

A Rare Case of Concurrent 2q34q36 Duplication and 2q37 Deletion in a Neonate with Syndromic Features

 ,
,  ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
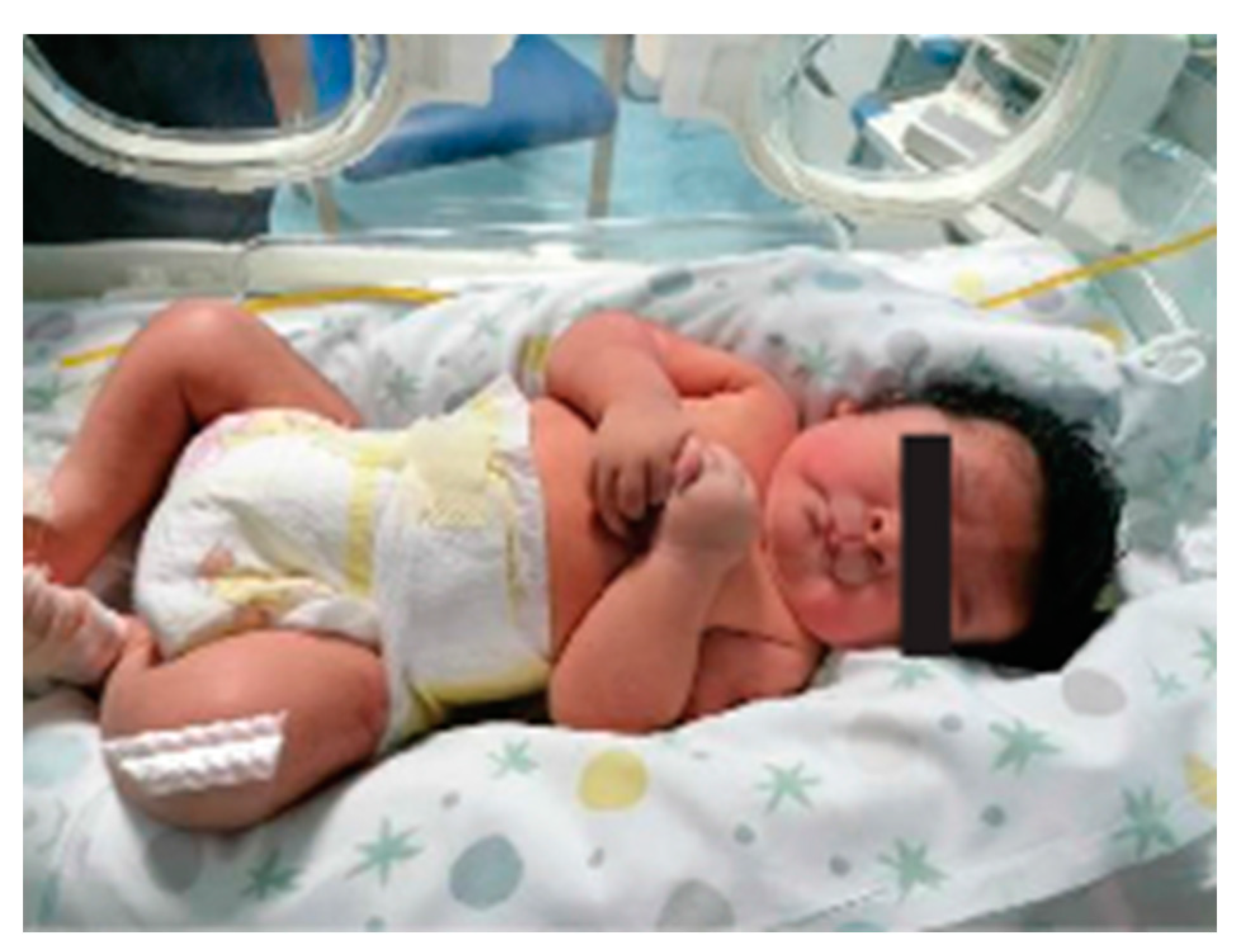
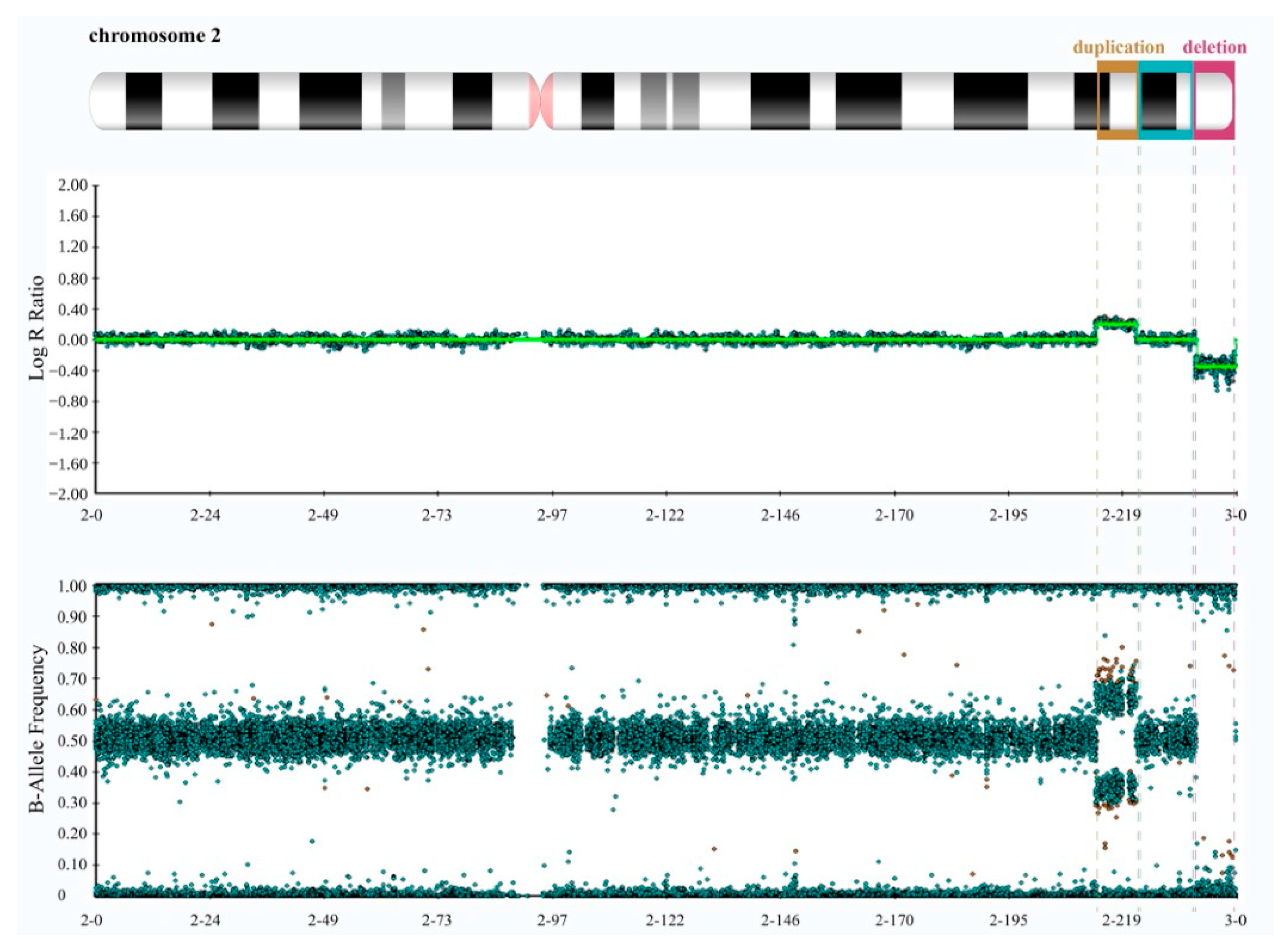
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Burssed, B.; Zamariolli, M.; Bellucco, F.T.; Melaragno, M.I. Mechanisms of structural chromosomal rearrangement formation. Mol. Cytogenet. 2022, 15, 23. [Google Scholar] [CrossRef]
- Leroy, C.; Landais, E.; Briault, S.; David, A.; Tassy, O.; Gruchy, N.; Delobel, B.; Grégoire, M.J.; Leheup, B.; Taine, L.; et al. The 2q37-deletion syndrome: An update of the clinical spectrum including overweight, brachydactyly and behavioural features in 14 new patients. Eur. J. Hum. Genet. 2013, 21, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Gavril, E.C.; Nucă, I.; Pânzaru, M.C.; Ivanov, A.V.; Mihai, C.T.; Antoci, L.M.; Ciobanu, C.G.; Rusu, C.; Popescu, R. Genotype-Phenotype Correlations in 2q37-Deletion Syndrome: An Update of the Clinical Spectrum and Literature Review. Genes 2023, 14, 465. [Google Scholar] [CrossRef]
- Aldred, M.A.; Sanford, R.O.; Thomas, N.S.; Barrow, M.A.; Wilson, L.C.; Brueton, L.A.; Bonaglia, M.C.; Hennekam, R.C.; Eng, C.; Dennis, N.R.; et al. Molecular analysis of 20 patients with 2q37.3 monosomy: Definition of minimum deletion intervals for key phenotypes. J. Med. Genet. 2004, 41, 433–439. [Google Scholar] [CrossRef]
- Elbracht, M.; Roos, A.; Schönherr, N.; Busse, S.; Damen, R.; Zerres, K.; Rudnik-Schöneborn, S.; Schüler, H.M. Pure distal trisomy 2q: A rare chromosomal abnormality with recognizable phenotype. Am. J. Med. Genet. A 2009, 149A, 2547–2550. [Google Scholar] [CrossRef] [PubMed]
- Bird, L.M.; Mascarello, J.T. Chromosome 2q duplications: Case report of a de novo interstitial duplication and review of the literature. Am. J. Med. Genet. 2001, 100, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Seidahmed, M.Z.; Rooney, D.E.; Salih, M.A.; Basit, O.B.; Shaheed, M.M.; Abdullah, M.A.; Abomelha, A. Case of partial trisomy 2q3 with clinical manifestations of Marshall-Smith syndrome. Am. J. Med. Genet. 1999, 85, 185–188. [Google Scholar] [CrossRef]
