
Genetic and Transcriptome Analyses of Callus Browning in Chaling Common Wild Rice (Oryza rufipogon Griff.)

Abstract
:1. Introduction
2. Results
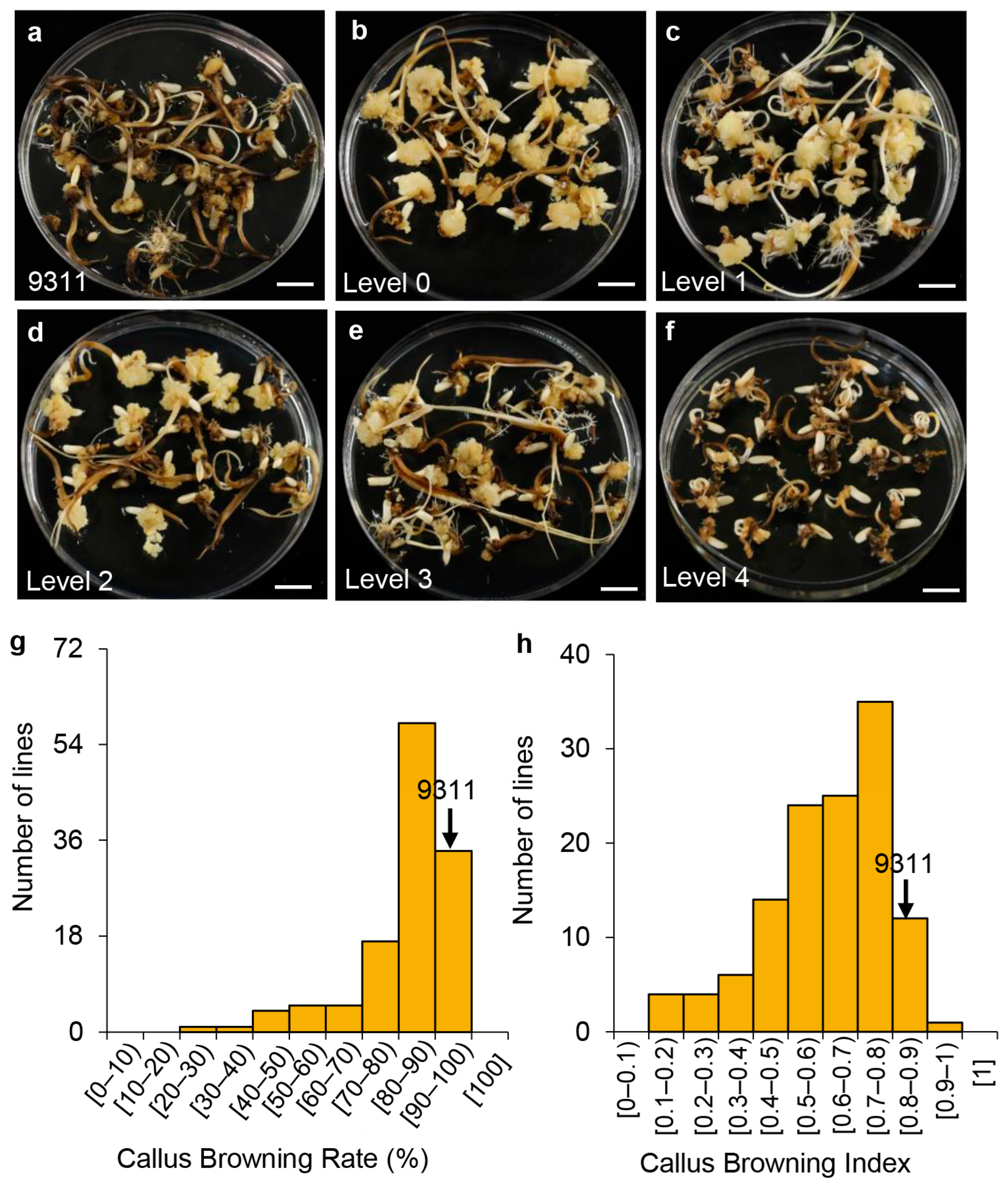
2.1. Callus Browning Phenotypes in the Introgression Lines and the Recurrent Parent 9311
2.2. QTL Analysis of CBR and CBI
2.3. Identification of Coexpression Network and Hub Genes
2.4. Candidate Gene Analysis with Significant Loci in the Intervals Involved in Callus Browning
3. Discussion
3.1. Accuracy of Phenotypic Identification
3.2. QTLs Identification of Callus Browning
3.3. Integrating Linkage Analysis and WGCNA to Identify Key Genes for Callus Browning
3.4. Three Candidate Genes Have Potential Applications for Rice Biotechnology
4. Materials and Methods
4.1. Plant Materials
4.2. Callus Induction and Proliferation Medium
4.3. Cultivation Process
4.4. Phenotypic Identification and Analysis
4.5. QTL Analysis
4.6. Transcriptome Analysis
4.7. WGCNA
4.8. qRT-PCR
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Altpeter, F.; Springer, N.M.; Bartley, L.E.; Blechl, A.E.; Brutnell, T.P.; Citovsky, V.; Conrad, L.J.; Gelvin, S.B.; Jackson, D.P.; Kausch, A.P.; et al. Advancing crop transformation in the era of genome editing. Plant Cell 2016, 28, 1510–1520. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Qi, B.; Mou, C.; Wang, L.; Jiao, Y.; Dou, Y.; Zheng, H. BREVIPEDICELLUS and ERECTA control the expression of AtPRX17 to prevent Arabidopsis callus browning. J. Exp. Bot. 2022, 73, 1516–1532. [Google Scholar] [CrossRef]
- Zhang, K.; Su, J.; Xu, M.; Zhou, Z.; Zhu, X.; Ma, X.; Hou, J.; Tan, L.; Zhu, Z.; Cai, H.; et al. A common wild rice-derived BOC1 allele reduces callus browning in indica rice transformation. Nat. Commun. 2020, 11, 443. [Google Scholar] [CrossRef]
- Deng, X.; Huang, J.; Zhang, M.; Wei, X.; Song, H.; Wang, Y.; Xin, J.; Sun, H.; Liu, J.; Yang, D.; et al. Metabolite profiling and screening of callus browning-related genes in lotus (Nelumbo nucifera). Physiol. Plant. 2023, 175, e14027. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, X.; Xu, G.; Ye, X.; Ji, Y.; Lou, X.; Su, J.; Sun, C.; Fu, Y.; Zhang, K. Mapping quantitative trait loci associated with callus browning in Dongxiang common wild rice (Oryza rufipogon Griff.). Mol. Biol. Rep. 2023, 50, 3129–3140. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Li, H.; Qin, R.; Xu, R.; Li, J.; Li, L.; Wei, P.; Yang, J. Plant phosphomannose isomerase as a selectable marker for rice transformation. Sci. Rep. 2016, 6, 25921. [Google Scholar] [CrossRef]
