


Genome-Wide Characterization and Expression Profiling of Phytosulfokine Receptor Genes (PSKRs) in Triticum aestivum with Docking Simulations of Their Interactions with Phytosulfokine (PSK): A Bioinformatics Study


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. TaPSKR Gene Family Identification
2.2. Chromosomal Localization and Nomenclature
2.3. Phylogenetic Tree Construction
2.4. RNA-Seq Data Collection
2.5. Quality Control
2.6. Read Mapping
2.7. Statistical Analysis and Visualization
2.8. Docking Screening Analysis
3. Results
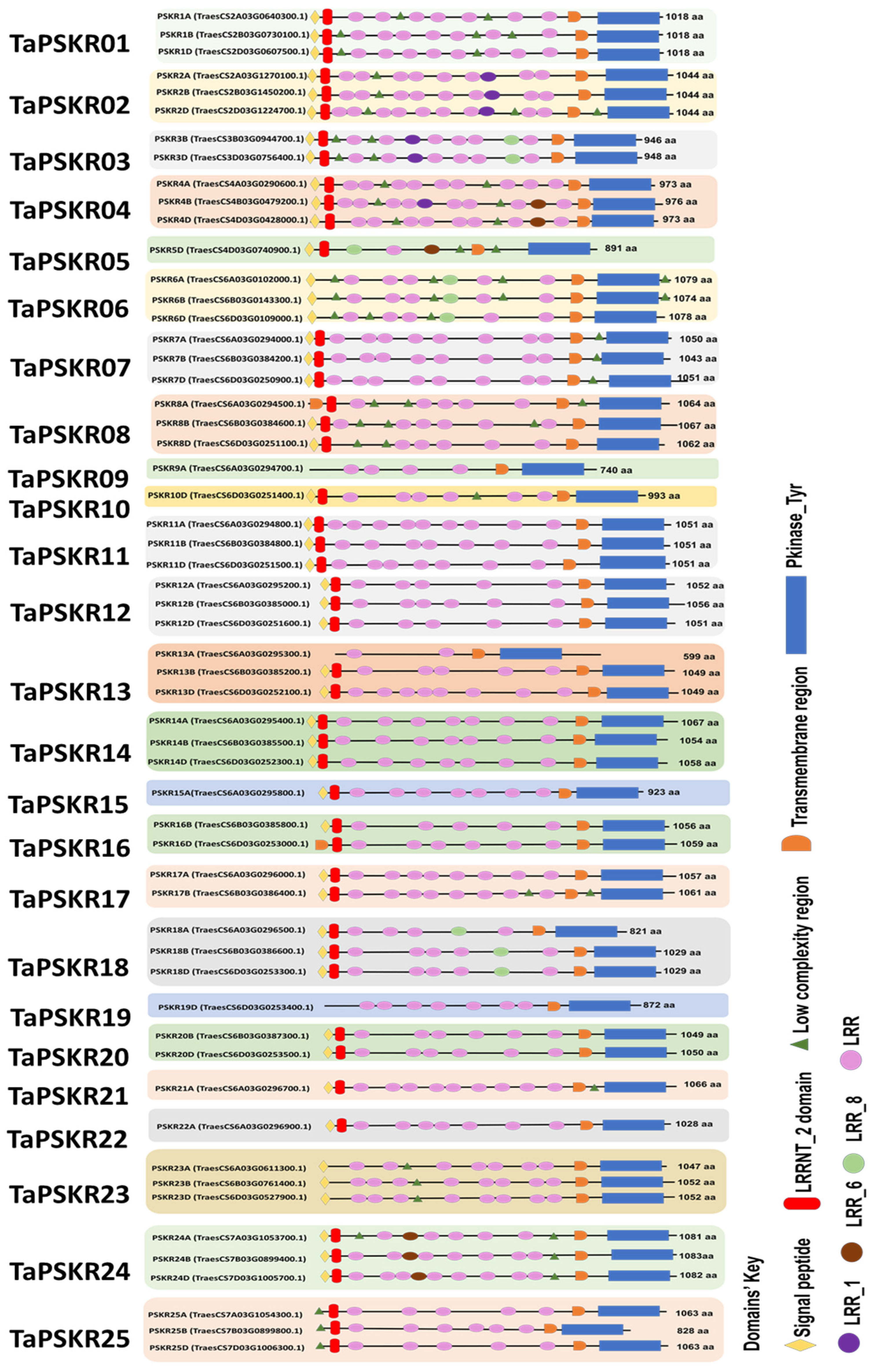
3.1. Genome-Wide Analysis of TaPSKR
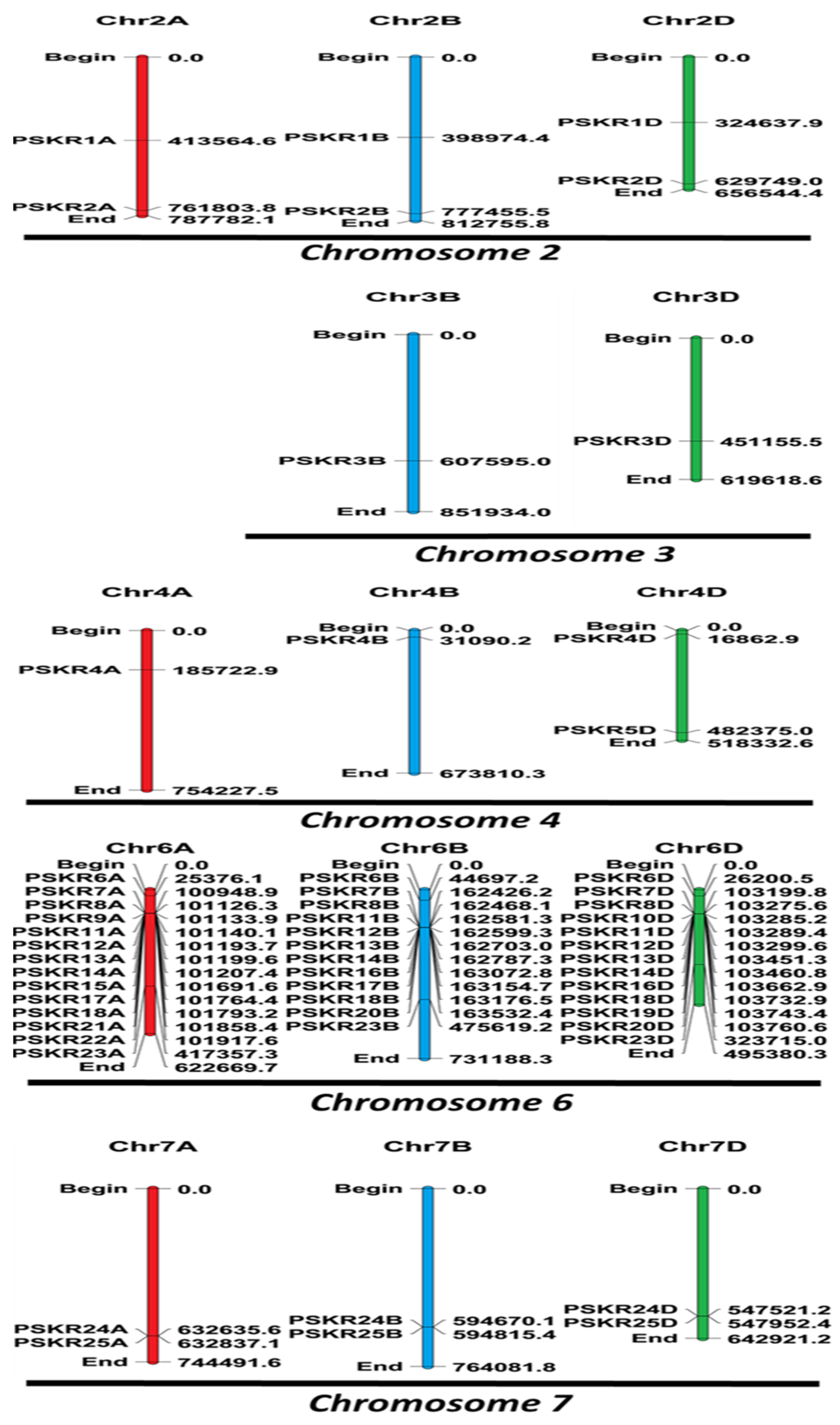
3.2. Chromosomal Assignment of TaPSKR
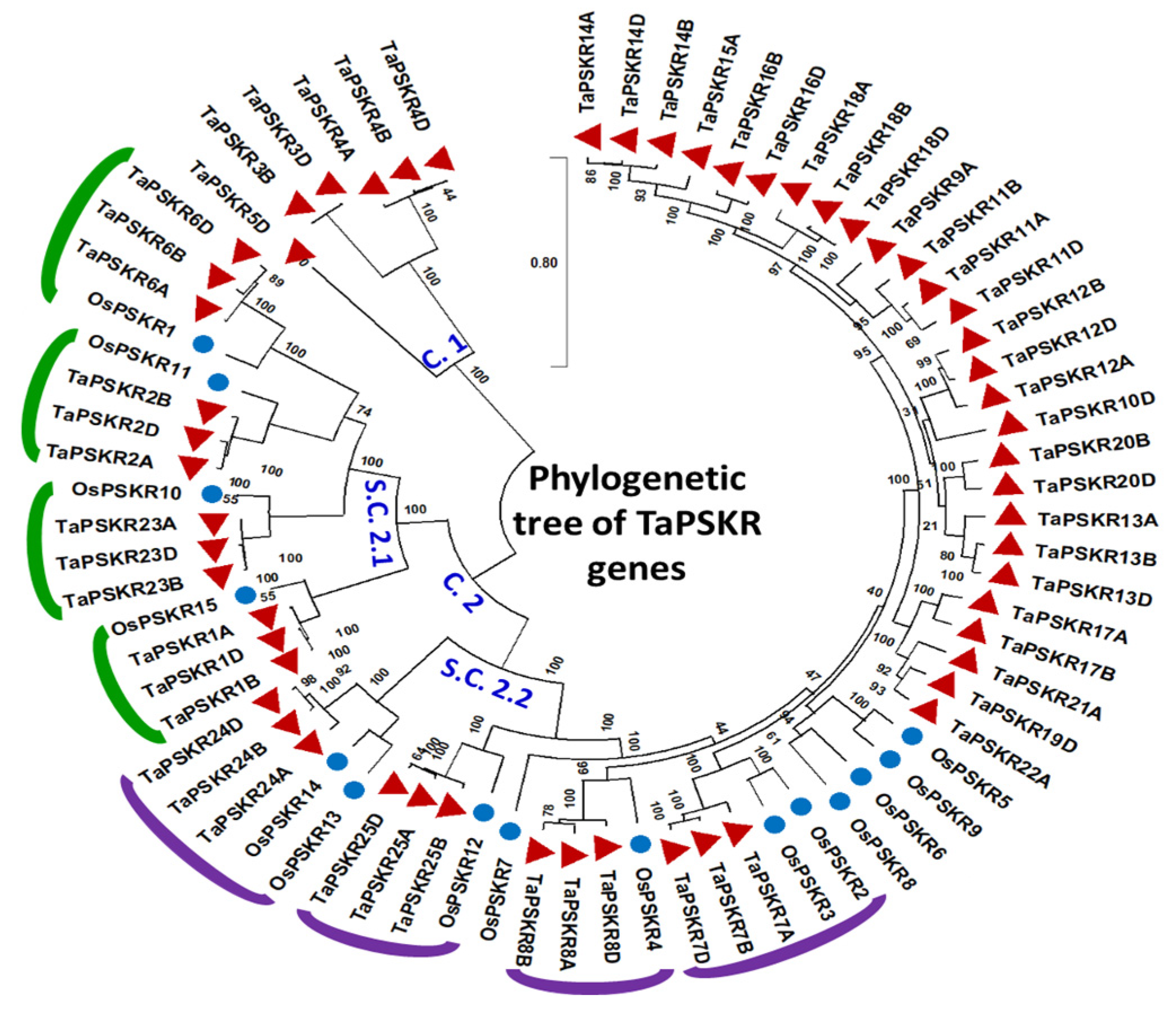
3.3. Evolution of TaPSKR Gene Family
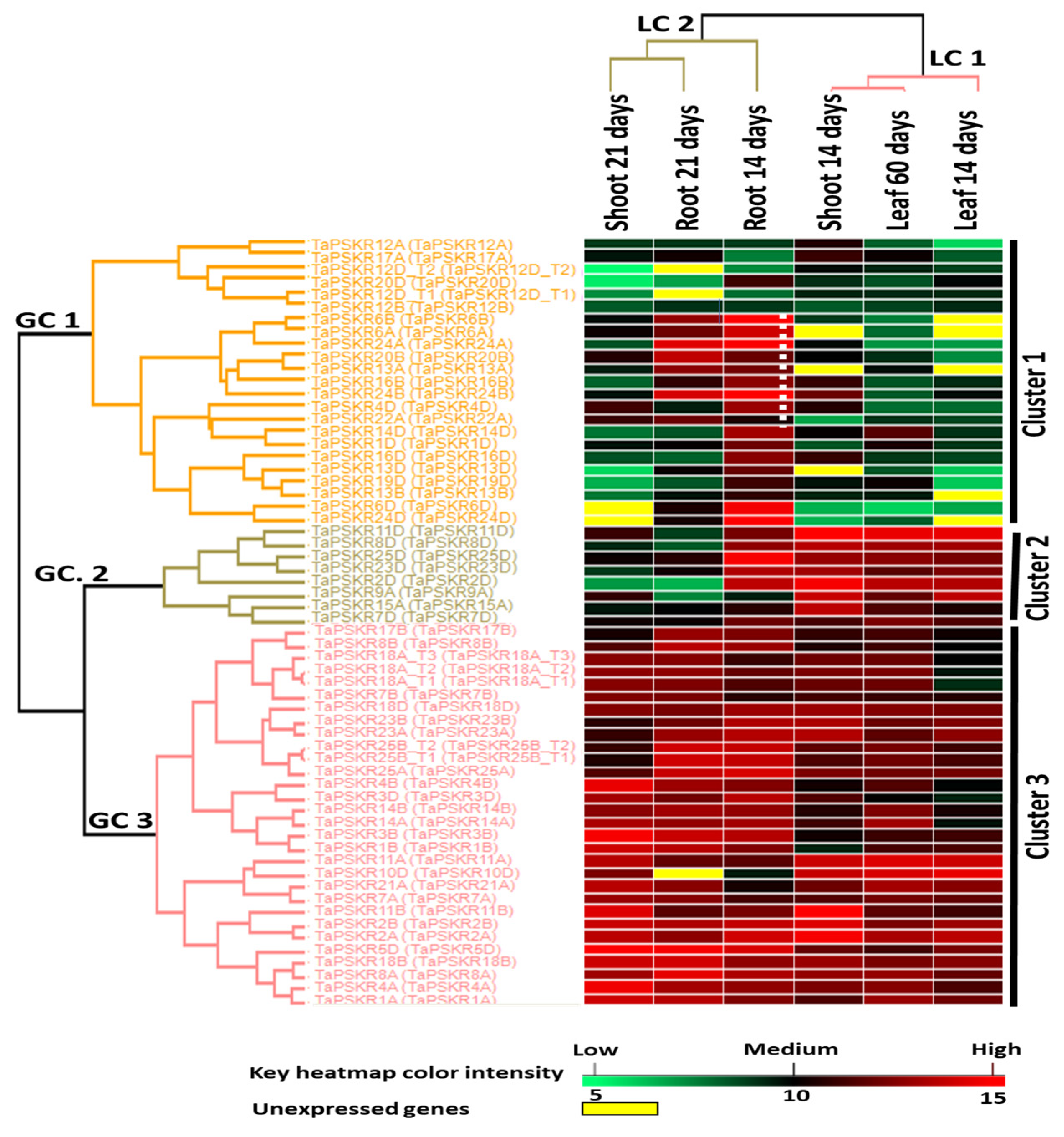
3.4. Expression Profile of the TaPSKRs
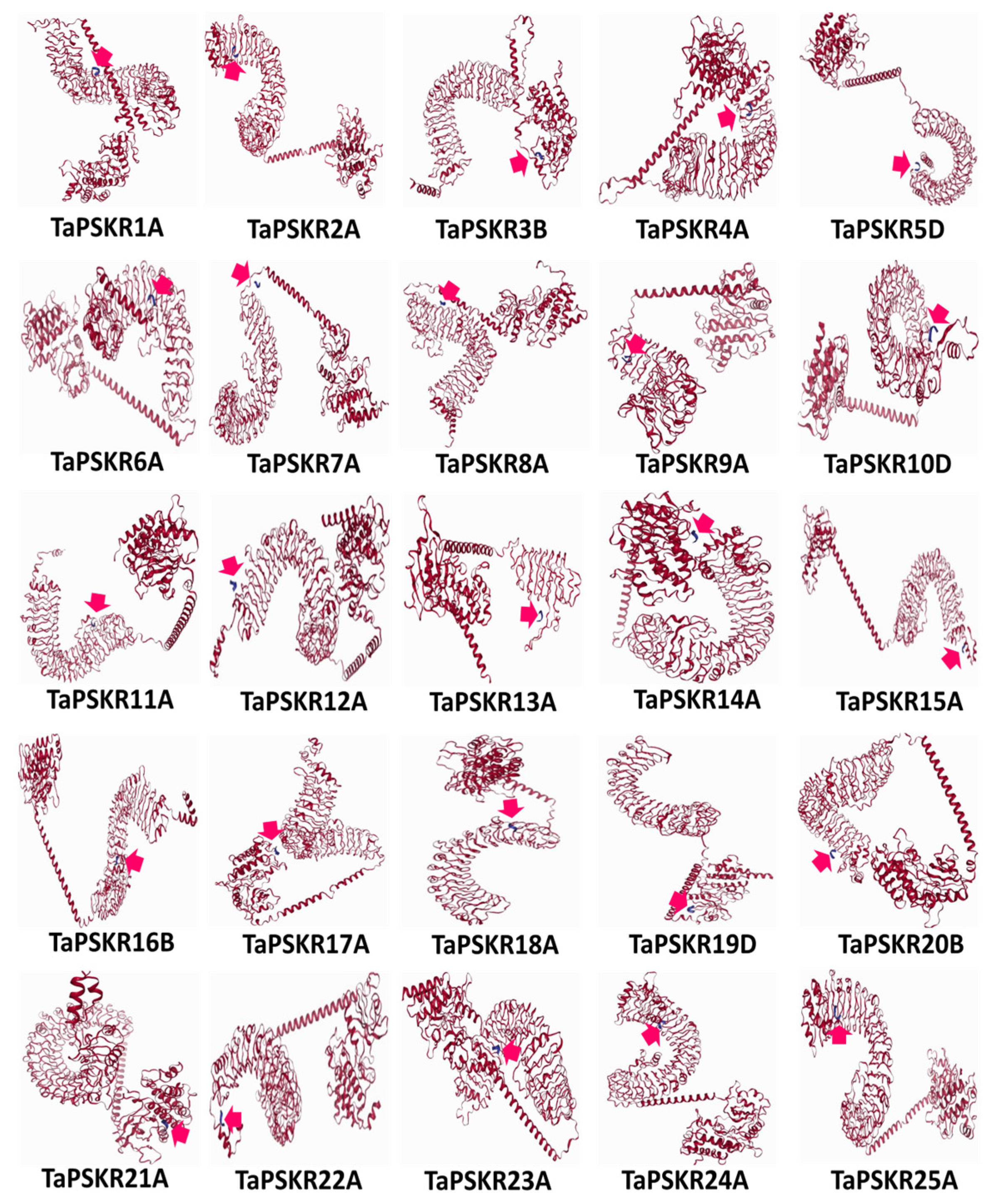
3.5. Molecular Docking Simulations of Phytosulfokines and Phytosulfokine Receptors
4. Discussion
5. Conclusions
6. Perspectives
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Viana, C.M.; Freire, D.; Abrantes, P.; Rocha, J.; Pereira, P. Agricultural land systems importance for supporting food security and sustainable development goals: A systematic review. Sci. Total Environ. 2022, 806, 150718. [Google Scholar] [CrossRef] [PubMed]
- Roychowdhury, R.; Ballén-Taborda, C.; Chaturvedi, P. Characterizing and improving traits for resilient crop development. Front. Plant Sci. 2023, 14, 1307327. [Google Scholar] [CrossRef] [PubMed]