- Angle, B.; Hersh, J.H.; Yen, F.; Christensen, K.M. Case of partial duplication 2q3 with characteristic phenotype: Rare occurrence of an unbalanced offspring resulting from a parental pericentric inversion. Am. J. Med. Genet. 2000, 91, 126–130. [Google Scholar] [CrossRef]
- Slavotinek, A.M.; Boles, D.; Lacbawan, F. A female infant with duplication of chromosome 2q33 to 2q37.3. Clin. Dysmorphol. 2003, 12, 251–256. [Google Scholar] [CrossRef]
- Zuffardi, O.; Bonaglia, M.; Ciccone, R.; Giorda, R. Inverted duplications deletions: Underdiagnosed rearrangements? Clin. Genet. 2009, 75, 505–513. [Google Scholar] [CrossRef]
- Vera-Carbonell, A.; López-Expósito, I.; Bafalliu, J.A.; Ballesta-Martínez, M.; Glóver, G.; Llópis, C.; Moya-Quiles, R.; Suela, J.; Fernández, A.; Guillén-Navarro, E. Molecular characterization of a new patient with a non-recurrent inv dup del 2q and review of the mechanisms for this rearrangement. Am. J. Med. Genet. A 2010, 152A, 2670–2680. [Google Scholar] [CrossRef] [PubMed]
- Bonaglia, M.C.; Kurtas, N.E.; Errichiello, E.; Bertuzzo, S.; Beri, S.; Mehrjouy, M.M.; Provenzano, A.; Vergani, D.; Pecile, V.; Novara, F.; et al. De novo unbalanced translocations have a complex history/aetiology. Hum. Genet. 2018, 137, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Hermetz, K.E.; Newman, S.; Conneely, K.N.; Martin, C.L.; Ballif, B.C.; Shaffer, L.G.; Cody, J.D.; Rudd, M.K. Large inverted duplications in the human genome form via a fold-back mechanism. PLoS Genet. 2014, 10, e1004139. [Google Scholar] [CrossRef]
- Caspersson, T.; Zech, L.; Johansson, C.; Modest, E.J. Identification of human chromosomes by DNA-binding fluorescent agents. Chromosoma 1970, 30, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Lichter, P.; Tang, C.J.; Call, K.; Hermanson, G.; Evans, G.A.; Housman, D.; Ward, D.C. High-resolution mapping of human chromosome 11 by in situ hybridization with cosmid clones. Science 1990, 247, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Strande, N.T.; Riggs, E.R.; Buchanan, A.H.; Ceyhan-Birsoy, O.; DiStefano, M.; Dwight, S.S.; Goldstein, J.; Ghosh, R.; Seifert, B.A.; Sneddon, T.P.; et al. Evaluating the Clinical Validity of Gene-Disease Associations: An Evidence-Based Framework Developed by the Clinical Genome Resource. Am. J. Hum. Genet. 2017, 100, 895–906. [Google Scholar] [CrossRef]
- Rehm, H.L.; Berg, J.S.; Brooks, L.D.; Bustamante, C.D.; Evans, J.P.; Landrum, M.J.; Ledbetter, D.H.; Maglott, D.R.; Martin, C.L.; Nussbaum, R.L.; et al. ClinGen--the Clinical Genome Resource. N. Engl. J. Med. 2015, 372, 2235–2242. [Google Scholar] [CrossRef]
- Riggs, E.R.; Church, D.M.; Hanson, K.; Horner, V.L.; Kaminsky, E.B.; Kuhn, R.M.; Wain, K.E.; Williams, E.S.; Aradhya, S.; Kearney, H.M.; et al. Towards an evidence-based process for the clinical interpretation of copy number variation. Clin. Genet. 2012, 81, 403–412. [Google Scholar] [CrossRef]
- Heard, P.L.; Carter, E.M.; Crandall, A.C.; Sebold, C.; Hale, D.E.; Cody, J.D. High resolution genomic analysis of 18q- using oligo-microarray comparative genomic hybridization (aCGH). Am. J. Med. Genet. A 2009, 149A, 1431–1437. [Google Scholar] [CrossRef]
- Hulick, P.J.; Noonan, K.M.; Kulkarni, S.; Donovan, D.J.; Listewnik, M.; Ihm, C.; Stoler, J.M.; Weremowicz, S. Cytogenetic and array-CGH characterization of a complex de novo rearrangement involving duplication and deletion of 9p and clinical findings in a 4-month-old female. Cytogenet. Genome Res. 2009, 126, 305–312. [Google Scholar] [CrossRef]
- Lee, N.C.; Chang, S.P.; Chang, C.S.; Chen, C.H.; Lee, D.J.; Lin, C.C.; Hwu, W.L.; Chen, M. Cryptic subtelomeric deletion plus inverted duplication at chromosome 18q in a fetus: Molecular delineation by multicolor banding. Prenat. Diagn. 2009, 29, 1058–1060. [Google Scholar] [CrossRef] [PubMed]
