

KEGG_Extractor: An Effective Extraction Tool for KEGG Orthologs

Abstract
:1. Introduction
2. Materials and Methods
2.1. KEGG Orthology Search Tools
2.2. Gene Filter and Construction of the Non-Redundant Gene Dataset
2.3. Python3 and the Genomic Annotation Files
2.4. The Assembly Summary File
2.5. The Whole Genomic Annotation Sequences
2.6. Accuracy Assessment of the Constructed Database
3. Results and Discussion
3.1. Implementation and Performance Assessing
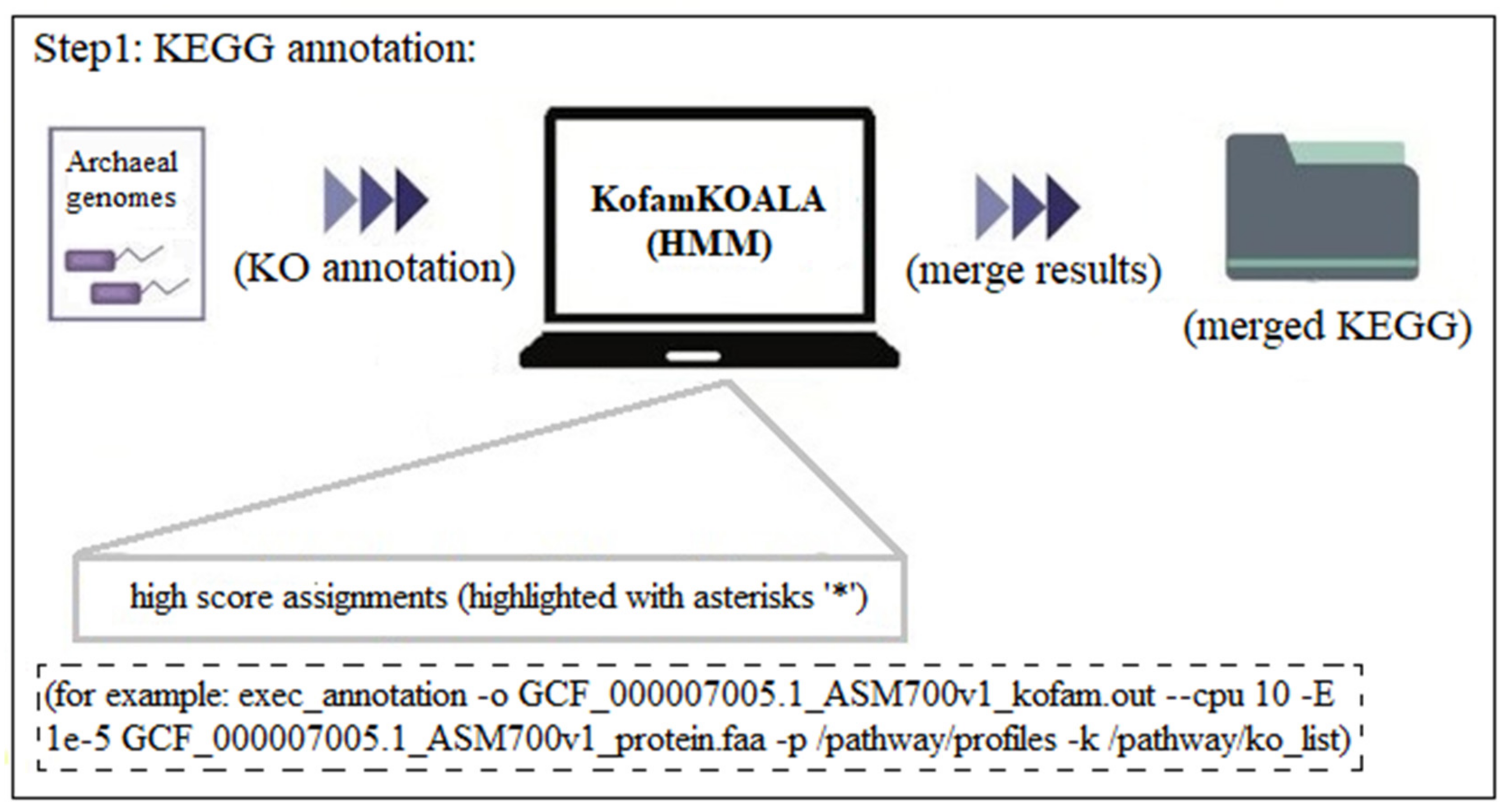
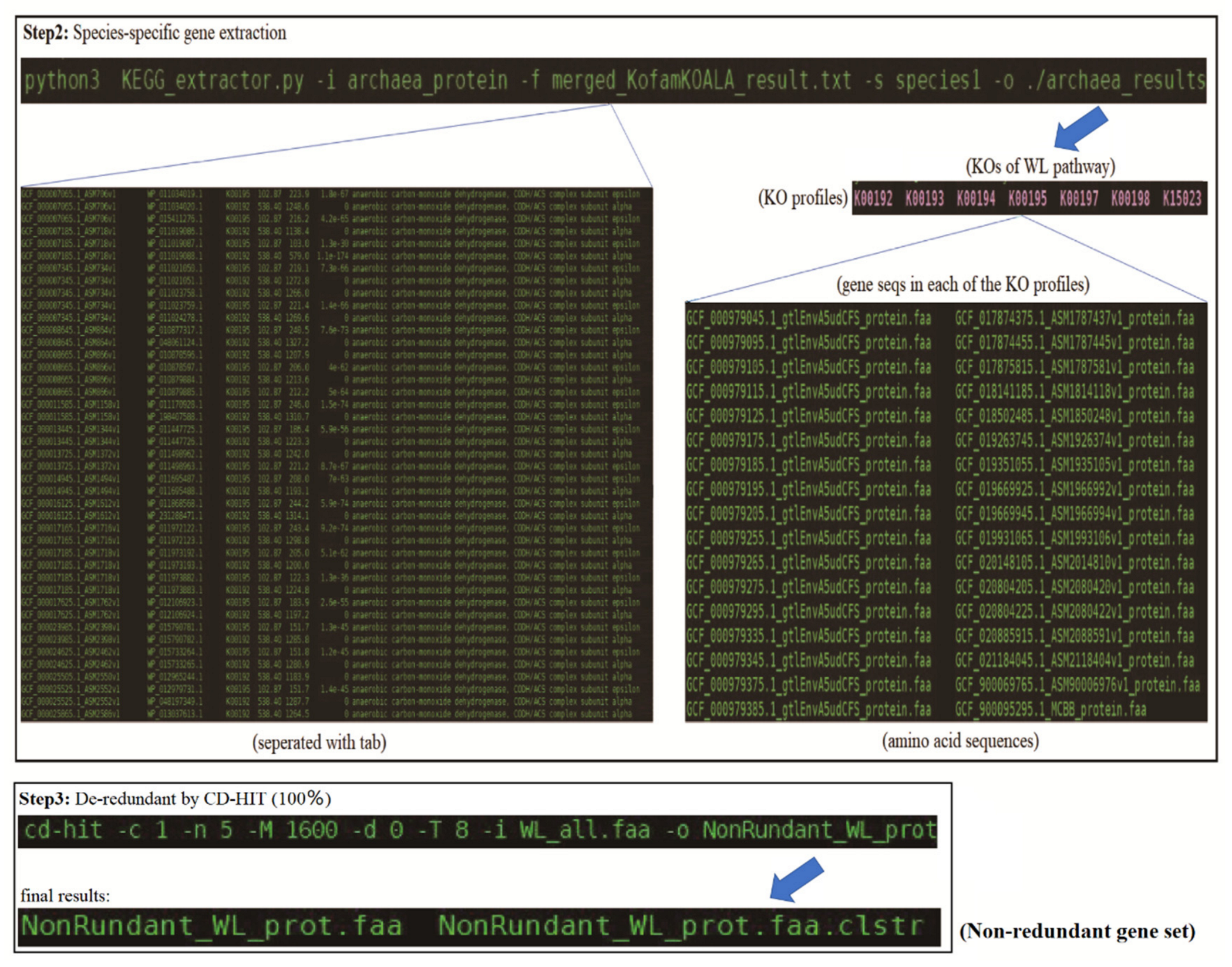
3.2. Step-By-Step Example of the Usage of KEGG_Extractor
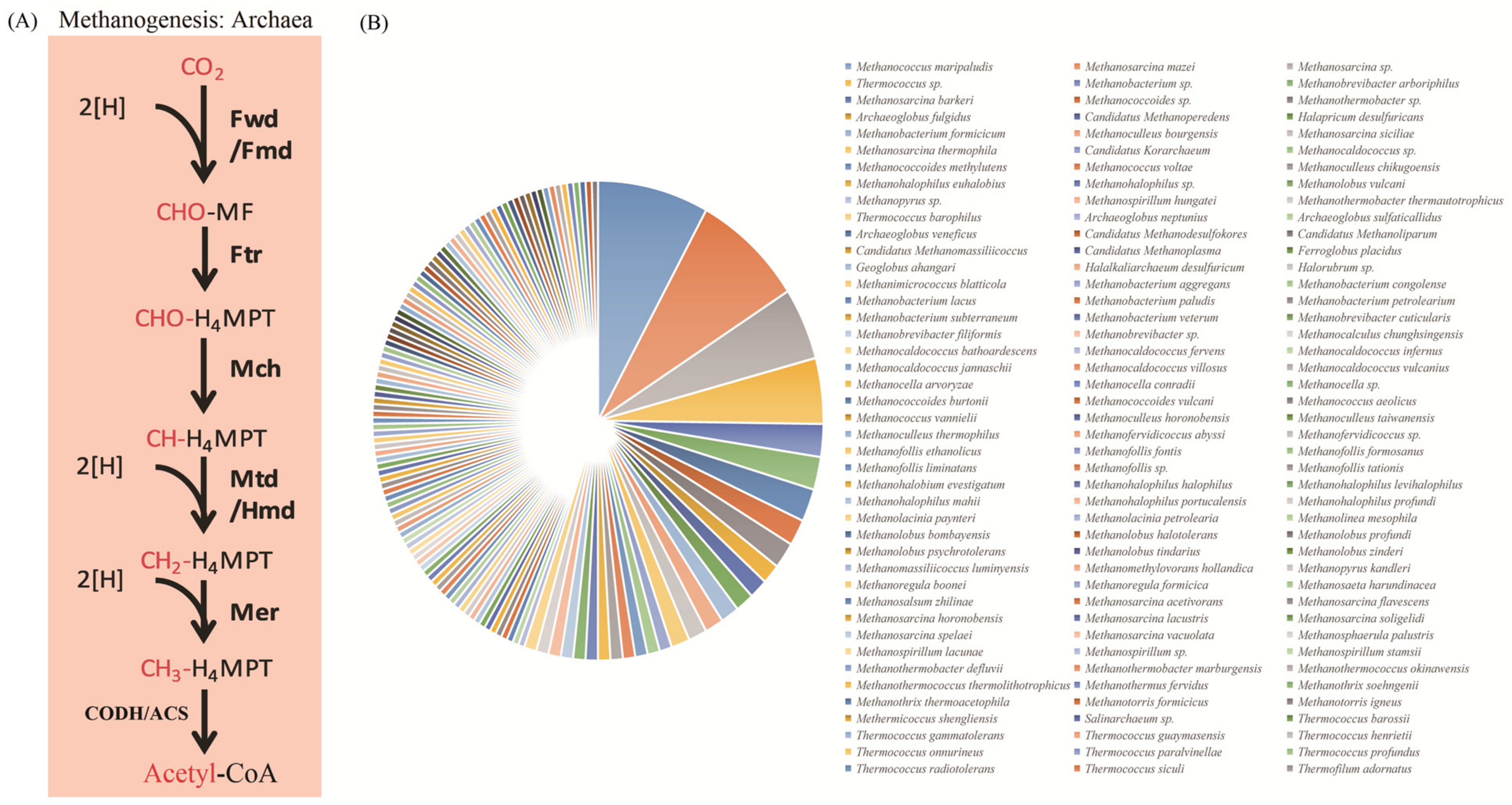
3.3. Applications of the KEGG_Extractor in Genomic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burian, A.N.; Zhao, W.; Lo, T.W.; Thurtle-Schmidt, D.M. Genome sequencing guide: An introductory toolbox to whole-genome analysis methods. Biochem. Mol. Biol. Educ. 2021, 49, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Land, M.; Hauser, L.; Jun, S.R.; Nookaew, I.; Leuze, M.R.; Ahn, T.H.; Karpinets, T.; Lund, O.; Kora, G.; Wassenaar, T.; et al. Insights from 20 years of bacterial genome sequencing. Funct. Integr. Genom. 2015, 15, 141–161. [Google Scholar] [CrossRef] [PubMed]
