
Combined MITOchondrial-NUCLEAR (MITO-NUCLEAR) Analysis for Mitochondrial Diseases Diagnosis: Validation and Implementation of a One-Step NGS Method


 ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples for Validation Experiments
2.2. DNA Purification
2.3. Samples Preparation
2.4. Next-Generation Sequencing
3. Results
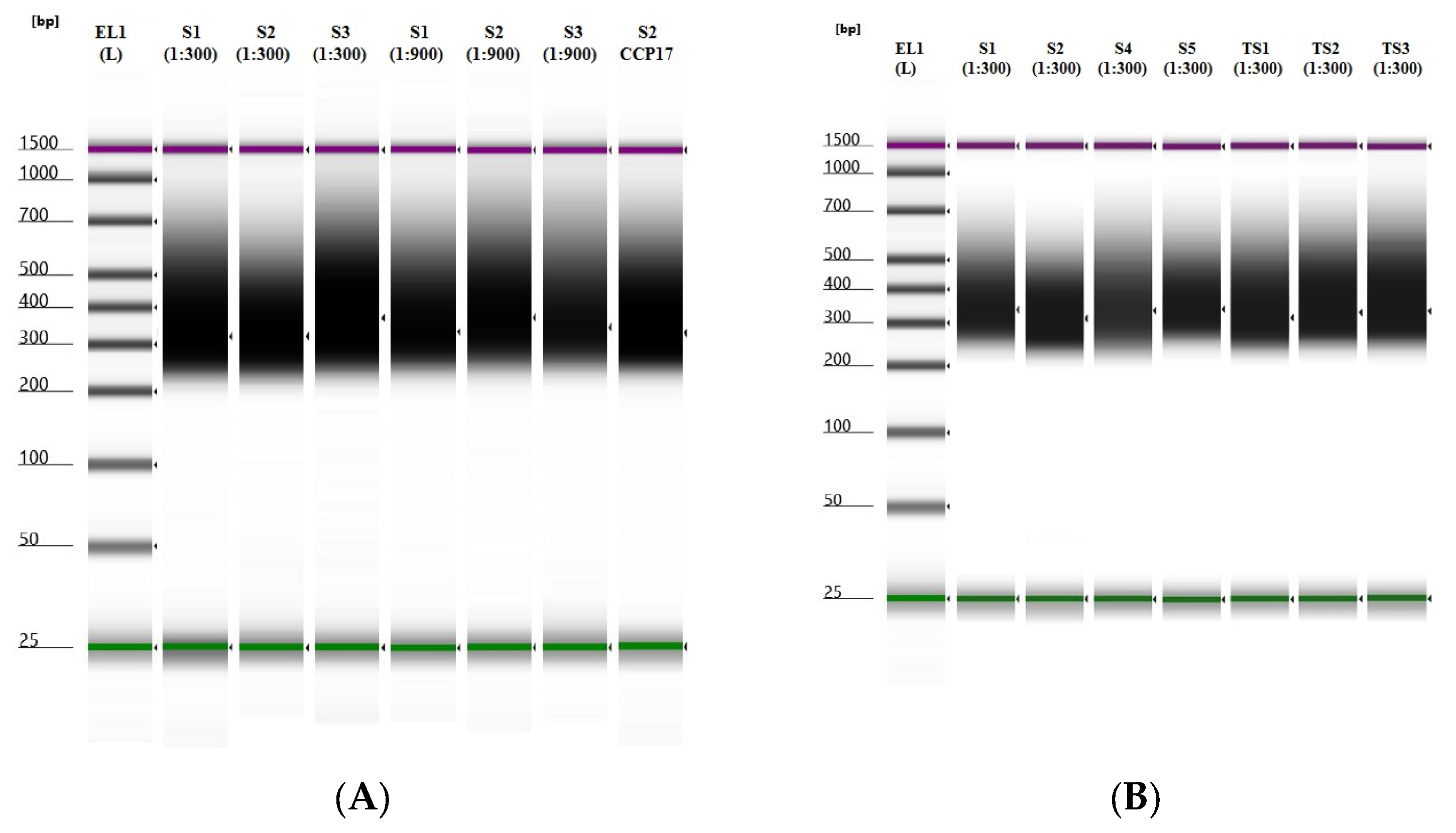
3.1. Libraries Samples Suitability
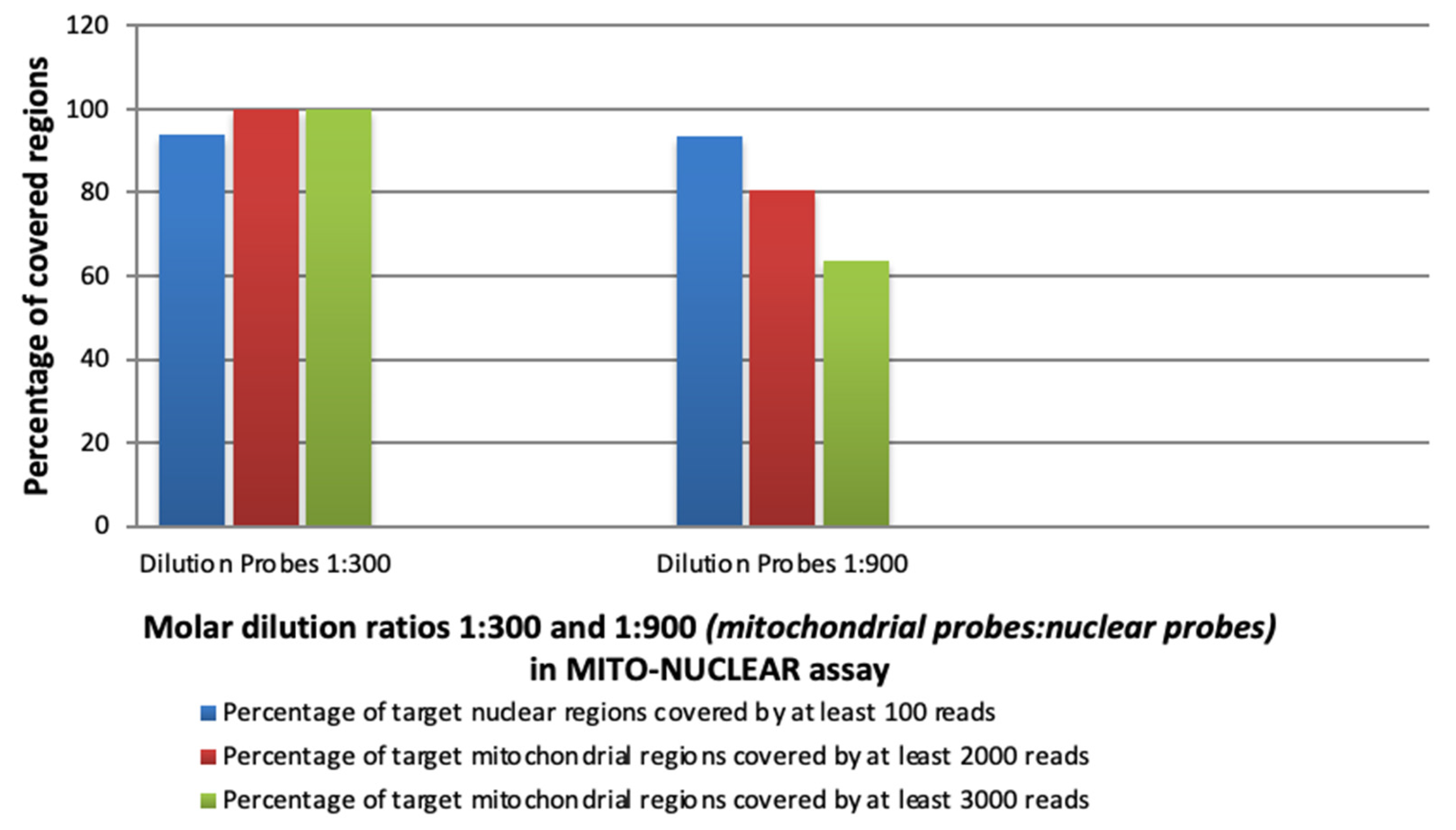
3.2. Experimental Evaluation of the Capture Efficiency of mtDNA (SSel Custom Constitutional Panel 16.708 Kbp) and Nuclear CCP17 (Custom Constitutional Panel; Design Size: 17 Mb) in Blended MITO-NUCLEAR Panel at Different Concentrations
3.3. Diagnostic Yield of MITO-NUCLEAR Assay
3.4. mtDNA Genome Heteroplasmy Detection
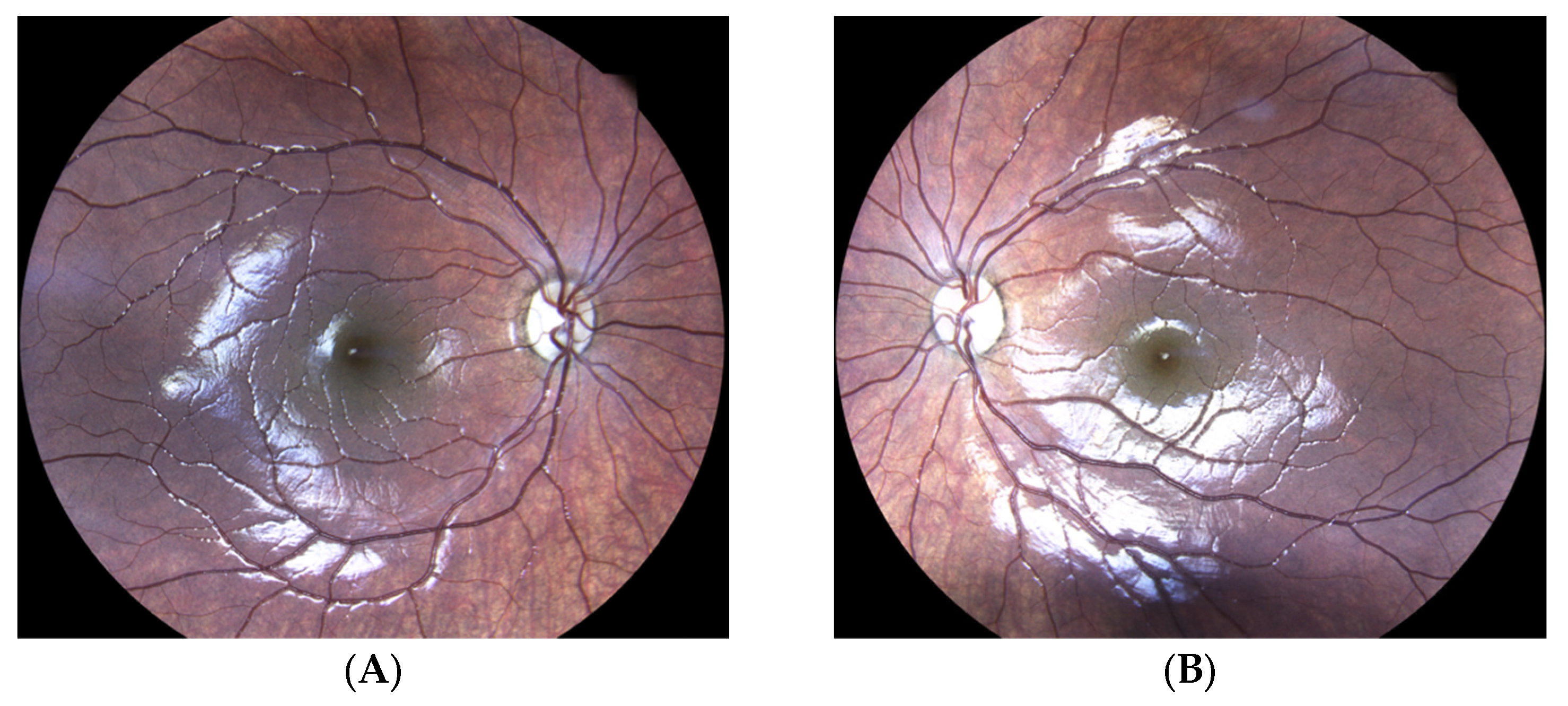
3.5. Molecular Diagnosis of Autosomal Dominant Optic Atrophy by MITO-NUCLEAR Investigation (Case Report)
4. Discussion
5. Study Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thompson, K.; Collier, J.J.; Glasgow, R.I.C.; Robertson, F.M.; Pyle, A.; Blakely, E.L.; Alston, C.L.; Olahova, M.; McFarland, R.; Taylor, R.W. Recent advances in understanding the molecular genetic basis of mitochondrial disease. J. Inherit. Metab. Dis. 2020, 43, 36–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craven, L.; Alston, C.L.; Taylor, R.W.; Turnbull, D.M. Recent Advances in Mitochondrial Disease. Annu. Rev. Genom. Hum. Genet. 2017, 18, 257–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alston, C.L.; Rocha, M.C.; Lax, N.Z.; Turnbull, D.M.; Taylor, R.W. The genetics and pathology of mitochondrial disease. J. Pathol. 2017, 241, 236–250. [Google Scholar] [CrossRef] [PubMed]
- Lioncino, M.; Monda, E.; Caiazza, M.; Fusco, A.; Cirillo, A.; Dongiglio, F.; Simonelli, V.; Sampaolo, S.; Ruggiero, L.; Scarano, G.; et al. Cardiovascular Involvement in mtDNA Disease: Diagnosis, Management, and Therapeutic Options. Heart Fail. Clin. 2022, 18, 51–60. [Google Scholar] [CrossRef]
