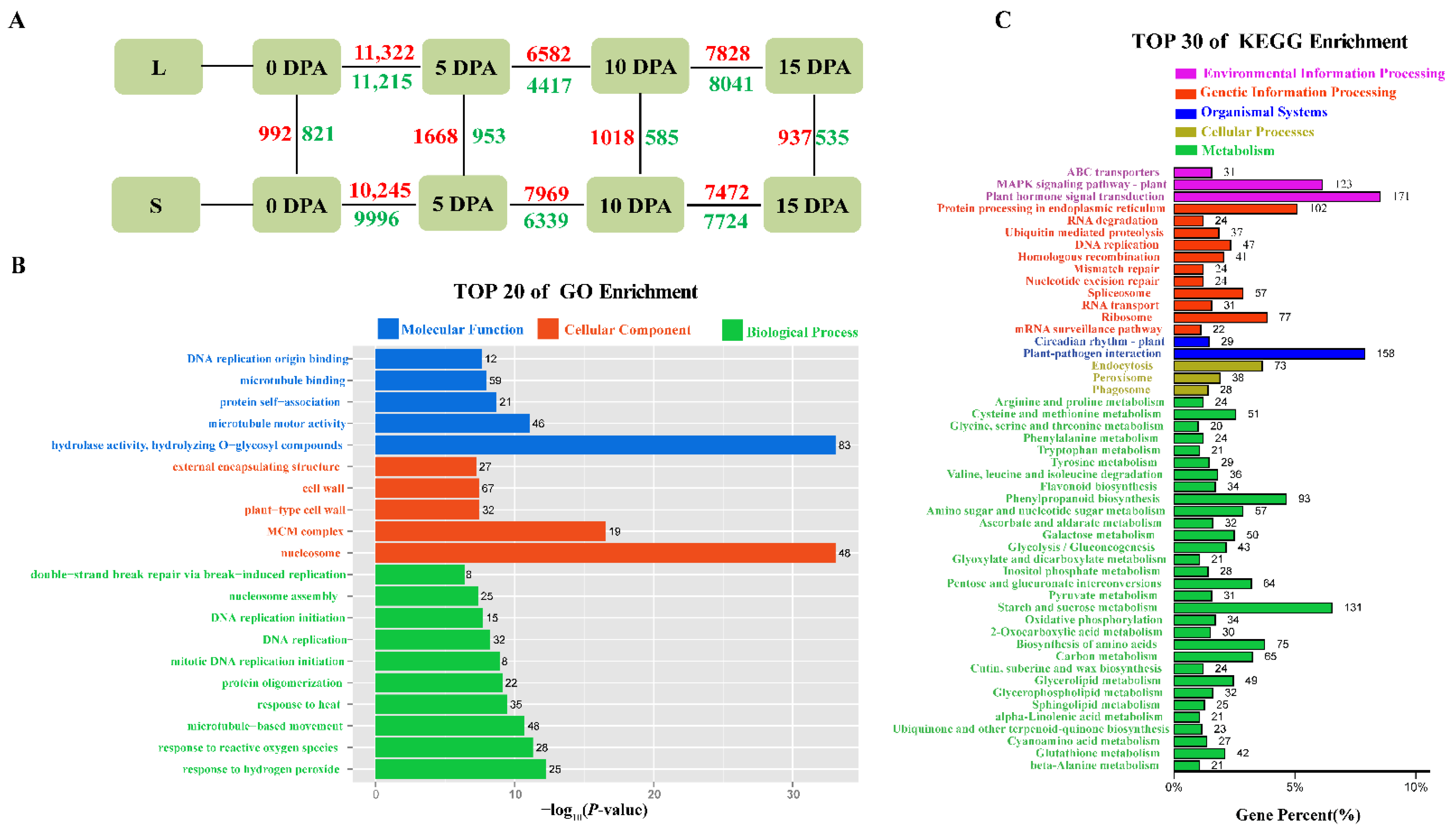
3.3. Analysis of Differentially Expressed Genes (DEGs)
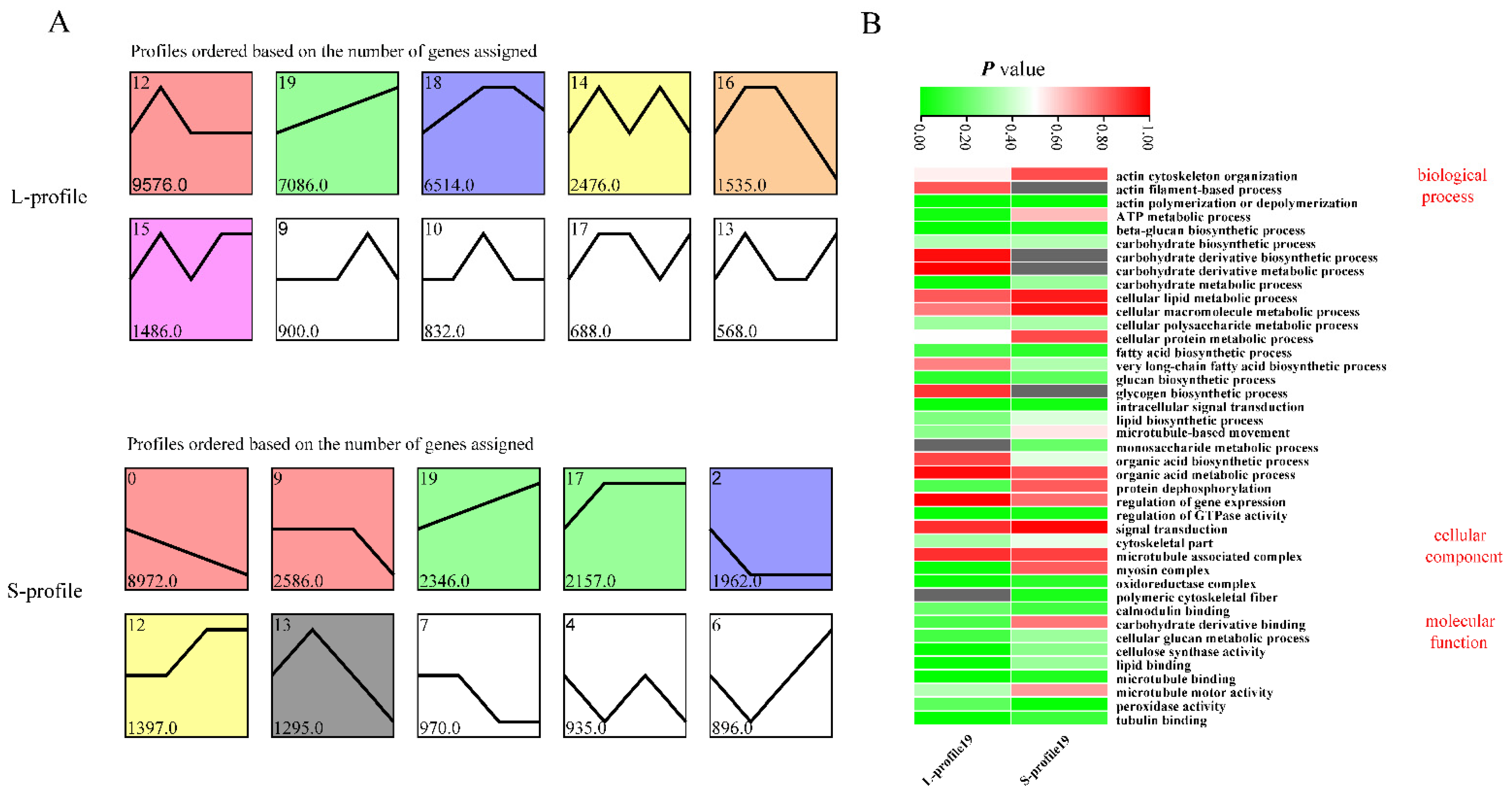
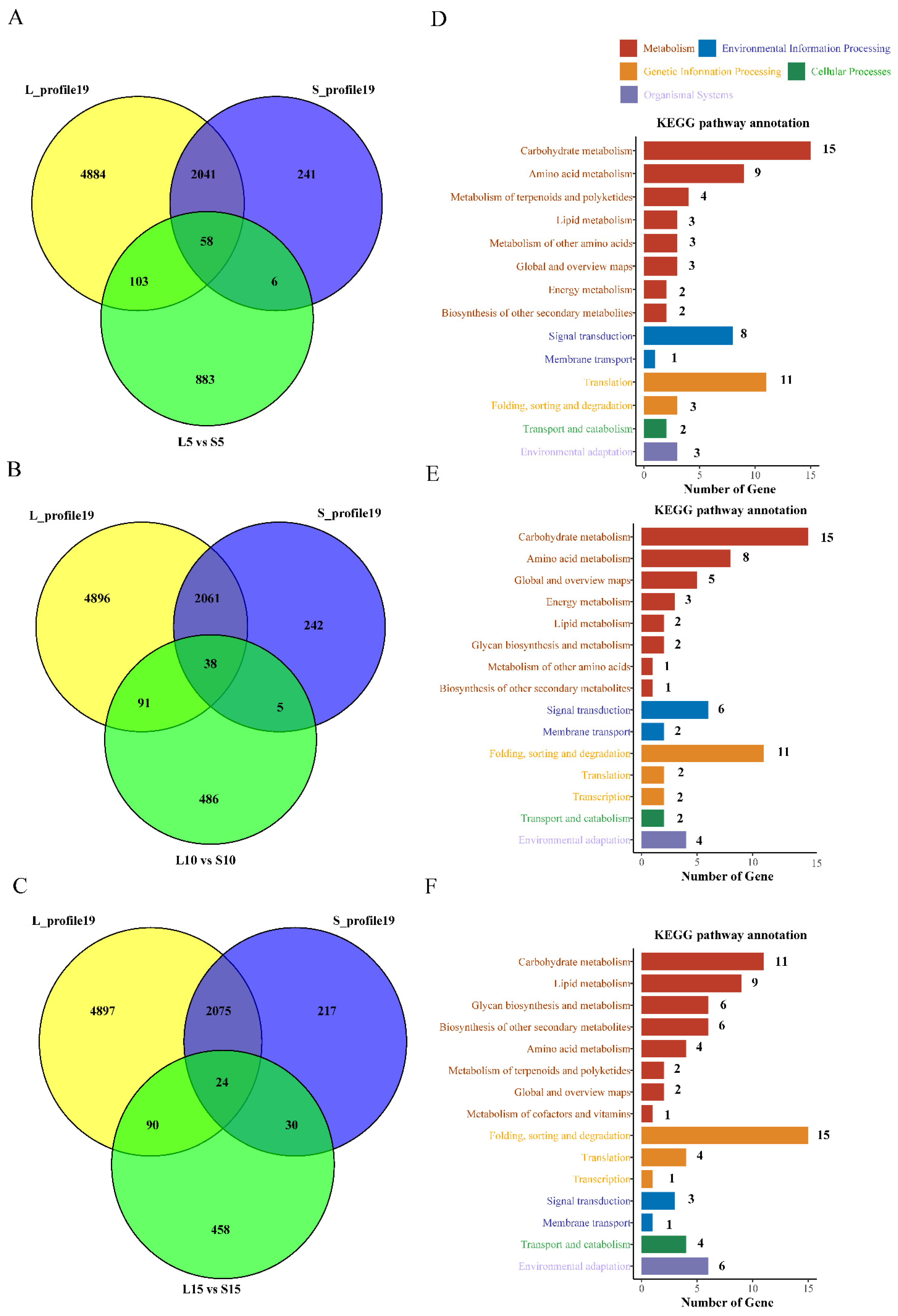
3.4. Analysis of Gene Expression Patterns
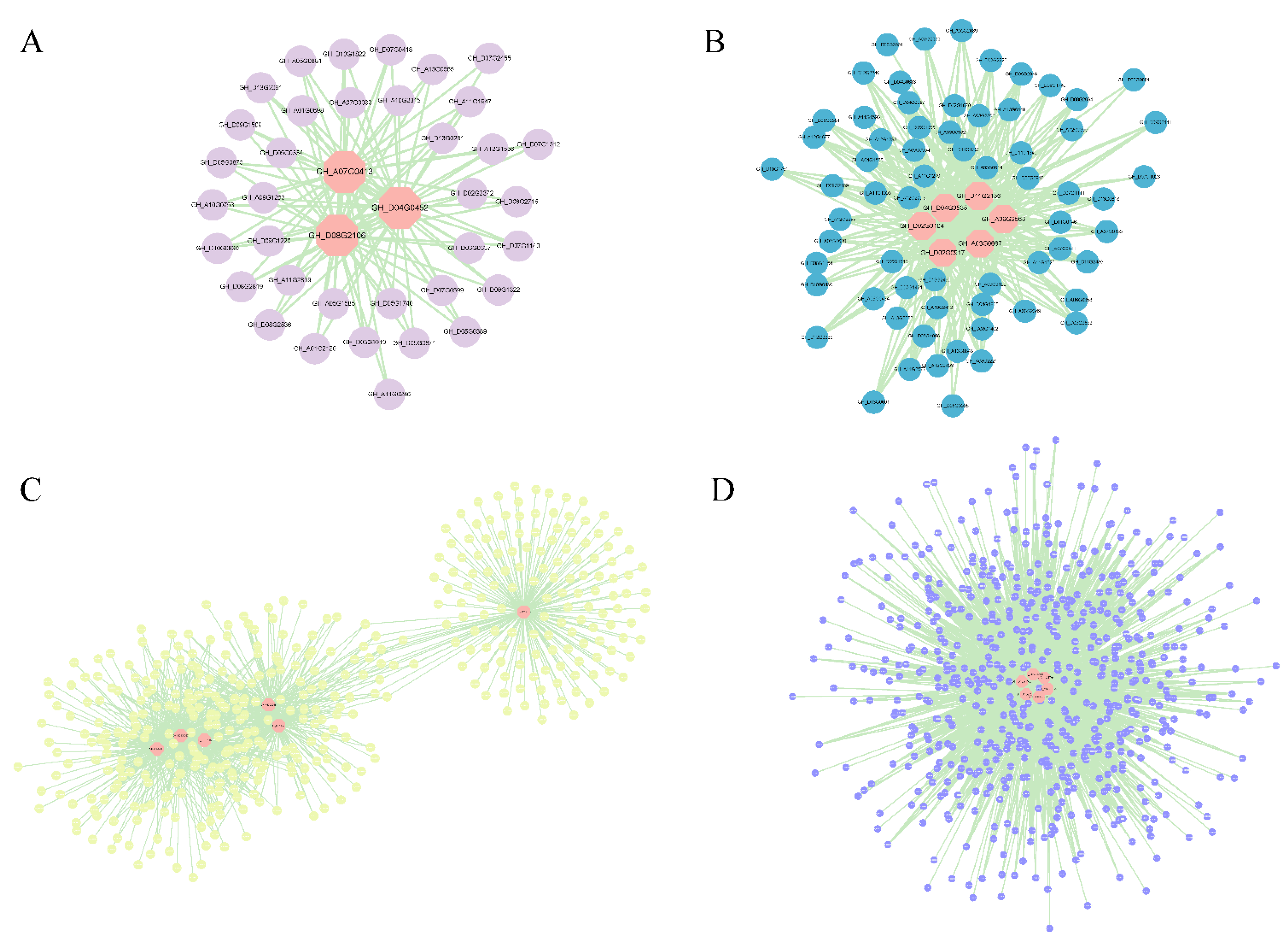
3.5. Coexpression Network Analysis and Identification of Hub Genes
4. Discussion
4.1. Transcriptome Sequencing of Backcross Offspring Provides New Insights to Explore the Expression Profile of the Fiber Elongation Stage
4.2. DEGs Reveal Transcriptional Differences in the Elongation Stage
4.3. The DEGs Identified by STEM May Have a Significant Effect on Fiber Length
4.4. The Hub Gene Identified by WGCNA May Have a Significant Effect on Fiber Length
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Wendel, J.F.; Cronn, R.C. Polyploidy and the evolutionary history of cotton. Adv. Agron. 2003, 78, 139–186. [Google Scholar] [CrossRef] [Green Version]
- Fryxell, P.A.; Stewart, L.A.C.M. A revision of Gossypium sect. Grandicalyx (Malvaceae), including the description of six new species. Syst. Bot. 1992, 17, 91–114. [Google Scholar] [CrossRef]
- Huang, G.; Huang, J.Q.; Chen, X.Y.; Zhu, Y.X. Recent advances and future perspectives in cotton research. Annu. Rev. Plant Biol. 2021, 72, 437–462. [Google Scholar] [CrossRef] [PubMed]
- Li, F.G.; Fan, G.Y.; Lu, C.R.; Xiao, G.H.; Zou, C.S.; Kohel, R.J.; Ma, Z.Y.; Shang, H.H.; Ma, X.F.; Wu, J.Y.; et al. Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biotechnol. 2015, 33, 524–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.Z.; Hu, Y.; Jiang, W.K.; Fang, L. sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.J.; Tu, L.L.; Yuan, D.J.; Zhu, D.; Shen, C.; Li, J.Y.; Liu, F.Y.; Pei, L.L.; Wang, P.C.; Zhao, G.N.; et al. Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense. Nat. Genet. 2019, 51, 224–229. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Chen, J.D.; Fang, L.; Zhang, Z.Y.; Ma, W.; Niu, Y.C.; Ju, L.Z.; Deng, J.Q.; Zhao, T.; Lian, J.M.; et al. Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat. Genet. 2019, 51, 739–748. [Google Scholar] [CrossRef] [Green Version]
- Huang, G.; Wu, Z.G.; Percy, R.G.; Bai, M.Z.; Zhu, Y.X. Genome sequence of Gossypium herbaceum and genome updates of Gossypium arboreum and Gossypium hirsutum provide insights into cotton A-genome evolution. Nat. Genet. 2020, 52, 516–524. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.Z.; Li, W.T.; Li, A.G.; Ge, R.H.; Zhang, B.C. Constructing a high-density linkage map for Gossypium hirsutum × Gossypium barbadense and identifying QTLs for lint percentage. J. Integr. Plant Biol. 2014, 57, 450–467. [Google Scholar] [CrossRef]
- Said, I.J.; Song, M.Z.; Wang, H.T.; Lin, Z.X.; Zhang, X.L. A comparative meta-analysis of QTL between intraspecific Gossypium hirsutum and interspecific G. hirsutum × G. barbadense populations. Mol. Genet. Genom. 2015, 290, 1003–1025. [Google Scholar] [CrossRef]
- Liu, X.; Zhao, B.; Zheng, H.; Hu, Y.; Lu, G.; Yang, C.; Chen, J.; Chen, J.; Chen, D.; Zhang, L.; et al. Gossypium barbadense genome sequence provides insight into the evolution of extra-long staple fiber and specialized metabolites. Sci. Rep. 2015, 5, 14139. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.J.; Tang, Z.H.; Wang, M.J.; Gao, W.H.; Tu, L.L.; Jin, X.; Chen, L.L.; He, Y.H.; Zhang, L.; Zhu, L.; et al. The genome sequence of Sea-Island cotton (Gossypium barbadense) provides insights into the allopolyploidization and development of superior spinnable fibres. Sci. Rep. 2015, 5, 17662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, M. A view of urgency in improving cotton quality from “the supply side”. China Fiber Insp. 2016, 2, 44–45. [Google Scholar] [CrossRef]
- Yang, X.L. Genetic Map Construction and QTL Mapping of Fiber Quality in Allotetraploid Cotton. Ph.D. Thesis, Hebei Agricultural University, Hebei, China, 2009. [Google Scholar]
- Paterson, A.H.; Saranga, Y.; Menz, M.; Jiang, C.X.; Wright, R.J. QTL analysis of genotype x environment interactions affecting cotton fiber quality. Theor. Appl. Genet. 2003, 106, 384–396. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, J.; Ma, J.; Tang, S.; Liu, D.; Teng, Z.; Liu, D.; Zhang, Z. Genetic mapping and quantitative trait locus analysis of fiber quality traits using a three-parent composite population in upland cotton (Gossypium hirsutum L.). Mol. Breed. 2012, 29, 335–348. [Google Scholar] [CrossRef]