- Borisjuk, N.; Kishchenko, O.; Eliby, S.; Schramm, C.; Anderson, P.; Jatayev, S.; Kurishbayev, A.; Shavrukov, Y. Genetic Modification for Wheat Improvement: From Transgenesis to Genome Editing. BioMed Res. Int. 2019, 2019, 6216304. [Google Scholar] [CrossRef] [PubMed]
- Ijaz, M.; Khan, F.; Ahmed, T.; Noman, M.; Zulfiqar, F.; Rizwan, M.; Chen, J.; Siddique, K.; Li, B. Nanobiotechnology to advance stress resilience in plants: Current opportunities and challenges. Mater. Today Bio 2023, 22, 100759. [Google Scholar] [CrossRef]
- Waadt, R.; Seller, C.A.; Hsu, P.K.; Takahashi, Y.; Munemasa, S.; Schroeder, J.I. Plant hormone regulation of abiotic stress responses. Nat. Rev. Mol. Cell Biol. 2022, 23, 516. [Google Scholar] [CrossRef]
- Matsubayashi, Y.; Sakagami, Y. Phytosulfokine, sulfated peptides that induce the proliferation of single mesophyll cells of Asparagus officinalis L. Proc. Natl. Acad. Sci. USA 1996, 93, 7623–7627. [Google Scholar] [CrossRef]
- Komori, R.; Amano, Y.; Ogawa-Ohnishi, M.; Matsubayashi, Y. Identification of tyrosylprotein sulfotransferase in Arabidopsis. Proc. Natl. Acad. Sci. USA 2009, 106, 15067–15072. [Google Scholar] [CrossRef]
- Matsubayashi, Y. Posttranslationally modified small-peptide signals in plants. Annu. Rev. Plant Biol. 2014, 65, 385–413. [Google Scholar] [CrossRef]
- Sauter, M. Phytosulfokine peptide signalling. J. Exp. Bot. 2015, 66, 5161–5169. [Google Scholar] [CrossRef]
- Stührwohldt, N.; Dahlke, R.I.; Steffens, B.; Johnson, A.; Sauter, M. Phytosulfokine-α controls hypocotyl length and cell expansion in Arabidopsis thaliana through phytosulfokine receptor 1. PLoS ONE 2011, 6, e21054. [Google Scholar] [CrossRef]
