
Novel Genetic and Phenotypic Expansion in GOSR2-Related Progressive Myoclonus Epilepsy
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Recruitment
2.2. Molecular Genetic Investigations
3. Results
3.1. Phenotype Expansion
3.2. Molecular Genetic Investigations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Corbett, M.A.; Schwake, M.; Bahlo, M.; Dibbens, L.M.; Lin, M.; Gandolfo, L.C.; Vears, D.F.; O’Sullivan, J.D.; Robertson, T.; Bayly, M.A.; et al. A Mutation in the Golgi Qb-SNARE Gene GOSR2 Causes Progressive Myoclonus Epilepsy with Early Ataxia. Am. J. Hum. Genet. 2011, 88, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Polet, S.S.; Anderson, D.G.; Koens, L.H.; van Egmond, M.E.; Drost, G.; Brusse, E.; Willemsen, M.A.; Sival, D.A.; Brouwer, O.F.; Kremer, H.P.; et al. A detailed description of the phenotypic spectrum of North Sea Progressive Myoclonus Epilepsy in a large cohort of seventeen patients. Park. Relat. Disord. 2020, 72, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.L.; Peter, F.; Subramaniam, V.N.; Wong, S.H.; Hong, W. A SNARE involved in protein transport through the Golgi apparatus. Nature 1997, 389, 881–884. [Google Scholar] [CrossRef] [PubMed]
- Dibbens, L.M.; Rubboli, G. GOSR2: A progressive myoclonus epilepsy gene. Epileptic Disord. 2016, 18, S111–S114. [Google Scholar] [CrossRef]
- Lambrechts, R.A.; Polet, S.S.; Hernandez-Pichardo, A.; van Ninhuys, L.; Gorter, J.A.; Grzeschik, N.A.; de Koning-Tijssen, M.A.J.; de Koning, T.J.; Sibon, O.C.M. North Sea Progressive Myoclonus Epilepsy is Exacerbated by Heat, A Phenotype Primarily Associated with Affected Glia. Neuroscience 2019, 423, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dafsari, H.S.; Pemberton, J.G.; Ferrer, E.A.; Yammine, T.; Farra, C.; Mohammadi, M.H.; Ghayoor Karimiani, E.; Hashemi, N.; Souaid, M.; Sabbagh, S.; et al. PI4K2A deficiency causes innate error in intracellular trafficking with developmental and epileptic-dyskinetic encephalopathy. Ann. Clin. Transl. Neurol. 2022, 9, 1345–1358. [Google Scholar] [CrossRef]
- Bayram, N.; Kaçar Bayram, A.; Daimagüler, H.S.; Dafsari, H.S.; Bamborschke, D.; Uyanik, G.; Erdogan, M.; Özsaygılı, C.; Pangal, E.; Yuvaci, İ.; et al. Genotype-phenotype correlations in ocular manifestations of Marinesco-Sjögren syndrome: Case report and literature review. Eur. J. Ophthalmol. 2021, 32, NP92–NP97. [Google Scholar] [CrossRef]
- Dafsari, H.S.; Kawalia, A.; Sprute, R.; Karakaya, M.; Malenica, A.; Herkenrath, P.; Nürnberg, P.; Motameny, S.; Thiele, H.; Cirak, S. Novel mutations in SLC6A5 with benign course in hyperekplexia. Cold Spring Harb. Mol. Case Stud. 2019, 5, a004465. [Google Scholar] [CrossRef]
- Dafsari, H.S.; Sprute, R.; Wunderlich, G.; Daimagüler, H.-S.; Karaca, E.; Contreras, A.; Becker, K.; Schulze-Rhonhof, M.; Kiening, K.; Karakulak, T.; et al. Novel mutations in KMT2B offer pathophysiological insights into childhood-onset progressive dystonia. J. Hum. Genet. 2019, 64, 803–813. [Google Scholar] [CrossRef]