- McCormick, E.M.; Zolkipli-Cunningham, Z.; Falk, M.J. Mitochondrial disease genetics update: Recent insights into the molecular diagnosis and expanding phenotype of primary mitochondrial disease. Curr. Opin. Pediatr. 2018, 30, 714–724. [Google Scholar] [CrossRef]
- Aldossary, A.M.; Tawfik, E.A.; Alomary, M.N.; Alsudir, S.A.; Alfahad, A.J.; Alshehri, A.A.; Almughem, F.A.; Mohammed, R.Y.; Alzaydi, M.M. Recent advances in mitochondrial diseases: From molecular insights to therapeutic perspectives. Saudi Pharm. J. 2022, 30, 1065–1078. [Google Scholar] [CrossRef]
- Iafusco, D.; Zanfardino, A.; Piscopo, A.; Curto, S.; Troncone, A.; Chianese, A.; Rollato, A.S.; Testa, V.; Iafusco, F.; Maione, G.; et al. Metabolic Treatment of Wolfram Syndrome. Int. J. Environ. Res. Public Health 2022, 19, 2755. [Google Scholar] [CrossRef]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080. [Google Scholar] [CrossRef]
- Calvo, S.E.; Mootha, V.K. The mitochondrial proteome and human disease. Annu. Rev. Genom. Hum. Genet. 2010, 11, 25–44. [Google Scholar] [CrossRef] [Green Version]
- Ng, Y.S.; Turnbull, D.M. Mitochondrial disease: Genetics and management. J. Neurol. 2016, 263, 179–191. [Google Scholar] [CrossRef] [Green Version]
- Mazzaccara, C.; Mirra, B.; Barretta, F.; Caiazza, M.; Lombardo, B.; Scudiero, O.; Tinto, N.; Limongelli, G.; Frisso, G. Molecular Epidemiology of Mitochondrial Cardiomyopathy: A Search Among Mitochondrial and Nuclear Genes. Int. J. Mol. Sci. 2021, 22, 5742. [Google Scholar] [CrossRef]
- Barretta, F.; Mirra, B.; Monda, E.; Caiazza, M.; Lombardo, B.; Tinto, N.; Scudiero, O.; Frisso, G.; Mazzaccara, C. The Hidden Fragility in the Heart of the Athletes: A Review of Genetic Biomarkers. Int. J. Mol. Sci. 2020, 21, 6682. [Google Scholar] [CrossRef]
- Mazzaccara, C.; Iafusco, D.; Liguori, R.; Ferrigno, M.; Galderisi, A.; Vitale, D.; Simonelli, F.; Landolfo, P.; Prisco, F.; Masullo, M.; et al. Mitochondrial diabetes in children: Seek and you will find it. PLoS ONE 2012, 7, e34956. [Google Scholar] [CrossRef]
- Vertika, S.; Singh, K.K.; Rajender, S. Mitochondria, spermatogenesis, and male infertility—An update. Mitochondrion 2020, 54, 26–40. [Google Scholar] [CrossRef]