- Ramessar, K.; Peremarti, A.; Gomez-Galera, S.; Naqvi, S.; Moralejo, M.; Munoz, P.; Capell, T.; Christou, P. Biosafety and risk assessment framework for selectable marker genes in transgenic crop plants: A case of the science not supporting the politics. Transgenic Res. 2007, 16, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Pontiroli, A.; Simonet, P.; Frostegard, A.; Vogel, T.M.; Monier, J.M. Fate of transgenic plant DNA in the environment. Environ. Biosaf. Res. 2007, 6, 15–35. [Google Scholar] [CrossRef]
- Rizzi, A.; Raddadi, N.; Sorlini, C.; Nordgrd, L.; Nielsen, K.M.; Daffonchio, D. The stability and degradation of dietary DNA in the gastrointestinal tract of mammals: Implications for horizontal gene transfer and the biosafety of GMOs. Crit. Rev. Food Sci. Nutr. 2012, 52, 142–161. [Google Scholar] [CrossRef]
- Huang, C.; Zhang, J.; Zhou, D.; Huang, Y.; Su, L.; Yang, G.; Luo, W.; Chen, Z.; Wang, H.; Guo, T. Identification and candidate gene screening of qCIR9.1, a novel QTL associated with anther culturability in rice (Oryza sativa L.). Theor. Appl. Genet. 2021, 134, 2097–2111. [Google Scholar] [CrossRef]
- Nishimura, A.; Ashikari, M.; Lin, S.; Takashi, T.; Angeles, E.R.; Yamamoto, T.; Matsuoka, M. Isolation of a rice regeneration quantitative trait loci gene and its application to transformation systems. Proc. Natl. Acad. Sci. USA 2005, 102, 11940–11944. [Google Scholar] [CrossRef]
- Li, Z.; Duan, S.; Kong, J.; Li, S.; Li, Y.; Zhu, Y. A single genetic locus in chromosome 1 controls conditional browning during the induction of calli from mature seeds of Oryza sativa ssp. indica. Plant Cell Tiss. Org. 2007, 89, 237–245. [Google Scholar] [CrossRef]
- Li, S.; Yan, S.; Wang, A.; Zou, G.; Huang, X.; Han, B.; Qian, Q.; Tao, Y. Identification of QTLs associated with tissue culture response through sequencing-based genotyping of RILs derived from 93-11 x Nipponbare in rice (Oryza sativa). Plant Cell Rep. 2013, 32, 103–116. [Google Scholar] [CrossRef]
- TaguchiShiobara, F.; Lin, S.Y.; Tanno, K.; Komatsuda, T.; Yano, M.; Sasaki, T.; Oka, S. Mapping quantitative trait loci associated with regeneration ability of seed callus in rice, Oryza sativa L. Theor. Appl. Genet. 1997, 95, 828–833. [Google Scholar] [CrossRef]
- Taguchi-Shiobara, F.; Yamamoto, T.; Yano, M.; Oka, S. Mapping QTLs that control the performance of rice tissue culture and evaluation of derived near-isogenic lines. Theor. Appl. Genet. 2006, 112, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, Y.; Abe, T.; Sasahara, T. RFLP mapping of QTLs influencing shoot regeneration from mature seed-derived calli in rice. Crop Sci. 2000, 40, 245–247. [Google Scholar] [CrossRef]