- Boolchandani, M.; D’Souza, A.W.; Dantas, G. Sequencing-based methods and resources to study antimicrobial resistance. Nat. Rev. Genet. 2019, 20, 356–370. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Borrel, G.; Adam, P.S.; Gribaldo, S. Methanogenesis and the Wood-Ljungdahl Pathway: An Ancient, Versatile, and Fragile Association. Genome Biol. Evol. 2016, 8, 1706–1711. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.G. Whole-genome sequencing in the prediction of antimicrobial resistance. Expert. Rev. Anti. Infect. Ther. 2016, 14, 617–619. [Google Scholar] [CrossRef]
- Bräuer, S.L.; Cadillo-Quiroz, H.; Ward, R.J.; Yavitt, J.B.; Zinder, S.H. Methanoregula boonei gen. nov., sp. nov., an acidiphilic methanogen isolated from an acidic peat bog. Int. J. Syst. Evol. Microbiol. 2011, 61, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Yashiro, Y.; Sakai, S.; Ehara, M.; Miyazaki, M.; Yamaguchi, T.; Imachi, H. Methanoregula formicica sp. nov., a methane-producing archaeon isolated from methanogenic sludge. Int. J. Syst. Evol. Microbiol. 2011, 61, 53–59. [Google Scholar] [CrossRef]
- Sakai, S.; Conrad, R.; Liesack, W.; Imachi, H. Methanocella arvoryzae sp. nov., a hydrogenotrophic methanogen isolated from rice field soil. Int. J. Syst. Evol. Microbiol. 2010, 60, 2918–2923. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M. The KEGG database. Novartis Found Symp. 2002, 247, 91–101, discussion 101–103, 119–128, 244–252. [Google Scholar]