- Baszynska-Wilk, M.; Moszczynska, E.; Szarras-Czapnik, M.; Wysocka-Mincewicz, M.; Watrobinska, U.; Kozlowska, A.; Szalecki, M. Endocrine disorders in a patient with a suspicion of a mitochondrial disease, MELAS syndrome—A case report and literature review. Pediatr. Endocrinol. Diabetes Metab. 2021, 27, 213–218. [Google Scholar] [CrossRef]
- Al-Gadi, I.S.; Haas, R.H.; Falk, M.J.; Goldstein, A.; McCormack, S.E. Endocrine Disorders in Primary Mitochondrial Disease. J. Endocr. Soc. 2018, 2, 361–373. [Google Scholar] [CrossRef]
- Carroll, C.J.; Brilhante, V.; Suomalainen, A. Next-generation sequencing for mitochondrial disorders. Br. J. Pharmacol. 2014, 171, 1837–1853. [Google Scholar] [CrossRef] [Green Version]
- Protasoni, M.; Zeviani, M. Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions. Int. J. Mol. Sci. 2021, 22, 586. [Google Scholar] [CrossRef]
- Frazier, A.E.; Thorburn, D.R.; Compton, A.G. Mitochondrial energy generation disorders: Genes, mechanisms, and clues to pathology. J. Biol. Chem. 2019, 294, 5386–5395. [Google Scholar] [CrossRef] [Green Version]
- Mahmud, S.; Biswas, S.; Afrose, S.; Mita, M.A.; Hasan, M.R.; Shimu, M.S.S.; Paul, G.K.; Chung, S.; Saleh, M.A.; Alshehri, S.; et al. Use of Next-Generation Sequencing for Identifying Mitochondrial Disorders. Curr. Issues Mol. Biol. 2022, 44, 1127–1148. [Google Scholar] [CrossRef]
- Lombardo, B.; D’Argenio, V.; Monda, E.; Vitale, A.; Caiazza, M.; Sacchetti, L.; Pastore, L.; Limongelli, G.; Frisso, G.; Mazzaccara, C. Genetic analysis resolves differential diagnosis of a familial syndromic dilated cardiomyopathy: A new case of Alstrom syndrome. Mol. Genet. Genom. Med. 2020, 8, e1260. [Google Scholar] [CrossRef] [PubMed]
- Legati, A.; Reyes, A.; Nasca, A.; Invernizzi, F.; Lamantea, E.; Tiranti, V.; Garavaglia, B.; Lamperti, C.; Ardissone, A.; Moroni, I.; et al. New genes and pathomechanisms in mitochondrial disorders unraveled by NGS technologies. Biochim. Biophys. Acta 2016, 1857, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Mazzaccara, C.; Lombardi, R.; Mirra, B.; Barretta, F.; Esposito, M.V.; Uomo, F.; Caiazza, M.; Monda, E.; Losi, M.A.; Limongelli, G.; et al. Next-Generation Sequencing Gene Panels in Inheritable Cardiomyopathies and Channelopathies: Prevalence of Pathogenic Variants and Variants of Unknown Significance in Uncommon Genes. Biomolecules 2022, 12, 1417. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, G.; Nunziato, M.; Mazzaccara, C.; Intrieri, M.; D’argenio, V.; Esposito, M.V.; Monda, E.; Di Maggio, F.; Frisso, G.; Salvatore, F. Genotype-Phenotype Correlation: A Triple DNA Mutational Event in a Boy Entering Sport Conveys an Additional Pathogenicity Risk. Genes 2020, 11, 524. [Google Scholar] [CrossRef]
- Monda, E.; Sarubbi, B.; Russo, M.G.; Caiazza, M.; Mazzaccara, C.; Magrelli, J.; Rubino, M.; Esposito, A.; Perna, A.; Passariello, A.; et al. Unexplained sudden cardiac arrest in children: Clinical and genetic characteristics of survivors. Eur. J. Prev. Cardiol. 2020, 28, 1134–1137. [Google Scholar] [CrossRef]
- Calvo, S.E.; Compton, A.G.; Hershman, S.G.; Lim, S.C.; Lieber, D.S.; Tucker, E.J.; Laskowski, A.; Garone, C.; Liu, S.; Jaffe, D.B.; et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci. Transl. Med. 2012, 4, 118ra110. [Google Scholar] [CrossRef] [Green Version]
- Falk, M.J.; Pierce, E.A.; Consugar, M.; Xie, M.H.; Guadalupe, M.; Hardy, O.; Rappaport, E.F.; Wallace, D.C.; LeProust, E.; Gai, X.W. Mitochondrial Disease Genetic Diagnostics: Optimized Whole-Exome Analysis for All MitoCarta Nuclear Genes and the Mitochondrial Genome. Discov Med. 2012, 14, 389-U140. [Google Scholar]
- Liu, Q.; Guo, Y.; Li, J.; Long, J.; Zhang, B.; Shyr, Y. Steps to ensure accuracy in genotype and SNP calling from Illumina sequencing data. BMC Genom. 2012, 13 (Suppl. 8), S8. [Google Scholar] [CrossRef] [Green Version]
- Picardi, E.; Pesole, G. Mitochondrial genomes gleaned from human whole-exome sequencing. Nat. Methods 2012, 9, 523–524. [Google Scholar] [CrossRef]
- Vasta, V.; Merritt, J.L., 2nd; Saneto, R.P.; Hahn, S.H. Next-generation sequencing for mitochondrial diseases: A wide diagnostic spectrum. Pediatr. Int. Off. J. Jpn. Pediatr. Soc. 2012, 54, 585–601. [Google Scholar] [CrossRef]