- Zhang, K.; Yin, Z.; Xu, X.; Pu, C.; Li, Q.; Wu, D.; Sun, C.; Fu, Y. Quantitative trait loci for mature embryo culturability traits from Yuanjiang common wild rice (Oryza rufipogon Griff.). Indian J. Genet. Plant Breed. 2016, 76, 167–172. [Google Scholar] [CrossRef]
- Zhao, L.; Zhou, H.; Lu, L.; Liu, L.; Li, X.; Lin, Y.; Yu, S. Identification of quantitative trait loci controlling rice mature seed culturability using chromosomal segment substitution lines. Plant Cell Rep. 2009, 28, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Zakizadeh, H.; Stummann, B.M.; Luetken, H.; Mueller, R. Isolation and characterization of four somatic embryogenesis receptor-like kinase (RhSERK) genes from miniature potted rose (Rosa hybrida cv. Linda). Plant Cell Tiss. Org. 2010, 101, 331–338. [Google Scholar] [CrossRef]
- Lotan, T.; Ohto, M.; Yee, K.M.; West, M.A.; Lo, R.; Kwong, R.W.; Yamagishi, K.; Fischer, R.L.; Goldberg, R.B.; Harada, J.J. Arabidopsis LEAFY COTYLEDON1 is sufficient to induce embryo development in vegetative cells. Cell 1998, 93, 1195–1205. [Google Scholar] [CrossRef]
- Stone, S.L.; Kwong, L.W.; Yee, K.M.; Pelletier, J.; Lepiniec, L.; Fischer, R.L.; Goldberg, R.B.; Harada, J.J. LEAFY COTYLEDON2 encodes a B3 domain transcription factor that induces embryo development. Proc. Natl. Acad. Sci. USA 2001, 98, 11806–11811. [Google Scholar] [CrossRef]
- Boutilier, K.; Offringa, R.; Sharma, V.K.; Kieft, H.; Ouellet, T.; Zhang, L.; Hattori, J.; Liu, C.M.; van Lammeren, A.A.; Miki, B.L.; et al. Ectopic expression of BABY BOOM triggers a conversion from vegetative to embryonic growth. Plant Cell 2002, 14, 1737–1749. [Google Scholar] [CrossRef] [PubMed]
- Bouchabke-Coussa, O.; Obellianne, M.; Linderme, D.; Montes, E.; Maia-Grondard, A.; Vilaine, F.; Pannetier, C. Wuschel overexpression promotes somatic embryogenesis and induces organogenesis in cotton (Gossypium hirsutum L.) tissues cultured in vitro. Plant Cell Rep. 2013, 32, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Shi, L.; Liang, X.; Zhao, P.; Wang, W.; Liu, J.; Chang, Y.; Hiei, Y.; Yanagihara, C.; Du, L.; et al. The gene TaWOX5 overcomes genotype dependency in wheat genetic transformation. Nat. Plants 2022, 8, 110–117. [Google Scholar] [CrossRef]
- Liu, X.; Bie, X.M.; Lin, X.; Li, M.; Wang, H.; Zhang, X.; Yang, Y.; Zhang, C.; Zhang, X.S.; Xiao, J. Uncovering the transcriptional regulatory network involved in boosting wheat regeneration and transformation. Nat. Plants 2023, 9, 908–925. [Google Scholar] [CrossRef] [PubMed]
- Murata, M.; Nishimura, M.; Murai, N.; Haruta, M.; Homma, S.; Itoh, Y. A transgenic apple callus showing reduced polyphenol oxidase activity and lower browning potential. Biosci. Biotechnol. Biochem. 2001, 65, 383–388. [Google Scholar] [CrossRef]