- Saffari, A.; Lau, T.; Tajsharghi, H.; Karimiani, E.G.; Kariminejad, A.; Efthymiou, S.; Zifarelli, G.; Sultan, T.; Toosi, M.B.; Sedighzadeh, S.; et al. The clinical and genetic spectrum of autosomal-recessive TOR1A-related disorders. Brain 2023, 146, 3273–3288. [Google Scholar] [CrossRef]
- Becker, L.L.; Dafsari, H.S.; Schallner, J.; Abdin, D.; Seifert, M.; Petit, F.; Smol, T.; Bok, L.; Rodan, L.; Krapels, I.; et al. The clinical-phenotype continuum in DYNC1H1-related disorders—Genomic profiling and proposal for a novel classification. J. Hum. Genet. 2020, 65, 1003–1017. [Google Scholar] [CrossRef]
- Sprute, R.; Jergas, H.; Ölmez, A.; Alawbathani, S.; Karasoy, H.; Dafsari, H.S.; Becker, K.; Daimagüler, H.S.; Nürnberg, P.; Muntoni, F.; et al. Genotype-phenotype correlation in seven motor neuron disease families with novel ALS2 mutations. Am. J. Med. Genet. A 2021, 185, 344–354. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.-J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- de Sainte Agathe, J.M.; Filser, M.; Isidor, B.; Besnard, T.; Gueguen, P.; Perrin, A.; Van Goethem, C.; Verebi, C.; Masingue, M.; Rendu, J.; et al. SpliceAI-visual: A free online tool to improve SpliceAI splicing variant interpretation. Hum. Genom. 2023, 17, 7. [Google Scholar] [CrossRef]
- Davydov, E.V.; Goode, D.L.; Sirota, M.; Cooper, G.M.; Sidow, A.; Batzoglou, S. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 2010, 6, e1001025. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef]
- Dafsari, H.S.; Becker, L.; von der Hagen, M.; Cirak, S. Genomic profiling in neuronal dyneinopathies and updated classifications. Am. J. Med. Genet. A 2021, 185, 2607–2610. [Google Scholar] [CrossRef] [PubMed]
- Tsai, L.; Schwake, M.; Corbett, M.A.; Gecz, J.; Berkovic, S.; Shieh, P.B. P. 1.20 GOSR2: A novel form of Congenital Muscular Dystrophy. Neuromuscul. Disord. 2013, 23, 748. [Google Scholar] [CrossRef]
- Praschberger, R.; Balint, B.; Mencacci, N.E.; Hersheson, J.; Rubio-Agusti, I.; Kullmann, D.M.; Bettencourt, C.; Bhatia, K.; Houlden, H. Expanding the Phenotype and Genetic Defects Associated with the GOSR2 Gene. Mov. Disord. Clin. Pract. 2015, 2, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Henige, H.; Kaur, S.; Pappas, K. Compound heterozygous variants in GOSR2 associated with congenital muscular dystrophy: A case report. Eur. J. Med. Genet. 2021, 64, 104184. [Google Scholar] [CrossRef]
- Boissé Lomax, L.; Bayly, M.A.; Hjalgrim, H.; Møller, R.S.; Vlaar, A.M.; Aaberg, K.M.; Marquardt, I.; Gandolfo, L.C.; Willemsen, M.; Kamsteeg, E.J.; et al. “North Sea” Progressive Myoclonus Epilepsy: Phenotype of Subjects with GOSR2 Mutation. Brain 2013, 136, 1146–1154. [Google Scholar] [CrossRef]
- van Egmond, M.E.; Verschuuren-Bemelmans, C.C.; Nibbeling, E.A.; Elting, J.W.J.; Sival, D.A.; Brouwer, O.F.; de Vries, J.J.; Kremer, H.P.; Sinke, R.J.; Tijssen, M.A.; et al. Ramsay Hunt Syndrome: Clinical Characterization of Progressive Myoclonus Ataxia Caused by GOSR2 Mutation. Mov. Disord. 2014, 29, 139–143. [Google Scholar] [CrossRef]