- Dames, S.; Chou, L.S.; Xiao, Y.; Wayman, T.; Stocks, J.; Singleton, M.; Eilbeck, K.; Mao, R. The development of next-generation sequencing assays for the mitochondrial genome and 108 nuclear genes associated with mitochondrial disorders. J. Mol. Diagn. 2013, 15, 526–534. [Google Scholar] [CrossRef]
- Abicht, A.; Scharf, F.; Kleinle, S.; Schon, U.; Holinski-Feder, E.; Horvath, R.; Benet-Pages, A.; Diebold, I. Mitochondrial and nuclear disease panel (Mito-aND-Panel): Combined sequencing of mitochondrial and nuclear DNA by a cost-effective and sensitive NGS-based method. Mol. Genet. Genom. Med. 2018, 6, 1188–1198. [Google Scholar] [CrossRef]
- Davis, R.L.; Kumar, K.R.; Puttick, C.; Liang, C.; Ahmad, K.E.; Edema-Hildebrand, F.; Park, J.S.; Minoche, A.E.; Gayevskiy, V.; Mallawaarachchi, A.C.; et al. Use of Whole-Genome Sequencing for Mitochondrial Disease Diagnosis. Neurology 2022, 99, e730–e742. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- World Medical Association. World Medical Association Declaration of Helsinki: Ethical principles for medical research involving human subjects. JAMA 2013, 310, 2191–2194. [Google Scholar] [CrossRef] [Green Version]
- Koopman, W.J.; Willems, P.H.; Smeitink, J.A. Monogenic mitochondrial disorders. N. Engl. J. Med. 2012, 366, 1132–1141. [Google Scholar] [CrossRef] [Green Version]
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016, 44, D1251–D1257. [Google Scholar] [CrossRef] [Green Version]
- Duan, M.; Tu, J.; Lu, Z. Recent Advances in Detecting Mitochondrial DNA Heteroplasmic Variations. Molecules 2018, 23, 323. [Google Scholar] [CrossRef] [Green Version]
- Liguori, R.; Mazzaccara, C.; Pasanisi, F.; Buono, P.; Oriani, G.; Finelli, C.; Contaldo, F.; Sacchetti, L. The mtDNA 15497 G/A polymorphism in cytochrome b in severe obese subjects from Southern Italy. Nutr. Metab. Cardiovasc. Dis. 2006, 16, 466–470. [Google Scholar] [CrossRef]