- Habib, D.; Chaudhary, M.F.; Zia, M. The study of ascorbate peroxidase, catalase and peroxidase during in vitro regeneration of Argyrolobium roseum. Appl. Biochem. Biotech. 2014, 172, 1070–1084. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Fukuoka, H.; Yano, H.; Ohkawa, Y. Relationships between nitrite reductase activity and genotype-dependent callus growth in rice cell cultures. Plant Cell Rep. 1999, 18, 576–581. [Google Scholar] [CrossRef]
- Adkins, S.W.; Shiraishi, T.; Mccomb, J.A. Rice callus physiology—Identification of volatile emissions and their effects on culture—Growth. Physiol. Plant. 1990, 78, 526–531. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Fukui, H.; Tabata, M. Effect of carbon dioxide and ethylene on berberine production and cell browning in Thalictrum minus cell cultures. Plant Cell Rep. 1991, 9, 496–499. [Google Scholar] [CrossRef]
- Liang, T.; Hu, Y.; Xi, N.; Zhang, M.; Zou, C.; Ge, F.; Yuan, G.; Gao, S.; Zhang, S.; Pan, G.; et al. GWAS across multiple environments and WGCNA suggest the involvement of ZmARF23 in embryonic callus induction from immature maize embryos. Theor. Appl. Genet. 2023, 136, 93. [Google Scholar] [CrossRef] [PubMed]
- Serra, T.S.; Figueiredo, D.D.; Cordeiro, A.M.; Almeida, D.M.; Lourenco, T.; Abreu, I.A.; Sebastian, A.; Fernandes, L.; Contreras-Moreira, B.; Oliveira, M.M.; et al. OsRMC, a negative regulator of salt stress response in rice, is regulated by two AP2/ERF transcription factors. Plant Mol. Biol. 2013, 82, 439–455. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ni, L.; Liu, Y.; Wang, Y.; Zhang, A.; Tan, M.; Jiang, M. The C2H2-type zinc finger protein ZFP182 is involved in abscisic acid-induced antioxidant defense in rice. J. Integr. Plant Biol. 2012, 54, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Mun, B.G.; Shahid, M.; Lee, G.S.; Hussain, A.; Yun, B.W. A novel RHS1 locus in rice attributes seed-pod shattering by the regulation of endogenous S-nitrosothiols. Int. J. Mol. Sci. 2022, 23, 13225. [Google Scholar] [CrossRef]
- Kirungu, J.N.; Magwanga, R.O.; Pu, L.; Cai, X.; Xu, Y.; Hou, Y.; Zhou, Y.; Cai, Y.; Hao, F.; Zhou, Z.; et al. Knockdown of Gh_A05G1554 (GhDHN_03) and Gh_D05G1729 (GhDHN_04) dehydrin genes, reveals their potential role in enhancing osmotic and salt tolerance in cotton. Genomics 2020, 112, 1902–1915. [Google Scholar] [CrossRef]
- Cui, L.G.; Shan, J.X.; Shi, M.; Gao, J.P.; Lin, H.X. DCA1 acts as a transcriptional co-activator of DST and contributes to drought and salt tolerance in rice. PLoS Genet. 2015, 11, e1005617. [Google Scholar] [CrossRef]