- Anderson, D.G.; Németh, A.H.; Fawcett, K.A.; Sims, D.; Miller, J.; Krause, A. Deep Brain Stimulation in Three Related Cases of North Sea Progressive Myoclonic Epilepsy from South Africa. Mov. Disord. Clin. Pract. 2016, 4, 249–253. [Google Scholar] [CrossRef]
- Larson, A.A.; Baker, P.R.; Milev, M.P.; Press, C.A.; Sokol, R.J.; Cox, M.O.; Lekostaj, J.K.; Stence, A.A.; Bossler, A.D.; Mueller, J.M.; et al. TRAPPC11 and GOSR2 Mutations Associate with Hypoglycosylation of α-Dystroglycan and Muscular Dystrophy. Skelet. Muscle 2018, 8, 17. [Google Scholar] [CrossRef]
- Stemmerik, M.G.; Borch, J.d.S.; Dunø, M.; Krag, T.; Vissing, J. Myopathy Can Be a Key Phenotype of Membrin (GOSR2) Deficiency. Hum. Mutat. 2021, 42, 1101–1106. [Google Scholar] [CrossRef]
- Hay, J.C.; Klumperman, J.; Oorschot, V.; Steegmaier, M.; Kuo, C.S.; Scheller, R.H. Localization, dynamics, and protein interactions reveal distinct roles for ER and Golgi SNAREs. J. Cell Biol. 1998, 141, 1489–1502. [Google Scholar] [CrossRef]
- Lee, M.C.S.; Miller, E.A.; Goldberg, J.; Orci, L.; Schekman, R. Bi-directional protein transport between the ER and Golgi. Annu. Rev. Cell Dev. Biol. 2004, 20, 87–123. [Google Scholar] [CrossRef]
- Mancias, J.D.; Goldberg, J. Structural basis of cargo membrane protein discrimination by the human COPII coat machinery. EMBO J. 2008, 27, 2918. [Google Scholar] [CrossRef]
- Dafsari, H.S.; Kocaturk, N.M.; Daimagüler, H.-S.; Brunn, A.; Dötsch, J.; Weis, J.; Deckert, M. Bi-allelic mutations in uncoordinated mutant number-45 myosin chaperone B are a cause for congenital myopathy. Acta Neuropathol. Commun. 2019, 7, 211. [Google Scholar] [CrossRef]
- Zhao, S.; Li, C.M.; Luo, X.M.; Siu, G.K.; Gan, W.J.; Zhang, L.; Wu, W.K.; Chan, H.C.; Yu, S. Mammalian TRAPPIII Complex positively modulates the recruitment of Sec13/31 onto COPII vesicles. Sci. Rep. 2017, 7, 43207. [Google Scholar] [CrossRef] [PubMed]
- Stanga, D.; Zhao, Q.; Milev, M.P.; Saint-Dic, D.; Jimenez-Mallebrera, C.; Sacher, M. TRAPPC11 functions in autophagy by recruiting ATG2B-WIPI4/WDR45 to preautophagosomal membranes. Traffic 2019, 20, 325–345. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Zhao, H.; Chen, D.; Zhang, H.; Zhao, Y.G. β-propeller proteins WDR45 and WDR45B regulate autophagosome maturation into autolysosomes in neural cells. Curr. Biol. 2021, 31, 1666–1677.e6. [Google Scholar] [CrossRef]
- Saffari, A.; Schröter, J.; Garbade, S.F.; Alecu, J.E.; Ebrahimi-Fakhari, D.; Hoffmann, G.F.; Kölker, S.; Ries, M.; Syrbe, S. Quantitative retrospective natural history modeling of WDR45-related developmental and epileptic encephalopathy—A systematic cross-sectional analysis of 160 published cases. Autophagy 2022, 18, 1715–1727. [Google Scholar] [CrossRef]
- Dafsari, H.S.; Ebrahimi-Fakhari, D.; Saffari, A.; Deneubourg, C.; Fanto, M.; Jungbluth, H. EPG5-Related Disorder. 2022. Available online: http://www.ncbi.nlm.nih.gov/pubmed/29227033 (accessed on 20 September 2023).