- Kanehisa, M.; Sato, Y. KEGG Mapper for inferring cellular functions from protein sequences. Protein Sci. 2020, 29, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M. KEGG mapping tools for uncovering hidden features in biological data. Protein Sci. 2022, 31, 47–53. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 2014, 42, D199–D205. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Suzuki, S.; Ishida, T.; Ohue, M.; Kakuta, M.; Akiyama, Y. GHOSTX: A Fast Sequence Homology Search Tool for Functional Annotation of Metagenomic Data. Methods Mol. Biol. 2017, 1611, 15–25. [Google Scholar]
- Aramaki, T.; Blanc-Mathieu, R.; Endo, H.; Ohkubo, K.; Kanehisa, M.; Goto, S.; Ogata, H. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 2020, 36, 2251–2252. [Google Scholar] [CrossRef]
- Salam, L.B.; Obayori, O.S. Remarkable shift in structural and functional properties of an animal charcoal-polluted soil accentuated by inorganic nutrient amendment. J. Genet. Eng. Biotechnol. 2020, 18, 70. [Google Scholar] [CrossRef] [PubMed]
- Salam, L.B.; Obayori, O.S. Functional characterization of the ABC transporters and transposable elements of an uncultured Paracoccus sp. recovered from a hydrocarbon-polluted soil metagenome. Folia Microbiol 2022, 26, 241–248. [Google Scholar] [CrossRef]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinform. 2014, 47, 11.12.1–11.12.34. [Google Scholar] [CrossRef]