- Rowe, L.R.; Lee, J.Y.; Rector, L.; Kaminsky, E.B.; Brothman, A.R.; Martin, C.L.; South, S.T. U-type exchange is the most frequent mechanism for inverted duplication with terminal deletion rearrangements. J. Med. Genet. 2009, 46, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Córdova-Fletes, C.; Sáinz-González, E.; Avendaño-Gálvez, R.I.; Ramírez-Velazco, A.; Rivera, H.; Ortiz-López, R.; Arámbula-Meraz, E.; Picos-Cárdenas, V.J. De novo dir dup/del of 18q characterized by SNP arrays and FISH in a girl child with mixed phenotypes. J. Genet. 2014, 93, 869–873. [Google Scholar] [CrossRef] [PubMed]
- Fritz, B.; Müller-Navia, J.; Hillig, U.; Köhler, M.; Aslan, M.; Rehder, H. Trisomy 2q35-q37 due to insertion of 2q material into 17q25: Clinical, cytogenetic, and molecular cytogenetic characterization. Am. J. Med. Genet. 1999, 87, 297–301. [Google Scholar] [CrossRef]
- Hermsen, M.A.; Tijssen, M.; Acero, I.H.; Meijer, G.A.; Ylstra, B.; Toral, J.F. High resolution microarray CGH and MLPA analysis for improved genotype/phenotype evaluation of two childhood genetic disorder cases: Ring chromosome 19 and partial duplication 2q. Eur. J. Med. Genet. 2005, 48, 310–318. [Google Scholar] [CrossRef]
- Dahoun-Hadorn, S.; Bretton-Chappuis, B. de novo inversion-duplication of 2q35-2qter without growth retardation. Ann. Genet. 1992, 35, 55–57. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 | P9 | Our Patient |
|---|---|---|---|---|---|---|---|---|---|---|
| Sex | M | F | F | F | M | F | M | F | F | M |
| Age | 30 years | 8 years | 4 years | 12 years | 9 years | 2 years | 18 years | 15 years | 12 years | 9 months |
| ID | mild | moderate | - | moderate | profound | moderate | severe | moderate | moderate | moderate |
| Hypotonia | + | + | - | - | + | - | + | + | - | + |
| Behavioral problems | stereotipies | - | - | ADHD | - | - | autism | aggressivity | laught crises | stereotipies |
| IUGR | - | - | - | - | - | + | - | - | - | - |
| Brachydactyly E | +, MT4, 5, AI | +, MT3-5 | - | clinodactyly | +, tapering | Al | - | +, MC4, 5 | +, MC/MT4, 5 | - |
| Joint hypermobility | + | + | - | - | + | - | - | - | - | + |
| Low frontal hairline | +/- | high | - | - | + | - | +, widow’s peak | +, widow’s peak | high | - |
| Frontal bossing | - | + | + | - | + | - | - | - | - | + |