- Wang, Y.; Ning, Z.; Hu, Y.; Chen, J.; Zhao, R.; Chen, H.; Ai, N.; Guo, W.; Zhang, T.; Yin, T. Molecular Mapping of Restriction-Site Associated DNA Markers in Allotetraploid Upland Cotton. PLoS ONE 2015, 10, e124781. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Jin, X.; Zhang, B.; Shen, C.; Lin, Z. Enrichment of an intraspecific genetic map of upland cotton by developing markers using parental RAD sequencing. DNA Res. 2015, 22, 147–160. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J. Cotton Fiber Growth in planta and in vitro. Models for plant cell elongation and cell wall biogenesis. Plant Physiol. 2001, 127, 1361–1366. [Google Scholar] [CrossRef]
- Gao, W.; Saha, S.; Ma, D.P.; Guo, Y.; Stelly, D.M. A cotton-fiber-associated cyclin-dependent kinase a gene: Characterization and chromosomal location. Int. J. Plant Genom. 2012, 2012, 613812. [Google Scholar] [CrossRef]
- Liu, B.; Zhu, Y.; Zhang, T. The R3-MYB gene GhCPC negatively regulates cotton fiber elongation. PLoS ONE 2015, 10, e0116272. [Google Scholar] [CrossRef] [Green Version]
- Basra, A.S.; Malik, C.P. Development of the cotton fiber. Int. Review Cytol. 1984, 89, 65–113. [Google Scholar] [CrossRef]
- Zhang, S.; Jiang, Z.; Chen, J.; Han, Z.; Chi, J.; Li, X.; Yu, J.; Xing, C.; Song, M.; Wu, J.; et al. The cellulose synthase (CesA) gene family in four Gossypium species: Phylogenetics, sequence variation and gene expression in relation to fiber quality in Upland cotton. Mol. Genet. Genom. 2021, 296, 355–368. [Google Scholar] [CrossRef]
- Lee, J.J.; Woodward, A.W.; Chen, Z.J. Gene expression changes and early events in cotton fibre development. Ann. Bot.-Lond. 2007, 100, 1391–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansoor, S.; Paterson, A.H. Genomes for jeans: Cotton genomics for engineering superior fiber. Trends Biotechnol. 2012, 30, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Shang, H.; Ge, Q.; Zou, C.; Cai, J.; Wang, D.; Fan, S.; Zhang, Z.; Deng, X.; Tan, Y.; et al. Genome-wide identification, phylogeny, and expression analysis of pectin methylesterases reveal their major role in cotton fiber development. BMC Genom. 2016, 17, 1000. [Google Scholar] [CrossRef] [Green Version]
- Haigler, C.H.; Betancur, L.; Stiff, M.R.; Tuttle, J.R. Cotton fiber: A powerful single-cell model for cell wall and cellulose research. Front. Plant Sci. 2012, 3, 104. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Gong, J.; Zhang, J.; Zhang, Z.; Shi, Y.; Li, J.; Liu, A.; Gong, W.; Ge, Q.; Deng, X.; et al. Quantitative trait loci and transcriptome analysis reveal genetic basis of fiber quality traits in CCRI70 RIL population of Gossypium hirsutum. Front. Plant Sci. 2021, 12, 753755. [Google Scholar] [CrossRef]
- Li, H.; Peng, Z.; Yang, X.; Wang, W.; Fu, J.; Wang, J.; Han, Y.; Chai, Y.; Guo, T.; Yang, N.; et al. Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat. Genet. 2013, 45, 43–50. [Google Scholar] [CrossRef]
- Li, A.; Xia, T.; Xu, W.; Chen, T.; Peng, L. An integrative analysis of four CESA isoforms specific for fiber cellulose production between Gossypium hirsutum and Gossypium barbadense. Planta 2013, 237, 1585–1597. [Google Scholar] [CrossRef]
- Razzaq, A.; Zafar, M.M.; Ali, A.; Hafeez, A.; Sharif, F.; Guan, X.; Deng, X.; Pengtao, L.; Shi, Y.; Haroon, M.; et al. The pivotal role of major chromosomes of sub-genomes A and D in fiber quality traits of cotton. Front. Genet. 2022, 12, 642595. [Google Scholar] [CrossRef]
- Shang, X.; Zhu, L.; Duan, Y.; He, Q.; Zhao, M.; Yu, Y.; Guo, W. An Easy and rapid transformation protocol for transient expression in cotton fiber. Front. Plant Sci. 2022, 13, 837994. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Sun, Z.; Zhang, Y.; Ke, H.; Yang, J.; Li, Z.; Wu, L.; Zhang, G.; Wang, X.; Ma, Z. Development and utilization of functional kompetitive allele-specific PCR markers for key genes underpinning fiber length and strength in Gossypium hirsutum L. Front. Plant Sci. 2022, 13, 853827. [Google Scholar] [CrossRef]