- Mosher, S.; Seybold, H.; Rodriguez, P.; Stahl, M.; Davies, K.A.; Dayaratne, S.; Morillo, S.A.; Wierzba, M.; Favery, B.; Keller, H.; et al. The tyrosine-sulfated peptide receptors PSKR1 and PSY1R modify the immunity of Arabidopsis to biotrophic and necrotrophic pathogens in an antagonistic manner. Plant J. 2013, 73, 469–482. [Google Scholar] [CrossRef]
- Yang, H.; Matsubayashi, Y.; Nakamura, K.; Sakagami, Y. Oryza sativa PSK gene encodes a precursor of phytosulfokine-Alpha, a sulfated peptide growth factor found in plants. Proc. Natl. Acad. Sci. USA 1999, 96, 13560–13565. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Di, Q.; Luo, L.; Yu, L. Phytosulfokine peptides, their receptors, and functions. Front. Plant Sci. 2024, 14, 1326964. [Google Scholar] [CrossRef]
- Kutschmar, A.; Rzewuski, G.; Stührwohldt, N.; Beemster, G.T.S.; Inzé, D.; Sauter, M. PSK-α promotes root growth in Arabidopsis. New Phytol. 2009, 181, 820–831. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.; Liu, J.X.; Howell, S.H. Proteolytic processing of a precursor protein for a growth-promoting peptide by a subtilisin serine protease in Arabidopsis. Plant J. 2008, 56, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Ladwig, F.; Dahlke, R.I.; Stührwohldt, N.; Hartmann, J.; Harter, K.; Sauter, M. Phytosulfokine Regulates Growth in Arabidopsis through a Response Module at the Plasma Membrane That Includes CYCLIC NUCLEOTIDE-GATED CHANNEL17, H+-ATPase, and BAK1. Plant Cell 2015, 27, 1718–1729. [Google Scholar] [CrossRef]