- Byrne, S.; Dionisi-Vici, C.; Smith, L.; Gautel, M.; Jungbluth, H. Vici syndrome: A review. Orphanet J. Rare Dis. 2016, 11, 21. [Google Scholar] [CrossRef]
- Allen, N.M.; Dafsari, H.S.; Wraige, E.; Jungbluth, H. Neck-Tongue Syndrome: An Underrecognized Childhood Onset Cephalalgia. J. Child. Neurol. 2018, 33, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Nalbach, K.; Schifferer, M.; Bhattacharya, D.; Ho-Xuan, H.; Tseng, W.; Williams, L.A.; Stolz, A.; Lichtenthaler, S.F.; Elazar, Z.; Behrends, C. Spatial proteomics reveals secretory pathway disturbances caused by neuropathy-associated TECPR2. Nat. Commun. 2023, 14. [Google Scholar] [CrossRef] [PubMed]
- Hassan, Z.; Kumar, N.D.; Reggiori, F.; Khan, G. How Viruses Hijack and Modify the Secretory Transport Pathway. Cells 2021, 10, 2535. [Google Scholar] [CrossRef] [PubMed]
- Cattin-Ortolá, J.; Welch, L.G.; Maslen, S.L.; Papa, G.; James, L.C.; Munro, S. Sequences in the cytoplasmic tail of SARS-CoV-2 Spike facilitate expression at the cell surface and syncytia formation. Nat. Commun. 2021, 12, 5333. [Google Scholar] [CrossRef] [PubMed]
- Avula, K.; Singh, B.; Samantaray, S.; Syed, G.H. The Early Secretory Pathway Is Crucial for Multiple Aspects of the Hepatitis C Virus Life Cycle. J. Virol. 2023, 97, e0018023. [Google Scholar] [CrossRef] [PubMed]
- Kagan, J.C.; Stein, M.P.; Pypaert, M.; Roy, C.R. Legionella Subvert the Functions of Rab1 and Sec22b to Create a Replicative Organelle. J. Exp. Med. 2004, 199, 1201. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.; Wang, J.; Zhu, M.; Stahmer, K.; Lakshminarayan, R.; Ghassemian, M.; Jiang, Y.; Miller, E.A.; Ferro-Novick, S. Sec24 phosphorylation regulates autophagosome abundance during nutrient deprivation. eLife 2016, 5, e21167. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Hauk, P.J.; Zhang, N.; Elsegeiny, W.; Guardia, C.M.; Kullas, A.; Crosby, K.; Deterding, R.R.; Schedel, M.; Reynolds, P.; et al. Autophagy-associated immune dysregulation and hyperplasia in a patient with compound heterozygous mutations in ATG9A. Autophagy 2023, 19, 678–691. [Google Scholar] [CrossRef] [PubMed]
- De Matteis, M.A.; Godi, A. PI-loting membrane traffic. Nat. Cell Biol. 2004, 6, 487–492. [Google Scholar] [CrossRef]
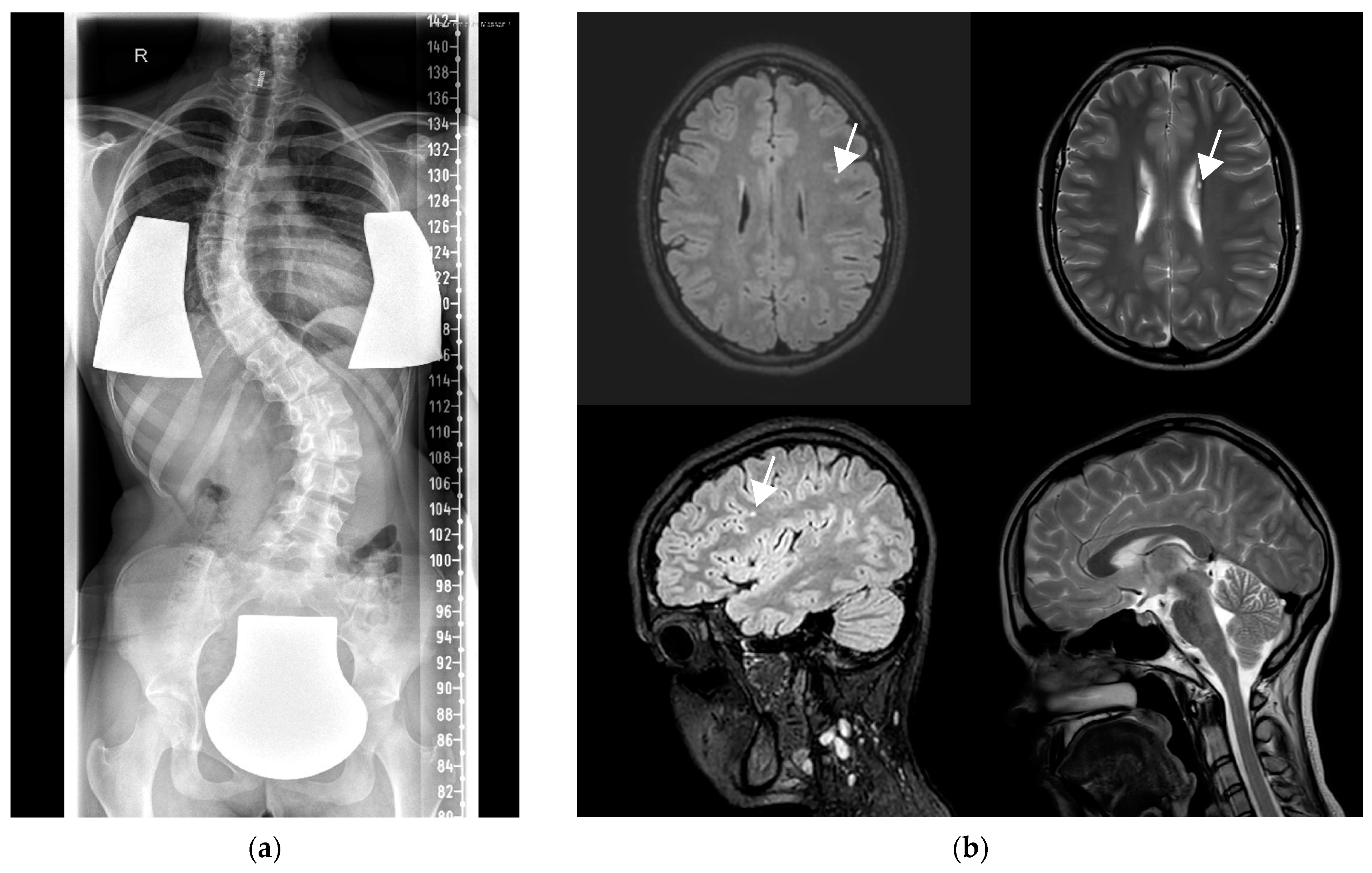
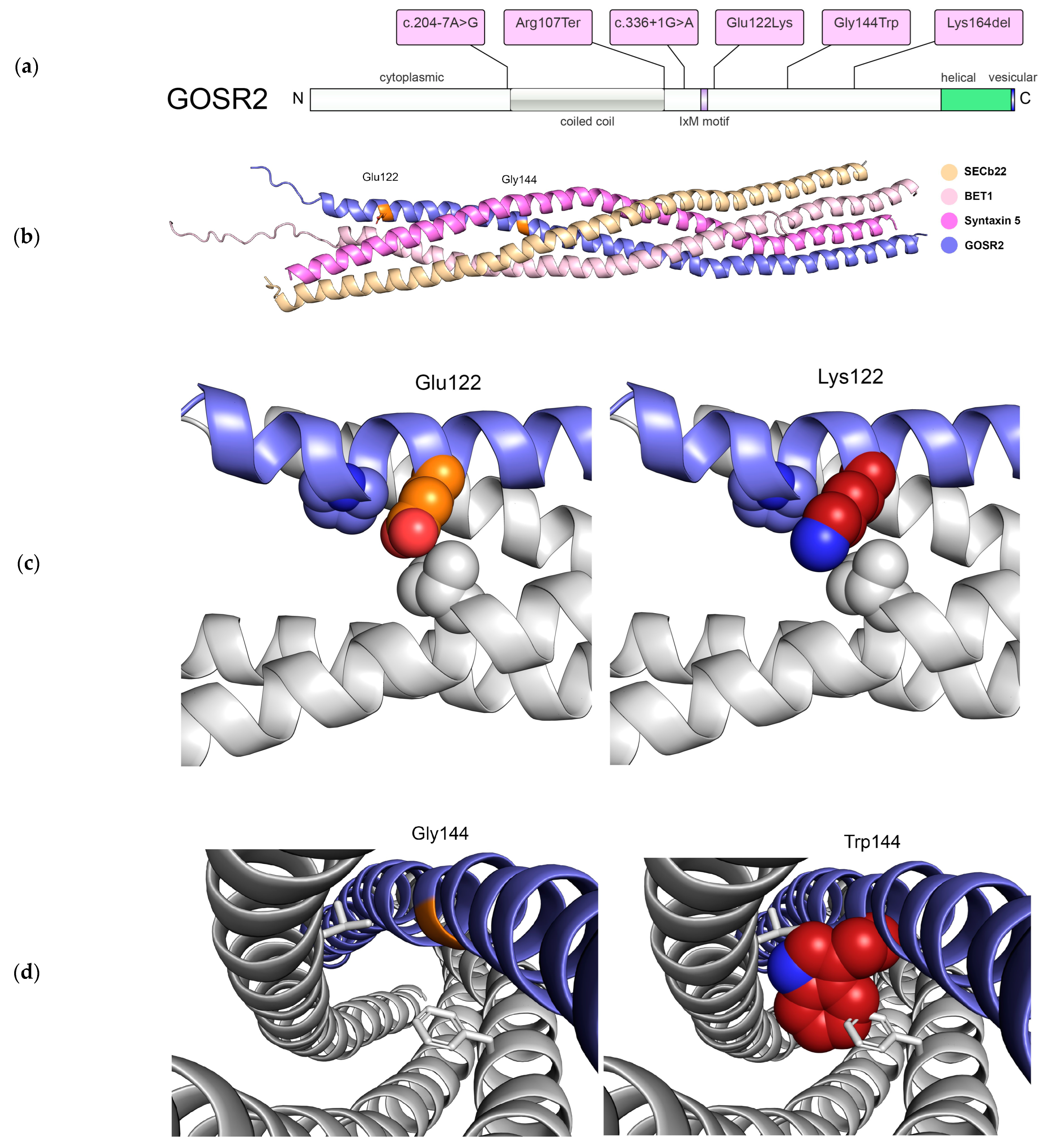

{kind=link}
{kind=link}
| Study | Case No. (Age Last Reported) | Genotype—Inheritance Mode, cDNA NM_001012511.3 (Protein NP_004278.2) | Ataxia (Age at Onset) | Myoclonus (Age at Onset) | Seizures (Age at Onset) | Others |
|---|---|---|---|---|---|---|
| Corbett et al., 2011 [1]; Boissé Lomax et al., 2013 [25] | #01 (32 y) | hom, c.430G>T (p.Gly144Trp) | + (2 y) | + (8 y) | + (absence 7y, GTCS with 13 y, drop attacks 13 y) | CD (25 y); pneumonia |
| #02 (17 y) | hom, c.430G>T (p.Gly144Trp) | + (1 y) | + (6 y) | + (drop attacks 13 y) | ID | |
| #03 (32 y) | hom, c.430G>T (p.Gly144Trp) | + (3 y) | + (6 y) | + (GTCS 14 y) | ||
| #04 (30 y) | hom, c.430G>T (p.Gly144Trp) | + (2 y) | + (10 y) | + (absence 6 y, GTCS 12 y, drop attacks 14 y) | CD (25 y) | |
| #05 (24 y) | hom, c.430G>T (p.Gly144Trp) | + (2 y) | + (6 y) | + (GTCS 21 y) | CD (30 y) | |