| Thin/arched evebrows | +, BE | +, BE | - | +, medial sparse | - | +, BE, medial sparse | +, BE | +, BE | +, medial sparse | +, medial sparse |
| Smooth philtrum | - | + | - | +/- | + | + | + | + | + | + |
| Thin upper lip | + | + | - | - | + | + | + | + | + | - |
| Microcephaly | +/- | +/- | - | - | + | ++ | +/- | +/- | Craniosynostosis | - |
| Short neck | + | + | + | + | + | + | + | + | + | + |
| Small/puffy hands/feet | + | - | + | - | - | - | + | + | +/- | + |
| Deletion size | 4.07 Mb | 4.07 Mb | 5.05 Mb | 8.14 Mb | 1.84 Mb | 2.48 Mb | 5.71 Mb | 5.71 Mb | 4.99 Mb | 8.5 Mb |
| Other CNV | no | no | no | no | Dup 2 (q32.1-q37.3) 42.1 Mb | Dup 2q37.3 1.01 Mb | Dup 9 (q34.11-q34.3) 6.94 Mb | Dup 9 (q34.11-q34.3) 6.94 Mb | Dup 11 (p15.5-p15.4) 1.06 Mb | Dup 2 (q34-q36.1) 8.6 Mb |
| Fritz et al. [24] | Elbracht et al. [5] | Hermsen et al. [25] | Dahoun-Hadorn and Bretton-Chappius [26] | Our Patient | |
|---|---|---|---|---|---|
| Duplicated segment | 2q35-2q37.1 | 2q35-2q37.3 | 2q35-2q37.3 | 2q35-2qter | 2q34-2q36.1 |
| Origin | de novo | ? | de novo | de novo | de novo |
| ins 17q25 | dup 2q | ins 2p | inv dup 2q | ||
| Age | 7 years | 16 years | 9 years | 7 years | 9 months |
| Sex | M | F | F | M | M |
| Birth measurements | Delivery at term | Delivery at term | Delivery at term | Delivery at term | Delivery at term |
| Weight | 3200 g (50° centile) | 3210 g (25–50° centile) | Not reported | 4000 g (90° centile) | 3780 g (75° centile) |
| Lenght | 51 cm (75° centile) | 56 cm (97° centile) | Not reported | Not reported | 50 cm (50° centile) |
| Occipito-frontal Circumference | Not reported | 34 cm (25° centile) | Not reported | Not reported | 37 cm (25° centile) |
| Body measurements at report | |||||
| Weight | 19.7 kg (10° centile) | 177.6 cm (97°centile) | 24.6 kg (10° centile) | 26 kg (75–90° centile) | 10 kg (75° centile) |
| Length | 120 cm (25° centile) | 56.4 cm (97° centile) | 131.6 cm (35°centile) | 124 cm (50° centile) | 73 cm (50–75° centile) |
| Occipito-frontal circumference | 49.9 cm (10–25°centile) | Not reported | 50 cm (8° centile) | 56 cm (>97° centile) | 47 cm (75–90° centile) |