- Pertea, G.; Pertea, M. GFF Utilities: GffRead and GffCompare. F1000Res 2020, 9, 304. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, 545–551. [Google Scholar] [CrossRef]
- Esposito, A.; Tamburini, S.; Triboli, L.; Ambrosino, L.; Chiusano, M.L.; Jousson, O. Insights into the genome structure of four acetogenic bacteria with specific reference to the Wood-Ljungdahl pathway. Microbiologyopen 2019, 8, e938. [Google Scholar] [CrossRef]
- Adam, P.S.; Borrel, G.; Gribaldo, S. Evolutionary history of carbon monoxide dehydrogenase/acetyl-CoA synthase, one of the oldest enzymatic complexes. Proc. Natl. Acad. Sci. USA 2018, 115, E1166–E1173. [Google Scholar] [CrossRef]
- Martin, W.; Russell, M.J. On the origin of biochemistry at an alkaline hydrothermal vent. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2007, 362, 1887–1925. [Google Scholar] [CrossRef] [PubMed]
- Ragsdale, S.W. Life with carbon monoxide. Crit. Rev. Biochem. Mol. Biol. 2004, 39, 165–195. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019, 47, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. Enzyme Annotation and Metabolic Reconstruction Using KEGG. Methods Mol Biol. 2017, 1611, 135–145. [Google Scholar]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 2005, 21, 3787–3793. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Types | KOfamScan | BlastKOALA | GhostKOALA | KAAS |
|---|---|---|---|---|