| #06 (29 y) | hom, c.430G>T (p.Gly144Trp) | + (2 y) | + (5 y) | + (GTCS 24 y, absence 24 y) | ||
| Boissé Lomax et al., 2013 [25] | #07 (10 y) | hom, c.430G>T (p.Gly144Trp) | + (1 y) | + (4.5 y) | + (focal seizures 14 m, GTCS 8.5 y) | |
| #08 (29 y) | hom, c.430G>T (p.Gly144Trp) | + (3 y) | + (5 y) | + (drop attacks 2 y, absence 2 y, GTCS 21 y) | LD; febrile seizures | |
| #09 (27 y) | hom, c.430G>T (p.Gly144Trp) | + (3.5 y) | + (4 y) | + (absence 5 y, GTCS 8 y) | ||
| #10 (18 y) | hom, c.430G>T (p.Gly144Trp) | + (2 y) | + (6 y) | + (GTCS 10 y, absence) | ||
| #11 (37 y) | hom, c.430G>T (p.Gly144Trp) | + (3 y) | + (12 y) | + (absence 3 y, GTCS 3 y) | ||
| #12 (13 y) | hom, c.430G>T (p.Gly144Trp) | + (2.3 y) | + (6 y) | + (GTCS 12 y) | ||
| Tsai et al., 2013 [22] | #13 (36 w) | comp het, c.430G>T (p.Gly144Trp), c.336+1G>A | − | − | − | Muscular dystrophy |
| van Egmond et al., 2014 [26] | #14 (19 y) | hom, c.430G>T (p.Gly144Trp) | + (3 y) | + (5 y) | + (tonic seizures 9 y) | |
| #15 (26 y) | hom, c.430G>T (p.Gly144Trp) | + (3 y) | + (6 y) | + (GTCS 11 y) | Mild LD | |
| #16 (20 y) | hom, c.430G>T (p.Gly144Trp) | + (2 y) | + (6 y) | + (GTCS 6 y) | Mild LD | |
| #17 (12 y) | hom, c.430G>T (p.Gly144Trp) | + (5 y) | + (8 y) | + (clonic seizures 3 y) | ||
| #18 (7 y) | hom, c.430G>T (p.Gly144Trp) | + (3 y) | + (6 y) | NA | ||
| Praschberger et al., 2015 [23] | #19 (61 y) | comp het, c.430G>T (p.Gly144Trp), c.491_493delAGA (p.Lys164del) | + (2 y) | + (14 y) | + (14 y) | Mild CD |
| Anderson et al., 2016 [27] | #20 (10 y) | hom, c.430G>T (p.Gly144Trp) | + (ND) | + (5 y) | + (GTCS 6 y) | DBS |