- Nardelli, C.; Labruna, G.; Liguori, R.; Mazzaccara, C.; Ferrigno, M.; Capobianco, V.; Pezzuti, M.; Castaldo, G.; Farinaro, E.; Contaldo, F.; et al. Haplogroup T is an obesity risk factor: Mitochondrial DNA haplotyping in a morbid obese population from southern Italy. BioMed Res. Int. 2013, 2013, 631082. [Google Scholar] [CrossRef] [Green Version]
- Levy, M.A.; Kerkhof, J.; Belmonte, F.R.; Kaufman, B.A.; Bhai, P.; Brady, L.; Bursztyn, L.; Tarnopolsky, M.; Rupar, T.; Sadikovic, B. Validation and clinical performance of a combined nuclear-mitochondrial next-generation sequencing and copy number variant analysis panel in a Canadian population. Am. J. Med. Genet. Part A 2021, 185, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Matthijs, G.; Souche, E.; Alders, M.; Corveleyn, A.; Eck, S.; Feenstra, I.; Race, V.; Sistermans, E.; Sturm, M.; Weiss, M.; et al. Guidelines for diagnostic next-generation sequencing. Eur. J. Hum. Genet. 2016, 24, 1515. [Google Scholar] [CrossRef] [PubMed]
- Jennings, L.J.; Arcila, M.E.; Corless, C.; Kamel-Reid, S.; Lubin, I.M.; Pfeifer, J.; Temple-Smolkin, R.L.; Voelkerding, K.V.; Nikiforova, M.N. Guidelines for Validation of Next-Generation Sequencing-Based Oncology Panels: A Joint Consensus Recommendation of the Association for Molecular Pathology and College of American Pathologists. J. Mol. Diagn. 2017, 19, 341–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenaers, G.; Reynier, P.; Elachouri, G.; Soukkarieh, C.; Olichon, A.; Belenguer, P.; Baricault, L.; Ducommun, B.; Hamel, C.; Delettre, C. OPA1 functions in mitochondria and dysfunctions in optic nerve. Int. J. Biochem. Cell Biol. 2009, 41, 1866–1874. [Google Scholar] [CrossRef] [Green Version]
- Lenaers, G.; Hamel, C.; Delettre, C.; Amati-Bonneau, P.; Procaccio, V.; Bonneau, D.; Reynier, P.; Milea, D. Dominant optic atrophy. Orphanet J. Rare Dis. 2012, 7, 46. [Google Scholar] [CrossRef] [Green Version]
- Al Othman, B.; Ong, J.E.; Dumitrescu, A.V. Biallelic Optic Atrophy 1 (OPA1) Related Disorder-Case Report and Literature Review. Genes 2022, 13, 1005. [Google Scholar] [CrossRef]