- Huang, X.; Kurata, N.; Wei, X.; Wang, Z.X.; Wang, A.; Zhao, Q.; Zhao, Y.; Liu, K.; Lu, H.; Li, W.; et al. A map of rice genome variation reveals the origin of cultivated rice. Nature 2012, 490, 497–501. [Google Scholar] [CrossRef]
- Manly, K.F.; Cudmore, R.H.; Meer, J.M. Map manager QTX, cross-platform software for genetic mapping. Mamm. Genome 2001, 12, 930–932. [Google Scholar] [CrossRef]
- Meng, L.; Li, H.; Zhang, L.; Wang, J. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J. 2015, 3, 269–283. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Bardou, P.; Mariette, J.; Escudie, F.; Djemiel, C.; Klopp, C. Jvenn: An interactive Venn diagram viewer. BMC Bioinform. 2014, 15, 293. [Google Scholar] [CrossRef]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. 2005, 4, 1–45. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | Source of Variation | SS | df | MS | F | p-Value |
|---|---|---|---|---|---|---|
| CBR | Replication | 0.456 | 90 | 0.005 | ||
| total | 4.623 | 212 | ||||
| line | 4.167 | 122 | 0.034 | 6.746 ** | 1.31 × 10−18 | |
| CBI | Replication | 0.592 | 90 | 0.007 | ||
| total | 7.907 | 212 | ||||
| line | 7.315 | 122 | 0.06 | 9.122 ** | 2.405 × 10−23 |
| CBR | CBI | |
|---|---|---|
| CBR | 1 | |
| CBI | 0.792 ** | 1 |
| Trait | Chr. a | Locus b | QTL | PV(%) c | P d | Add e |
|---|---|---|---|---|---|---|
| CBR | 4 | 10,720,051 | qBR4-1 | 10 | 0.00031 | −5.35 |
| 7 | 24,345,190 | qBR7-1 | 10 | 0.00038 | −5.36 | |
| 7 | 24,345,332 | qBR7-2 | 8 | 0.00101 | −9.67 | |
| 9 | 16,692,442 | qBR9-1 | 16 | 0.00001 | −6.94 | |
| 9 | 12,618,887 | qBR9-2 | 13 | 0.00003 | −5.92 | |
| 9 | 15,132,139 | qBR9-3 | 13 | 0.00002 | −6.22 | |
| 9 | 16,504,549 | qBR9-4 | 12 | 0.00009 | −9.51 | |
| 9 | 97,807,851 | qBR9-5 | 9 | 0.00087 | −5.49 | |
| 9 | 10,486,633 | qBR9-6 | 9 | 0.00076 | −7.53 | |
| 9 | 18,382,296 | qBR9-7 | 9 | 0.00057 | −5.13 | |
| 9 | 17,104,635 | qBR9-8 | 8 | 0.00105 | −5.51 | |
| 9 | 97,806,991 | qBR9-9 | 8 | 0.00143 | −7 | |
| 10 | 14,087,592 | qBR10-1 | 10 | 0.00037 | −5.47 | |
| 10 | 13,528,182 | qBR10-2 | 9 | 0.00045 | −5.52 | |
| CBI | 2 | 46,187,291 | qBI2-1 | 9 | 0.00071 | −0.08 |
| 4 | 10,720,051 | qBI4-1 | 11 | 0.00014 | −0.08 | |
| 7 | 24,345,190 | qBI7-1 | 13 | 0.00003 | −0.08 | |
| 7 | 24,345,332 | qBI7-2 | 11 | 0.00017 | −0.15 | |
| 7 | 88,806,091 | qBI7-3 | 10 | 0.00038 | −0.07 | |
| 7 | 77,390,231 | qBI7-4 | 9 | 0.00072 | −0.07 | |
| 7 | 12,434,577 | qBI7-5 | 9 | 0.00054 | −0.07 | |
| 7 | 15,404,459 | qBI7-6 | 8 | 0.00179 | −0.06 | |
| 7 | 18,251,259 | qBI7-7 | 8 | 0.00134 | −0.1 | |
| 7 | 22,320,372 | qBI7-8 | 8 | 0.0017 | −0.1 | |