| #21 (30 y) | hom, c.430G>T (p.Gly144Trp) | + (ND) | + (ND) | + (GTCS 8 y) | DBS | |
| #22 (27 y) | ND | + (ND) | + (3 y) | + (GTCS 5 y) | DBS | |
| Larson et al., 2018 [28] | #23 (died at 5 y) | ND | − | − | + (absence 2 y) | Muscular dystrophy |
| #24 (6 y) | comp het, c.430G>T (p.Gly144Trp), c.2T>G (p.Met1Arg) | − | − | + (2.5 y) | Muscular dystrophy | |
| Polet et al., 2020 [2] | #25 (5 y) | hom, c.430G>T (p.Gly144Trp) | + (4 y) | + (4 y) | NA | |
| #26 (8 y) | hom, c.430G>T (p.Gly144Trp) | + (2 y) | + (5 y) | + (7 y) | ||
| #27 (8 y) | hom, c.430G>T (p.Gly144Trp) | + (2 y) | + (3 y) | + (7 y) | ||
| #28 (13 y) | hom, c.430G>T (p.Gly144Trp) | + (3 y) | + (6 y) | NA | ||
| #29 (18 y) | hom, c.430G>T (p.Gly144Trp) | + (5 y) | + (7 y) | + (3 y) | DBS | |
| #30 (19 y) | hom, c.430G>T (p.Gly144Trp) | + (4 y) | + (7 y) | + (9 y) | DBS | |
| #31 (20 y) | hom, c.430G>T (p.Gly144Trp) | + (5 y) | + (5 y) | + (13 y) | ||
| #32 (26 y) | hom, c.430G>T (p.Gly144Trp) | + (3 y) | + (5 y) | + (9 y) | ||
| #33 (26 y) | hom, c.430G>T (p.Gly144Trp) | + (2 y) | + (6 y) | + (8 y) | ||
| #34 (31 y) | hom, c.430G>T (p.Gly144Trp) | + (5 y) | + (8 y) | + (8 y) | ||
| #35 (31 y) | hom, c.430G>T (p.Gly144Trp) | + (8 y) | + (9 y) | + (8 y) | ||
| #36 (32 y) | hom, c.430G>T (p.Gly144Trp) | + (3 y) | + (6 y) | + (11 y) | ||
| #37 (35 y) | hom, c.430G>T (p.Gly144Trp) | + (5 y) | + (ND) | + (ND) | DBS | |
| #38 (36 y) | hom, c.430G>T (p.Gly144Trp) | + (ND) | + (ND) | + (6 y) | ||
| #39 (37 y) | hom, c.430G>T (p.Gly144Trp) | + (5 y) | + (ND) | + (8 y) | DBS | |