- Zhuang, J.; Cao, Z.; Zhu, Y.; Liu, L.; Tong, Y.; Chen, X.; Wang, Y.; Lu, C.; Ma, X.; Yang, J. Mutation screening of crystallin genes in Chinese families with congenital cataracts. Mol. Vis. 2019, 25, 427–437. [Google Scholar]
- Cohn, A.C.; Toomes, C.; Potter, C.; Towns, K.V.; Hewitt, A.W.; Inglehearn, C.F.; Craig, J.E.; Mackey, D.A. Autosomal dominant optic atrophy: Penetrance and expressivity in patients with OPA1 mutations. Am. J. Ophthalmol. 2007, 143, 656–662.e1. [Google Scholar] [CrossRef]
- Rose, G.; Passarino, G.; Scornaienchi, V.; Romeo, G.; Dato, S.; Bellizzi, D.; Mari, V.; Feraco, E.; Maletta, R.; Bruni, A.; et al. The mitochondrial DNA control region shows genetically correlated levels of heteroplasmy in leukocytes of centenarians and their offspring. BMC Genom. 2007, 8, 293. [Google Scholar] [CrossRef] [Green Version]
- Tsiatis, A.C.; Norris-Kirby, A.; Rich, R.G.; Hafez, M.J.; Gocke, C.D.; Eshleman, J.R.; Murphy, K.M. Comparison of Sanger sequencing, pyrosequencing, and melting curve analysis for the detection of KRAS mutations: Diagnostic and clinical implications. J. Mol. Diagn. JMD 2010, 12, 425–432. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| ID | Tissue Type | Sample Type | Dilution mtDNA Probes | Bed Analysed | Average Reading Depth (Coverage) | |
|---|---|---|---|---|---|---|
| RUN 1 | S1 | Blood | MITO-NUCLEAR | 1.300 | Nuclear CCP17 | 126 |
| MITO-NUCLEAR | 1.300 | mtDNA | 5655 | |||
| S1 | Blood | MITO-NUCLEAR | 1.900 | Nuclear CCP17 | 142 | |
| MITO-NUCLEAR | 1.900 | mtDNA | 3131 | |||
| S2 | Blood | MITO-NUCLEAR | 1.300 | Nuclear CCP17 | 165 | |
| MITO-NUCLEAR | 1.300 | mtDNA | 3846 | |||
| S2 | Blood | MITO-NUCLEAR | 1.900 | Nuclear CCP17 | 143 | |
| MITO-NUCLEAR | 1.900 | mtDNA | 1616 | |||
| S3 | Blood | MITO-NUCLEAR | 1.300 | Nuclear CCP17 | 150 | |
| MITO-NUCLEAR | 1.300 | mtDNA | 4607 | |||
| S3 | Blood | MITO-NUCLEAR | 1.900 | Nuclear CCP17 | 156 | |
| MITO-NUCLEAR | 1.900 | mtDNA | 2329 | |||
| S2 | Blood | NUCLEAR CCP17 | ND | Nuclear CCP17 | 155 | |
| RUN 2 | S1 | Blood | MITO-NUCLEAR | 1.300 | Nuclear CCP17 | 140 |
| MITO-NUCLEAR | 1.300 | mtDNA | 7000 | |||
| S2 | Blood | MITO-NUCLEAR | 1.300 | Nuclear CCP17 | 158 | |