- Heyman, J.; Cools, T.; Vandenbussche, F.; Heyndrickx, K.S.; Van Leene, J.; Vercauteren, I.; Vanderauwera, S.; Vandepoele, K.; De Jaeger, G.; Van Der Straeten, D.; et al. ERF115 controls root quiescent center cell division and stem cell replenishment. Science 2013, 342, 860–863. [Google Scholar] [CrossRef]
- Stührwohldt, N.; Dahlke, R.I.; Kutschmar, A.; Peng, X.; Sun, M.X.; Sauter, M. Phytosulfokine peptide signaling controls pollen tube growth and funicular pollen tube guidance in Arabidopsis thaliana. Physiol. Plant. 2015, 153, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Stührwohldt, N.; Bühler, E.; Sauter, M.; Schaller, A. Phytosulfokine (PSK) precursor processing by subtilase SBT3.8 and PSK signaling improve drought stress tolerance in Arabidopsis. J. Exp. Bot. 2021, 72, 3427–3440. [Google Scholar] [CrossRef]
- Saini, S.; Kaur, N.; Marothia, D.; Singh, B.; Singh, V.; Gantet, P.; Pati, P.K. Morphological Analysis, Protein Profiling and Expression Analysis of Auxin Homeostasis Genes of Roots of Two Contrasting Cultivars of Rice Provide Inputs on Mechanisms Involved in Rice Adaptation towards Salinity Stress. Plants 2021, 10, 1544. [Google Scholar] [CrossRef]
- Igarashi, D.; Tsuda, K.; Katagiri, F. The peptide growth factor, phytosulfokine, attenuates pattern-triggered immunity. Plant J. 2012, 71, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Amano, Y.; Tsubouchi, H.; Shinohara, H.; Ogawa, M.; Matsubayashi, Y. Tyrosine-sulfated glycopeptide involved in cellular proliferation and expansion in Arabidopsis. Proc. Natl. Acad. Sci. USA 2007, 104, 18333–18338. [Google Scholar] [CrossRef]