- Prasad, P.; Khatoon, U.; Verma, R.K.; Aalam, S.; Kumar, A.; Mohapatra, D.; Bhattacharya, P.; Bag, S.K.; Sawant, S.V. Transcriptional landscape of cotton fiber development and its alliance with fiber-associated traits. Front. Plant Sci. 2022, 13, 811655. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Wang, W.; Jiang, K.; Grover, C.E.; Cheng, C.; Pan, Z.; Zhao, C.; Zhu, J.; Li, D.; Wang, M.; et al. A calmodulin-like gene (GbCML7) for fiber strength and yield improvement identified by resequencing core accessions of a pedigree in Gossypium barbadense. Front. Plant Sci. 2022, 12, 815648. [Google Scholar] [CrossRef]
- Jiang, Y.; Guo, W.; Zhu, H.; Ruan, Y.L.; Zhang, T. Overexpression of GhSusA1 increases plant biomass and improves cotton fiber yield and quality. Plant Biotechnol. J. 2012, 10, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Bing, X.; Gou, J.Y.; Li, F.G.; Shangguan, X.X.; Bo, Z.; Yang, C.Q.; Wang, L.J. A cotton BURP domain protein interacts with α-expansin and their co-expression promotes plant growth and fruit production. Mol. Plant 2013, 6, 945–958. [Google Scholar] [CrossRef] [Green Version]
- Li, D.D.; Ruan, X.M.; Zhang, J.; Wu, Y.J.; Wang, X.L. Cotton plasma membrane intrinsic protein 2s (PIP2s) selectively interact to regulate their water channel activities and are required for fibre development. New Phytol. 2013, 199, 695–707. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, Z.; Li, M.; Wei, X.; Li, X.; Li, B.; Li, X. Cotton (Gossypium hirsutum) 14-3-3 proteins participate in regulation of fibre initiation and elongation by modulating brassinosteroid signalling. Plant Biotechnol. J. 2015, 13, 269–280. [Google Scholar] [CrossRef]
- Liu, Z.H.; Chen, Y.; Wang, N.N.; Chen, Y.H.; Wei, N.; Lu, R.; Li, Y.; Li, X.B. A basic helix-loop-helix protein (GhFP1) promotes fibre elongation of cotton (Gossypium hirsutum) by modulating brassinosteroid biosynthesis and signalling. New Phytol. 2020, 225, 2439–2452. [Google Scholar] [CrossRef]
- Xiao, G.; Zhao, P.; Zhang, Y. A pivotal role of hormones in regulating cotton fiber development. Front. Plant Sci. 2019, 10, 87. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.M.; Hu, C.Y.; Pang, Y.; Kastaniotis, A.J.; Hiltunen, J.K.; Zhu, Y.X. Saturated very-long-chain f atty acids promote cotton fiber and Arabidopsis cell elongation by activating ethylene biosynthesis. Plant Cell Online 2007, 19, 3692–3704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salih, H.; He, S.; Li, H.; Peng, Z.; Du, X. Investigation of the EIL/EIN3 Transcription factor gene family members and their expression levels in the early stage of cotton fiber development. Plants 2020, 9, 128. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Tu, L.; Ye, Z.; Wang, M.; Gao, W.; Zhang, X. A cotton fiber-preferential promoter, PGbEXPA2, is regulated by GA and ABA in Arabidopsis. Plant Cell Rep. 2015, 34, 1539–1549. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.H.; Hinchliffe, D.J.; Triplett, B.A.; Jeffrey, C.Z.; Stelly, D.M.; Yeater, K.M.; Moon, H.S.; Gilbert, M.K.; Thyssen, G.N.; Turley, R.B. Phytohormonal networks promote differentiation of fiber initials on pre-anthesis cotton ovules grown in vitro and in planta. PLoS ONE 2015, 10, e125046. [Google Scholar] [CrossRef]
- Niu, Q.; Tan, K.; Zang, Z.; Xiao, Z.; Luo, M. Modification of phytosterol composition influences cotton fiber cell elongation and secondary cell wall deposition. BMC Plant Biol. 2019, 19, 208. [Google Scholar] [CrossRef]
- Zeng, J.; Zhang, M.; Hou, L.; Bai, W.; Yan, X.; Hou, N.; Wang, H.; Huang, J.; Zhao, J.; Pei, Y. Cytokinin inhibits cotton fiber initiation by disrupting PIN3a-mediated asymmetric accumulation of auxin in the ovule epidermis. J. Exp. Bot. 2019, 70, 3139–3151. [Google Scholar] [CrossRef]