| Craniofacial signs | |||||
| Prominent forehead | + | + | + | + | + |
| Broad nasal bridge | + | + | + | + | - |
| Overhanging nasal tip | + | + | + | + | - |
| Thin upper lip | + | + | + | + | + |
| Retrognathia | + | + | ? | + | + |
| Ears | Large/Low set | Large | Large/Low set | Large/Low set | Large/Low set |
| Minor skeletal abnormalities | |||||
| Clinodactyly of fifth finger | ? | + | + | + | - |
| Others | Incomplete syndactyly III/IV of both hands and II/III of both feet | joint hypermobility of ankles and wrists, hypotonia | |||
| Psychomotor retardation | + (no further information) | + (moderate) | + (reported as moderate) | + | + (moderate) |
| Other CNV | no | no | no | no | Del 2 (q37.1-q37.3) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riviello, F.N.; Daponte, A.; Ponzi, E.; Ficarella, R.; Orsini, P.; Bucci, R.; Ventura, M.; Antonacci, F.; Catacchio, C.R.; Gentile, M. A Rare Case of Concurrent 2q34q36 Duplication and 2q37 Deletion in a Neonate with Syndromic Features. Genes 2023, 14, 2194. https://doi.org/10.3390/genes14122194
Riviello FN, Daponte A, Ponzi E, Ficarella R, Orsini P, Bucci R, Ventura M, Antonacci F, Catacchio CR, Gentile M. A Rare Case of Concurrent 2q34q36 Duplication and 2q37 Deletion in a Neonate with Syndromic Features. Genes. 2023; 14(12):2194. https://doi.org/10.3390/genes14122194
Chicago/Turabian StyleRiviello, Francesco Nicola, Alessia Daponte, Emanuela Ponzi, Romina Ficarella, Paola Orsini, Roberta Bucci, Mario Ventura, Francesca Antonacci, Claudia Rita Catacchio, and Mattia Gentile. 2023. "A Rare Case of Concurrent 2q34q36 Duplication and 2q37 Deletion in a Neonate with Syndromic Features" Genes 14, no. 12: 2194. https://doi.org/10.3390/genes14122194
APA StyleRiviello, F. N., Daponte, A., Ponzi, E., Ficarella, R., Orsini, P., Bucci, R., Ventura, M., Antonacci, F., Catacchio, C. R., & Gentile, M. (2023). A Rare Case of Concurrent 2q34q36 Duplication and 2q37 Deletion in a Neonate with Syndromic Features. Genes, 14(12), 2194. https://doi.org/10.3390/genes14122194