- Hartmann, J.; Stührwohldt, N.; Dahlke, R.I.; Sauter, M. Phytosulfokine control of growth occurs in the epidermis, is likely to be non-cell autonomous and is dependent on brassinosteroids. Plant J. 2013, 73, 579–590. [Google Scholar] [CrossRef]
- Matsubayashi, Y.; Ogawa, M.; Morita, A.; Sakagami, Y. An LRR receptor kinase involved in perception of a peptide plant hormone, phytosulfokine. Science 2002, 296, 1470–1472. [Google Scholar] [CrossRef]
- Nagar, P.; Sharma, N.; Jain, M.; Sharma, G.; Prasad, M.; Mustafiz, A. OsPSKR15, a phytosulfokine receptor from rice enhances abscisic acid response and drought stress tolerance. Physiol. Plant. 2022, 174, e13569. [Google Scholar] [CrossRef]
- Hartmann, J.; Linke, D.; Bönniger, C.; Tholey, A.; Sauter, M. Conserved phosphorylation sites in the activation loop of the Arabidopsis phytosulfokine receptor PSKR1 differentially affect kinase and receptor activity. Biochem. J. 2015, 472, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Matsubayashi, Y.; Ogawa, M.; Kihara, H.; Niwa, M.; Sakagami, Y. Disruption and Overexpression of Arabidopsis Phytosulfokine Receptor Gene Affects Cellular Longevity and Potential for Growth. Plant Physiol. 2006, 142, 45–53. [Google Scholar] [CrossRef]
- Yang, W.; Zhang, B.; Qi, G.; Shang, L.; Liu, H.; Ding, X.; Chu, Z. Identification of the phytosulfokine receptor 1 (OsPSKR1) confers resistance to bacterial leaf streak in rice. Planta 2019, 250, 1603–1612. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.P.; Chen, T.; Jing, F.L.; Liu, Y.; Ma, J.F.; Tian, T.; Wang, P.; Yang, D.L. Cloning and expression analysis of TaPSKR1 gene in wheat. Acta Agric. Bor. Sin. 2023, 38, 1–9. [Google Scholar]