| MITO-NUCLEAR | 1.300 | mtDNA | 3920 | |||
| S4 | Swab | MITO-NUCLEAR | 1.300 | Nuclear CCP17 | 162 | |
| MITO-NUCLEAR | 1.300 | mtDNA | 3700 | |||
| S5 | Swab | MITO-NUCLEAR | 1.300 | Nuclear CCP17 | 149 | |
| MITO-NUCLEAR | 1.300 | mtDNA | 3280 | |||
| TS1 | Fresh Tissue | MITO-NUCLEAR | 1.300 | Nuclear CCP17 | 155 | |
| MITO-NUCLEAR | 1.300 | mtDNA | 7000 | |||
| TS2 | Tissue from slide | MITO-NUCLEAR | 1.300 | Nuclear CCP17 | 163 | |
| MITO-NUCLEAR | 1.300 | mtDNA | 5127 | |||
| TS3 | FFPE | MITO-NUCLEAR | 1.300 | Nuclear CCP17 | 125 | |
| MITO-NUCLEAR | 1.300 | mtDNA | 11,000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barretta, F.; Uomo, F.; Caldora, F.; Mocerino, R.; Adamo, D.; Testa, F.; Simonelli, F.; Scudiero, O.; Tinto, N.; Frisso, G.; et al. Combined MITOchondrial-NUCLEAR (MITO-NUCLEAR) Analysis for Mitochondrial Diseases Diagnosis: Validation and Implementation of a One-Step NGS Method. Genes 2023, 14, 1087. https://doi.org/10.3390/genes14051087
Barretta F, Uomo F, Caldora F, Mocerino R, Adamo D, Testa F, Simonelli F, Scudiero O, Tinto N, Frisso G, et al. Combined MITOchondrial-NUCLEAR (MITO-NUCLEAR) Analysis for Mitochondrial Diseases Diagnosis: Validation and Implementation of a One-Step NGS Method. Genes. 2023; 14(5):1087. https://doi.org/10.3390/genes14051087
Chicago/Turabian StyleBarretta, Ferdinando, Fabiana Uomo, Filomena Caldora, Rossella Mocerino, Daniela Adamo, Francesco Testa, Francesca Simonelli, Olga Scudiero, Nadia Tinto, Giulia Frisso, and et al. 2023. "Combined MITOchondrial-NUCLEAR (MITO-NUCLEAR) Analysis for Mitochondrial Diseases Diagnosis: Validation and Implementation of a One-Step NGS Method" Genes 14, no. 5: 1087. https://doi.org/10.3390/genes14051087
APA StyleBarretta, F., Uomo, F., Caldora, F., Mocerino, R., Adamo, D., Testa, F., Simonelli, F., Scudiero, O., Tinto, N., Frisso, G., & Mazzaccara, C. (2023). Combined MITOchondrial-NUCLEAR (MITO-NUCLEAR) Analysis for Mitochondrial Diseases Diagnosis: Validation and Implementation of a One-Step NGS Method. Genes, 14(5), 1087. https://doi.org/10.3390/genes14051087