- Xiao, G.H.; Wang, K.; Huang, G.; Zhu, Y.X. Genome-scale analysis of the cotton KCS gene family revealed a binary mode of action for gibberellin A regulated fiber growth. J. Integr. Plant Biol. 2016, 58, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Chen, L.; Qiu, X.; Wei, J.; Lu, H.; Sun, G.; Ma, X.; Yang, Z.; Zhu, C.; Hou, Y. AKR2A participates in the regulation of cotton fibre development by modulating biosynthesis of very-long-chain fatty acids. Plant Biotechnol. J. 2020, 18, 526–539. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.; Cheng, H.; Wang, Q.; Lv, L.; Zhang, Y.; Song, G.; Zuo, D. Identification of the cesA subfamily and functional analysis of GhMCesA35 in Gossypium hirsutum L. Genes 2022, 13, 292. [Google Scholar] [CrossRef]
- Zou, X.; Zhen, Z.; Ge, Q.; Fan, S.; Liu, A.; Gong, W.; Li, J.; Gong, J.; Shi, Y.; Wang, Y. Genome-wide identification and analysis of the evolution and expression patterns of the cellulose synthase gene superfamily in Gossypium species. Gene 2017, 646, 28–38. [Google Scholar] [CrossRef]
- Hu, H.; Zhang, R.; Tang, Y.; Peng, C.; Peng, L. Cotton CSLD3 restores cell elongation and cell wall integrity mainly by enhancing primary cellulose production in the Arabidopsis cesa6 mutant. Plant Mol. Biol. 2019, 101, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.M.; Zuo, D.Y.; Wang, X.F.; Zhang, Y.P.; Cheng, H.L.; Wang, Q.L.; Song, G.L.; Ma, Z.Y. Analysis and expression of GhEXPs related to fiber development in Gossypium hirsutum L. Cott. Sci. 2021, 33, 280–290. [Google Scholar]
- Pandey, D.K.; Chaudhary, B. Transcriptional loss of domestication-driven cytoskeletal GhPRF1 gene causes defective floral and fiber development in cotton (Gossypium). Plant Mol. Biol. 2021, 107, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.M.; Hu, C.Y.; Zhu, Y.X. The ascorbate peroxidase regulated by H2O2 and ethylene is involved in cotton fiber cell elongation by modulating ROS homeostasis. Plant Signal. Behav. 2014, 3, 194–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, X.; Li, X.; Wang, L.; Zeng, J.; Huang, L.; Xiong, L.; Song, S.; Zhao, J.; Hou, L.; Wang, F.; et al. Sucrose enhanced reactive oxygen species generation promotes cotton fibre initiation and secondary cell wall deposition. Plant Biotechnol. J. 2021, 19, 1092–1094. [Google Scholar] [CrossRef] [PubMed]
- Shangguan, X.; Yang, Q.; Wu, X.; Cao, J. Function analysis of a cotton R2R3 MYB transcription factor GhMYB3 in regulating plant trichome development. Plant Biol. 2021, 23, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, W.; Ren, Z.; Wang, X.; Liu, J.; Yang, Z.; Zhao, J.; Pei, X.; Liu, Y.; He, K. Glucose regulates cotton fiber elongation by interacting with brassinosteroid. J. Exp. Bot. 2022, 73, 711–726. [Google Scholar] [CrossRef]
- Feng, H.; Tian, X.; Liu, Y.; Li, Y.; Zhang, X.; Jones, B.J.; Sun, Y.; Sun, J. Analysis of flavonoids and the flavonoid structural genes in brown fiber of upland cotton. PLoS ONE 2013, 8, e58820. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.; Tu, L.; Deng, F.; Hu, H.; Nie, Y.; Zhang, X. A genetic and metabolic analysis revealed that cotton fiber cell development was retarded by flavonoid naringenin. Plant Physiol. 2013, 162, 86–95. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Yan, Q.; Ding, H.; Luo, M.; Hou, L.; Zhang, M.; Yao, D.; Liu, H.S.; Li, X.; Zhao, J. Transcriptome and biochemical analyses revealed a detailed proanthocyanidin biosynthesis pathway in brown cotton fiber. PLoS ONE 2014, 9, e86344. [Google Scholar] [CrossRef]
- Lu, Q.; Li, P.; Yang, R.; Xiao, X.; Li, Z.; Wu, Q.; Gong, J.; Ge, Q.; Liu, A.; Du, S. QTL mapping and candidate gene prediction for fiber yield and quality traits in a high-generation cotton chromosome substitution line with Gossypium barbadense segments. Mol. Genet. Genom. 2022, 297, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Du, W.; Yu, W.; Zhang, W.; Huang, F.; Cheng, H.; Yu, D. Genome-wide association analysis discovered new loci and candidate genes associated with low-phosphorus tolerance based on shoot mineral elements concentrations in soybean. Mol. Genet. Genom. 2022, 297, 843–858. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.H.; Qi, S.Y.; Yuan, M.; Zhao, D.Y.; Zhang, D.J.; Feng, J.Y.; Wang, J.N.; Li, W.; Song, C.X.; Wang, T.L.; et al. A genome-wide association study revealed the genetic variation and candidate genes for grain copper content in bread wheat (Triticum aestivum L.). Food Funct. 2022, 13, 5177–5188. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Li, X.; Gouker, F.; Hong, C. cDNA Transcriptome of Arabidopsis reveals various defense priming pnduced by a broad-spectrum biocontrol agent Burkholderia sp. SSG. Int. J. Mol. Sci. 2022, 23, 3151. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Du, J.; Ge, X.; Cao, D.; Hu, J. Leaf Size Development differences and comparative transcriptome analyses of two poplar genotypes. Genes 2021, 12, 1775. [Google Scholar] [CrossRef] [PubMed]