| #40 (41 y) | hom, c.430G>T (p.Gly144Trp) | + (ND) | + (5 y) | + (8 y) | ||
| #41 (46 y) | hom, c.430G>T (p.Gly144Trp) | + (3 y) | + (4 y) | + (6 y) | ||
| Stemmerik et al., 2021 [29] | #42 (48 y) | comp het, c.319C>T (p.Arg107Ter), c.204-7A>G (p.?) | + (13 y) | + (7 y) | NA | Mild CD (46 y) |
| This study | #43 (15 y) | comp het, c.364G>A (p.Glu122Lys), c.336+1G>A (p.?) | + (12 y) | + (8 y) | + (2 y) | Mild ID |
| #44 (21 y) | hom, c.430G>T (p.Gly144Trp) | + (16 y) | + (13 y) | + (GTCS 12y on urinary tract infection) | Gradual progression of movement disorders into dystonia on recurrent viral infections |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hentrich, L.; Parnes, M.; Lotze, T.E.; Coorg, R.; de Koning, T.J.; Nguyen, K.M.; Yip, C.K.; Jungbluth, H.; Koy, A.; Dafsari, H.S. Novel Genetic and Phenotypic Expansion in GOSR2-Related Progressive Myoclonus Epilepsy. Genes 2023, 14, 1860. https://doi.org/10.3390/genes14101860
Hentrich L, Parnes M, Lotze TE, Coorg R, de Koning TJ, Nguyen KM, Yip CK, Jungbluth H, Koy A, Dafsari HS. Novel Genetic and Phenotypic Expansion in GOSR2-Related Progressive Myoclonus Epilepsy. Genes. 2023; 14(10):1860. https://doi.org/10.3390/genes14101860
Chicago/Turabian StyleHentrich, Lea, Mered Parnes, Timothy Edward Lotze, Rohini Coorg, Tom J. de Koning, Kha M. Nguyen, Calvin K. Yip, Heinz Jungbluth, Anne Koy, and Hormos Salimi Dafsari. 2023. "Novel Genetic and Phenotypic Expansion in GOSR2-Related Progressive Myoclonus Epilepsy" Genes 14, no. 10: 1860. https://doi.org/10.3390/genes14101860