| Number of extracted genes | 40 | 37 | 39 | 34 |
| True positive rate | 100% | 100% | 100% | 100% |
| Recall | 1 | 0.925 | 0.975 | 0.85 |
| Pathway (Entry) | KOs | Key Enzymes | Substrate | Strains |
|---|---|---|---|---|
| Wood–Ljungdahl pathway (M00377) | K00198 | cooS, acsA; anaerobic carbon monoxide dehydrogenase catalytic subunit [EC:1.2.7.4] | CO2 | Bacteria (Firmicutes, Planctomycetes, Deltaproteobacteria, Spirochaeata); Archaea (Euryarchaeota) |
| K15023 | acsE; 5-methyltetrahydrofolate corrinoid/iron sulfur protein methyltransferase [EC:2.1.1.258] | |||
| K14138 | acsB; acetyl-CoA synthase [EC:2.3.1.169] | |||
| K00197 | cdhE, acsC; acetyl-CoA decarbonylase/synthase, CODH/ACS complex subunit γ [EC:2.1.1.245] | |||
| K00192-K00195 | cdhABCD; anaerobic carbon monoxide dehydrogenase, CODH/ACS complex subunit |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Chen, Z.; Zhang, M.; Jia, S. KEGG_Extractor: An Effective Extraction Tool for KEGG Orthologs. Genes 2023, 14, 386. https://doi.org/10.3390/genes14020386
Zhang C, Chen Z, Zhang M, Jia S. KEGG_Extractor: An Effective Extraction Tool for KEGG Orthologs. Genes. 2023; 14(2):386. https://doi.org/10.3390/genes14020386
Chicago/Turabian StyleZhang, Chao, Zhongwei Chen, Miming Zhang, and Shulei Jia. 2023. "KEGG_Extractor: An Effective Extraction Tool for KEGG Orthologs" Genes 14, no. 2: 386. https://doi.org/10.3390/genes14020386