- Geng, Y.; Jian, C.; Xu, W.; Liu, H.; Hao, C.; Hou, J.; Liu, H.; Zhang, X.; Li, T. miR164-targeted TaPSK5 encodes a phytosulfokine precursor that regulates root growth and yield traits in common wheat (Triticum aestivum L.). Plant Mol. Biol. 2020, 104, 615–628. [Google Scholar] [CrossRef]
- Thumuluri, V.; Almagro Armenteros, J.J.; Johansen, A.R.; Nielsen, H.; Winther, O. DeepLoc 2.0: Multi-label subcellular localization prediction using protein language models. Nucleic Acids Res. 2022, 50, W228–W234. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [PubMed]
- de Castro, E.; Sigrist, C.J.; Gattiker, A.; Bulliard, V.; Langendijk-Genevaux, P.S.; Gasteiger, E.; Bairoch, A.; Hulo, N. ScanProsite: Detection of PROSITE signature matches and Pro Rule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006, 34, W362–W365. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics: Cambridge, UK, 2010. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved recon-struction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Dessau, R.B.; Pipper, C.B. “R”—Project for statistical computing. Ugeskr. Laeger 2008, 170, 328–330. [Google Scholar]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, V.; Yang, Y.D.; Sael, L.; Kihara, D. Protein-protein docking using region-based 3D Zernike descriptors. BMC Bioinform. 2009, 10, 407. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Skolnick, J. GOAP: A Generalized Orientation-Dependent, All-Atom Statistical Potential for Protein Structure Prediction. Biophys. J. 2011, 101, 2043–2052. [Google Scholar] [CrossRef]