- Basnet, P.; Um, T.; Roy, N.S.; Cho, W.S.; Park, S.C.; Park, K.; Choi, I. Identification and characterization of key genes responsible for weedy and cultivar growth types in soybean. Front. Genet. 2022, 13, 805347. [Google Scholar] [CrossRef]
- Bundó, M.; Martín-Cardoso, H.; Pesenti, M.; Gómez-Ariza, J.; Castillo, L.; Frouin, J.; Serrat, X.; Nogués, S.; Courtois, B.; Grenier, C.; et al. Integrative approach for precise genotyping and transcriptomics of salt tolerant introgression rice lines. Front. Plant Sci. 2022, 12, 797141. [Google Scholar] [CrossRef]
- Kiseleva, A.A.; Bragina, M.K.; Muterko, A.F.; Salina, E.A. Functional characterization of genes with daily expression patterns in common wheat. Plant Mol. Biol. 2022, 109, 135–146. [Google Scholar] [CrossRef]
- Wang, K.; Wang, D.; Zheng, X.; Qin, A.; Zhou, J.; Guo, B.; Chen, Y.; Wen, X.; Ye, W.; Zhou, Y.; et al. Multi-strategic RNA-seq analysis reveals a high-resolution transcriptional landscape in cotton. Nat. Commun. 2019, 10, 4714. [Google Scholar] [CrossRef] [Green Version]
- Bowman, M.J.; Park, W.; Bauer, P.J.; Udall, J.A.; Page, J.T.; Raney, J.; Scheffler, B.E.; Jones, D.C.; Campbell, B.T. RNA-Seq transcriptome profiling of upland cotton (Gossypium hirsutum L.) root tissue under water-deficit stress. PLoS ONE 2013, 8, e82634. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Xiao, X.; Gong, J.; Li, P.; Zhao, Y.; Feng, J.; Peng, R.; Shi, Y.; Yuan, Y. Identification of candidate cotton genes associated with fiber length through quantitative trait loci mapping and RNA-sequencing using a chromosome segment substitution line. Front. Plant Sci. 2021, 12, 796722. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Sun, H.; Hao, P.; Wang, H.; Wang, C.; Ma, L.; Wei, H.; Yu, S. Transcriptome analysis reveals differences in the mechanisms of fiber initiation and elongation between long- and short-fiber cotton (Gossypium hirsutum L.) lines. BMC Genom. 2019, 20, 633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waititu, J.K.; Zhang, X.; Chen, T.; Zhang, C.; Zhao, Y.; Wang, H. Transcriptome analysis of tolerant and susceptible maize genotypes reveals novel insights about the molecular mechanisms underlying drought responses in leaves. Int. J. Mol. Sci. 2021, 22, 6980. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Gao, L.; Liu, X.; Zhang, X.; Wang, X.; Wang, Z. Comparative transcriptome analysis of fiber and nonfiber tissues to identify the genes preferentially expressed in fiber development in Gossypium hirsutum. Sci. Rep. 2021, 11, 22833. [Google Scholar] [CrossRef]
- Sun, S.; Xiong, X.; Zhu, Q.; Li, Y.; Sun, J. Transcriptome sequencing and metabolome analysis reveal genes involved in pigmentation of green-colored cotton fibers. Int. J. Mol. Sci. 2019, 20, 4838. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Myat, A.A.; Liang, C.; Meng, Z.; Guo, S.; Wei, Y.; Sun, G.; Wang, Y.; Zhang, R. Insights into MicroRNA-mediated regulation of flowering time in cotton through small RNA sequencing. Front. Plant Sci. 2022, 13, 761244. [Google Scholar] [CrossRef]
- Jiang, X.; Fan, L.; Li, P.; Zou, X.; Zhang, Z.; Fan, S.; Gong, J.; Yuan, Y.; Shang, H. Co-expression network and comparative transcriptome analysis for fiber initiation and elongation reveal genetic differences in two lines from upland cotton CCRI70 RIL population. PeerJ 2021, 9, e11812. [Google Scholar] [CrossRef]
- Feng, X.; Liu, S.; Cheng, H.; Zuo, D.; Zhang, Y.; Wang, Q.; Lv, L.; Song, G. Weighted gene co-expression network analysis reveals hub genes contributing to fuzz development in Gossypium arboreum. Genes 2021, 12, 753. [Google Scholar] [CrossRef]