| 7 | 24,690,967 | qBI7-9 | 8 | 0.0011 | −0.09 | |
| 9 | 15,132,139 | qBI9-1 | 9 | 0.00048 | −0.07 | |
| 9 | 16,692,442 | qBI9-2 | 9 | 0.00061 | −0.07 | |
| 9 | 12,618,887 | qBI9-3 | 8 | 0.00155 | −0.06 | |
| 10 | 14,087,592 | qBI10-1 | 15 | 0.00001 | −0.09 | |
| 10 | 13,528,182 | qBI10-2 | 13 | 0.00003 | −0.09 | |
| 10 | 17,073,302 | qBI10-3 | 10 | 0.00029 | −0.08 | |
| 10 | 13,727,931 | qBI10-4 | 9 | 0.00079 | −0.12 |
| Trait | Chr. | Left Marker | Right Marker | LOD | PV(%) | Add |
|---|---|---|---|---|---|---|
| CBR | 1 | 38,384,198 | 39,169,324 | 4.1653 | 1.5038 | −0.4482 |
| 2 | 3,991,537 | 5,258,744 | 6.8527 | 1.6944 | −15.3833 | |
| 2 | 6,478,167 | 7,064,337 | 4.2684 | 1.4617 | −0.2163 | |
| 3 | 1,305,123 | 2,984,348 | 4.0297 | 1.582 | −0.9482 | |
| 4 | 20,594,577 | 35,126,298 | 9.778 | 1.684 | −0.2971 | |
| 5 | 3,443,919 | 5,376,334 | 6.4935 | 1.6121 | −0.1325 | |
| 6 | 10,080,199 | 18,211,679 | 17.4229 | 2.9635 | −19.9732 | |
| 6 | 18,211,679 | 20,978,032 | 2.7674 | 1.5154 | −1.7639 | |
| 7 | 1,569,714 | 2,042,878 | 2.5546 | 1.3865 | −0.6964 | |
| 7 | 2,667,022 | 4,012,634 | 3.3743 | 1.4399 | −1.782 | |
| 7 | 6,813,655 | 7,739,024 | 7.2528 | 1.6284 | −1.1897 | |
| 7 | 7,739,071 | 8,880,501 | 4.2682 | 1.472 | −2.5481 | |
| 7 | 24,092,079 | 25,681,731 | 2.7068 | 1.547 | −12.9071 | |
| 7 | 26,562,840 | 28,917,404 | 6.0147 | 1.6942 | −14.8156 | |
| 8 | 15,795,968 | 19,682,615 | 8.5256 | 1.6275 | −0.9063 | |
| 8 | 19,682,615 | 21,333,828 | 6.8575 | 1.6091 | −1.0081 | |
| 8 | 24,878,660 | 26,386,382 | 6.3479 | 1.5513 | −3.6428 | |
| 9 | 8,714,345 | 15,691,090 | 6.6571 | 0.9438 | −0.2239 | |
| 10 | 10,588,708 | 17,676,090 | 9.2205 | 2.2374 | −17.1459 | |
| 11 | 2,496,055 | 4,037,176 | 7.016 | 1.8231 | −15.5316 | |
| 12 | 2,398,611 | 2,967,494 | 5.6647 | 1.601 | −0.6781 | |
| CBI | 7 | 24,092,079 | 25,681,731 | 3.9664 | 4.5413 | −0.134 |
| 10 | 10,588,708 | 17,676,090 | 3.0214 | 10.1809 | −0.154 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, L.; Su, J.; Fu, Y.; Zhang, K. Genetic and Transcriptome Analyses of Callus Browning in Chaling Common Wild Rice (Oryza rufipogon Griff.). Genes 2023, 14, 2138. https://doi.org/10.3390/genes14122138
Qiu L, Su J, Fu Y, Zhang K. Genetic and Transcriptome Analyses of Callus Browning in Chaling Common Wild Rice (Oryza rufipogon Griff.). Genes. 2023; 14(12):2138. https://doi.org/10.3390/genes14122138
Chicago/Turabian StyleQiu, Lingyi, Jingjing Su, Yongcai Fu, and Kun Zhang. 2023. "Genetic and Transcriptome Analyses of Callus Browning in Chaling Common Wild Rice (Oryza rufipogon Griff.)" Genes 14, no. 12: 2138. https://doi.org/10.3390/genes14122138