- Zhou, H.; Zhou, Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Sci. 2009, 11, 2714–2726. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-Y.; Zou, X. Statistical mechanics-based method to extract atomic distance-dependent potentials from protein structures. Proteins Struct. Funct. Bioinf. 2011, 79, 2648–2661. [Google Scholar] [CrossRef] [PubMed]
- Ng, A.; Xavier, R.J. Leucine-rich repeat (LRR) proteins: Integrators of pattern recognition and signaling in immunity. Autophagy. 2011, 7, 1082–1084. [Google Scholar] [CrossRef]
- Boden, S.A.; McIntosh, R.A.; Uauy, C.; Krattinger, S.G.; Dubcovsky, J.; Rogers, W.J.; Xia, X.C.; Badaeva, E.D.; Bentley, A.R.; Brown-Guedira, G.; et al. Wheat Initiative. Updated guidelines for gene nomenclature in wheat. TAG. Theor. Appl. Genet. 2023, 136, 72. [Google Scholar] [CrossRef]
- Nagar, P.; Kumar, A.; Jain, M.; Kumari, S.; Mustafiz, A. Genome-wide analysis and transcript profiling of PSKR gene family members in Oryza sativa. PLoS ONE 2020, 15, e0236349. [Google Scholar] [CrossRef]
- Zhang, Y.; Zeng, L. Crosstalk between Ubiquitination and Other Post-translational Protein Modifications in Plant Immunity. Plant Commun. 2020, 1, 100041. [Google Scholar] [CrossRef]
- Khalil, H.B.; Ehdaeivand, M.R.; Xu, Y.; Laroche, A.; Gulick, P.J. Identification and characterization of rye genes not expressed in allohexaploid triticale. BMC Genom. 2015, 16, 281. [Google Scholar] [CrossRef]