- Zhang, G.; Zhao, Z.; Ma, P.; Qu, Y.; Sun, G.; Chen, Q. Integrative transcriptomic and gene co-expression network analysis of host responses upon verticillium dahliae infection in Gossypium hirsutum. Sci. Rep. 2021, 11, 20586. [Google Scholar] [CrossRef]
- Jia, X.; Wang, H.; Pang, C.; Ma, Q.; Su, J.; Wei, H.; Song, M.; Fan, S.; Yu, S. QTL delineation for five fiber quality traits based on an intra-specific Gossypium hirsutum L. recombinant inbred line population. Mol. Genet. Genom. 2018, 293, 831–843. [Google Scholar] [CrossRef]
- Li, P.; Wang, M.; Lu, Q.; Ge, Q.; Rashid, M.H.O.; Liu, A.; Gong, J.; Shang, H.; Gong, W.; Li, J.; et al. Comparative transcriptome analysis of cotton fiber development of upland cotton (Gossypium hirsutum) and chromosome segment substitution lines from G. hirsutum × G. barbadense. BMC Genom. 2017, 18, 705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Jia, G. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florea, L.; Song, L.; Salzberg, S.L. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Research 2013, 2, 188. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Mccarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Biogeosciences 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Mccarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A new and powerful approach to multiple testing. J. R. Statist. Soc. B 1995, 57, 289–300. [Google Scholar]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Silva, J.C.D.; Oliveira, G.H.B.S.; Lodi, S.; Klusch, M. Clustering Distributed Short Time Series with Dense Patterns. In Proceedings of the 2017 16th IEEE International Conference on Machine Learning and Applications (ICMLA), Cancun, Mexico, 18–21 December 2017. [Google Scholar]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Priyam, A.; Woodcroft, B.J.; Rai, V.; Munagala, A.; Wurm, Y. Sequenceserver: A modern graphical user interface for custom BLAST databases. Mol. Biol. Evol. 2019, 36, 2922–2924. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR. Methods 2002, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Tian, R.P.; Chen, J.D.; Sen, W.; Xinghe, L.; Peng, W.; Tianzhen, Z.; Jinfa, Z. Transcriptomic analysis of fiber strength in upland cotton chromosome introgression lines carrying different Gossypium barbadense chromosomal segments. PLoS ONE 2014, 9, e94642. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Chen, Q.; Huang, L.; Luo, M. Advances about the roles of membranes in cotton fiber development. Membranes 2021, 11, 471. [Google Scholar] [CrossRef]
- Hamada, T. Microtubule organization and microtubule-associated proteins in plant cells. Int. Rev. Cel. Mol. Bio. 2014, 312, 1–52. [Google Scholar] [CrossRef]
- Mei, H.; Qi, B.; Han, Z.; Zhao, T.; Guo, M.; Han, J.; Zhang, J.; Guan, X.; Hu, Y.; Zhang, T.; et al. Subgenome bias and temporal postponement of gene expression contributes to the distinctions of fiber quality in Gossypium species. Front. Plant Sci. 2021, 12, 819679. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, N.; Zhang, J.; Zheng, Y.; Li, X. Genome-wide identification of the mitogen-activated protein kinase (MAPK) family in cotton (Gossypium hirsutum) reveals GhMPK6 involved in fiber elongation. Plant Mol. Biol. 2020, 103, 391–407. [Google Scholar] [CrossRef]
- Wang, M.Y.; Zhao, P.M.; Cheng, H.Q.; Han, L.B.; Wu, X.M.; Gao, P.; Wang, H.Y.; Yang, C.L.; Zhong, N.Q.; Zuo, J.R. The cotton transcription factor TCP14 functions in auxin-mediated epidermal cell differentiation and elongation. Plant Physiol. 2013, 162, 1669–1680. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.; Qanmber, G.; Lu, L.; Wang, L.; Li, J.; Yang, Z.; Liu, Z.; Li, Y.; Chen, Q.; Mendu, V. Genome-wide analysis of cotton GH3 subfamily II reveals functional divergence in fiber development, hormone response and plant architecture. BMC Plant Biol. 2018, 18, 350. [Google Scholar] [CrossRef]
- Zhang, M.; Cao, H.; Xi, J.; Zeng, J.; Pei, Y. Auxin directly upregulates GhRAC13 expression to promote the onset of secondary cell wall deposition in cotton fibers. Front. Plant Sci. 2020, 11, 581983. [Google Scholar] [CrossRef]