- Khalil, H.B.; Brunetti, S.C.; Pham, U.M.; Maret, D.; Laroche, A.; Gulick, P.J. Characterization of the caleosin gene family in the Triticeae. BMC Genom. 2014, 15, 239. [Google Scholar] [CrossRef]
- Torii, K.U. Leucine-rich repeat receptor kinases in plants: Structure, function, and signal transduction pathways. Int. Rev. Cytol. 2004, 234, 1–46. [Google Scholar]
- Kamel, A.M.; Metwally, K.; Sabry, M.; Albalawi, D.A.; Abbas, Z.K.; Darwish, D.B.E.; Al-Qahtani, S.M.; Al-Harbi, N.A.; Alzuaibr, F.M.; Khalil, H.B. The Expression of Triticum aestivum Cysteine-Rich Receptor-like Protein Kinase Genes during Leaf Rust Fungal Infection. Plants 2023, 12, 2932. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalil, H.B. Genome-Wide Characterization and Expression Profiling of Phytosulfokine Receptor Genes (PSKRs) in Triticum aestivum with Docking Simulations of Their Interactions with Phytosulfokine (PSK): A Bioinformatics Study. Genes 2024, 15, 1306. https://doi.org/10.3390/genes15101306
Khalil HB. Genome-Wide Characterization and Expression Profiling of Phytosulfokine Receptor Genes (PSKRs) in Triticum aestivum with Docking Simulations of Their Interactions with Phytosulfokine (PSK): A Bioinformatics Study. Genes. 2024; 15(10):1306. https://doi.org/10.3390/genes15101306
Chicago/Turabian StyleKhalil, Hala Badr. 2024. "Genome-Wide Characterization and Expression Profiling of Phytosulfokine Receptor Genes (PSKRs) in Triticum aestivum with Docking Simulations of Their Interactions with Phytosulfokine (PSK): A Bioinformatics Study" Genes 15, no. 10: 1306. https://doi.org/10.3390/genes15101306