- Wu, A.; Hao, P.; Wei, H.; Sun, H.; Yu, S. Genome-wide identification and characterization of glycosyltransferase family 47 in cotton. Front. Genet. 2019, 10, 824. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.X. The Cloning and Functional Characterization of Genes GhGRAM31 and GhTCP15 during Cotton Fiber Development. Ph.D. Thesis, Huazhong Agricultural University, Wuhan, China, 2021. [Google Scholar]
- Lefèvre, F.; Boutry, M. Towards identification of the substrates of ATP-binding cassette transporters. Plant Physiol. 2018, 178, 18–39. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.Q.; Xu, K.X.; Luo, B.; Wang, J.W. An ATP-binding cassette transporter GhWBC1 from elongating cotton fibers. Plant Physiol. 2003, 133, 580–588. [Google Scholar] [CrossRef] [Green Version]
- Sable, A.; Rai, K.M.; Choudhary, A.; Yadav, V.K.; Agarwal, S.K.; Sawant, S.V. Inhibition of heat shock proteins HSP90 and HSP70 induce oxidative stress, suppressing cotton fiber development. Sci. Rep. 2018, 8, 3620. [Google Scholar] [CrossRef]
- Li, Y.; Yuan, P.; Fan, S.; Zhai, B.; Jin, W.; Li, D.; Li, H.; Sun, G.; Han, R.; Liu, X.; et al. Weighted gene co-expression network indicates that the DYNLL2 is an important regulator of chicken breast muscle development and is regulated by miR-148a-3p. BMC Genom. 2022, 23, 258. [Google Scholar] [CrossRef]
- Yang, Z.; Dai, Z.; Chen, X.; Xie, D.; Su, J. Gene coexpression network analysis and tissue-specific profiling of gene expression in jute (Corchorus capsularis L.). BMC Genom. 2020, 21, 406. [Google Scholar] [CrossRef]
- Wang, X.; Kang, W.; Wu, F.; Miao, J.; Shi, S. Comparative transcriptome analysis reveals new insight of alfalfa (Medicago sativa L.) cultivars in response to abrupt freezing stress. Front. Plant Sci. 2022, 13, 798118. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, Y.; Shi, H.; Hu, H.; Yi, L.; Hou, J. Time-course transcriptome and WGCNA analysis revealed the drought response mechanism of two sunflower inbred lines. PLoS ONE 2022, 17, e265447. [Google Scholar] [CrossRef]
- Cheng, G.; Zhang, L.; Wang, H.; Lu, J.; Wei, H.; Yu, S. Transcriptomic profiling of young cotyledons response to chilling stress in two contrasting cotton (Gossypium hirsutum L.) genotypes at the seedling stage. Int. J. Mol. Sci. 2020, 21, 5095. [Google Scholar] [CrossRef]
- Yang, P.; Chang, Y.; Wang, L.; Wang, S.; Wu, J. Regulatory mechanisms of the resistance to common bacterial blight revealed by transcriptomic analysis in common bean (Phaseolus vulgaris L.). Front. Plant Sci. 2022, 12, 800535. [Google Scholar] [CrossRef]
- Dong, F.; Wang, Y.; Tang, M. Effects of laccaria bicolor on gene expression of populus trichocarpa root under poplar canker stress. J. Fungi 2021, 7, 1024. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Hao, P.; Gu, L.; Cheng, S.; Yu, S. Pectate lyase-like gene GhPEL76 regulates organ elongation in Arabidopsis and fiber elongation in cotton. Plant Sci. 2019, 293, 110395. [Google Scholar] [CrossRef] [PubMed]
- Dreiza, C.M.; Brophy, C.M.; Padmini, K.; Furnish, E.J.; Lokesh, J.; Pallero, M.A.; Murphy-Ullrich, J.E.; Moritz, V.R.; Ho, Y.; Bonnie, R. Transducible heat shock protein 20 (HSP20) phosphopeptide alters cytoskeletal dynamics. FASEB J. 2005, 19, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Fang, L.; Xiang, D.; Hu, Y.; Feng, H.; Chang, L.; Zhang, T. Transcriptome analysis of short fiber mutant ligon lintless-1 (Li1) reveals critical genes and key pathways in cotton fiber elongation and leaf development. PLoS ONE 2015, 10, e143503. [Google Scholar] [CrossRef] [Green Version]
- Seifert, G.J.; Roberts, K. The biology of arabinogalactan proteins. Annu. Rev. Plant Biol. 2007, 58, 137–161. [Google Scholar] [CrossRef]
- Li, Y.L.; Sun, J.; Xia, G.X. Cloning and characterization of a gene for an LRR receptor-like protein kinase associated with cotton fiber development. Mol. Genet. Genom. 2005, 273, 217–224. [Google Scholar] [CrossRef]
- Islam, M.S.; Zeng, L.; Thyssen, G.N.; Delhom, C.D.; Kim, H.J.; Li, P.; Fang, D.D. Mapping by sequencing in cotton (Gossypium hirsutum) line MD52ne identified candidate genes for fiber strength and its related quality attributes. Theor. Appl. Genet. 2016, 129, 1071–1086. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Materials | Grouping | Fiber Length (mm) | Fiber Strength (cN/tex) | Micronaire (Unit) | Uniformity (%) | Elongation |
|---|---|---|---|---|---|---|
| Line 9 | P1 | 28.67 ± 0.27 c | 30.28 ± 0.21 de | 4.69 ± 0.08 ab | 84.62 ± 0.20 bc | 4.33 ± 0.64 a |
| Xinhai16 | P2 | 35.20 ± 0.11 a | 49.45 ± 2.13 a | 4.05 ± 0.28 bc | 86.98 ± 0.82 a | 2.94 ± 0.66 bc |
| HL-9 | L | 30.40 ± 0.15 bc | 34.59 ± 0.75 bc | 4.10 ± 0.19 bc | 85.43 ± 0.42 ab | 2.38 ± 0.15 c |
| HL-62 | 30.16 ± 2.27 bc | 30.14 ± 0.49 de | 3.80 ± 0.48 c | 84.40 ± 2.03 bc | 3.56 ± 0.80 ab | |
| HL-194 | 31.62 ± 0.52 b | 35.51 ± 1.72 b | 3.60 ± 0.22 c | 86.35 ± 0.39 ab | 3.26 ± 0.74 abc | |
| HL-34 | S | 25.60 ± 0.30 d | 31.89 ± 0.62 cd | 4.60 ± 0.16 ab | 82.18 ± 0.43 d | 4.12 ± 0.28 a |
| HL-78 | 24.68 ± 1.16 d | 28.37 ± 2.81 ef | 5.31 ± 0.78 a | 81.96 ± 0.90 d | 3.96 ± 0.83 ab | |
| HL-159 | 25.86 ± 0.78 d | 26.98 ± 1.84 f | 4.96 ± 0.42 a | 83.18 ± 1.75 cd | 2.95 ± 0.30 bc |
| Module | Gene ID | Arabidopsis ID | Degree | Predicted Functions (Component) |
|---|---|---|---|---|
| magenta | GH_A07G0413 | AT4G38700 | 38 | Involved in lignification (indicator protein) |
| GH_D08G2106 | AT4G23365 | 37 | Involved in cell wall relaxation (pectinase) | |
| GH_D04G0452 | AT5G39280 | 36 | Involved in cell wall relaxation (expansin) | |
| black | GH_A03G0667 | AT5G50710 | 66 | Involved in cell wall skeleton regulation (actin) |
| GH_D11G2156 | AT4G37630 | 66 | Involved in microtubule formation (cyclin) | |
| GH_A09G2563 | AT5G49360 | 64 | Involved in secondary cell wall thickening (glycoside hydrolase) | |
| GH_D02G0917 | AT5G24080 | 64 | Involved in cytoskeletal rearrangement (receptor protein kinase) | |
| GH_D04G0535 | AT3G28345 | 64 | Involved in the differentiation of microtubule bundles (ABC transporter) | |
| GH_D02G0104 | AT3G59690 | 63 | Involvement in secondary wall deposition by regulating microtubules (IQ-calmodulin) | |
| brown | GH_A05G0924 | AT1G07400 | 170 | Involved in cytoskeletal structure (heat shock protein) |
| GH_D02G0408 | AT4G24000 | 165 | Involved in the hemicellulose backbone (Cellulose synthase) | |
| GH_A13G2053 | AT1G09380 | 159 | Involved in pectinase, cellulose regulation (transporter) | |
| GH_D09G2382 | AT1G55850 | 156 | Involved in the hemicellulose backbone (Cellulose synthase) | |
| GH_D03G0387 | AT1G22360 | 153 | Involved in glucose catalysis (glucotransferase) | |
| GH_D12G0897 | AT4G37790 | 152 | Involved in transcriptional regulation (homeodomain leucine zipper protein) | |
| blue | GH_D12G1588 | AT1G03870 | 479 | Involved in cell wall adhesion (Fasciclin-like arabinogalactan protein 9) |
| GH_A10G2524 | AT2G39890 | 454 | involved in cell wall proteins (proline protein) | |
| GH_A10G2000 | AT5G12380 | 435 | Involved in calcium channel formation (annexin) | |
| GH_D11G1764 | AT1G66880 | 429 | Involved in cell wall pectin binding (receptor kinase) | |
| GH_A07G0137 | AT4G29810 | 425 | Involved in hormone signal transduction (mitogen-activated protein kinase) | |
| GH_D06G1380 | AT5G49720 | 423 | Involved in cell wall assembly and xylem formation (endoglucanase) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Geng, S.; Pang, B.; Zhao, J.; Huang, Y.; Rui, C.; Cui, J.; Jiao, Y.; Zhang, R.; Gao, W. Revealing Genetic Differences in Fiber Elongation between the Offspring of Sea Island Cotton and Upland Cotton Backcross Populations Based on Transcriptome and Weighted Gene Coexpression Networks. Genes 2022, 13, 954. https://doi.org/10.3390/genes13060954
Li S, Geng S, Pang B, Zhao J, Huang Y, Rui C, Cui J, Jiao Y, Zhang R, Gao W. Revealing Genetic Differences in Fiber Elongation between the Offspring of Sea Island Cotton and Upland Cotton Backcross Populations Based on Transcriptome and Weighted Gene Coexpression Networks. Genes. 2022; 13(6):954. https://doi.org/10.3390/genes13060954
Chicago/Turabian StyleLi, Shengmei, Shiwei Geng, Bo Pang, Jieyin Zhao, Yajie Huang, Cun Rui, Jinxin Cui, Yang Jiao, Ru Zhang, and Wenwei Gao. 2022. "Revealing Genetic Differences in Fiber Elongation between the Offspring of Sea Island Cotton and Upland Cotton Backcross Populations Based on Transcriptome and Weighted Gene Coexpression Networks" Genes 13, no. 6: 954. https://doi.org/10.3390/genes13060954