
Mapping of Candidate Genes for Nitrogen Uptake and Utilization in Japonica Rice at Seedling Stage
 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material and Genotyping
2.2. Phenotypic Data
2.3. GWAS
2.4. Linkage Disequilibrium (LD) Analysis
2.5. Haplotype Analysis
2.6. RNA-seq
2.7. Candidate Gene Prediction
2.8. Quantitative Real-Time PCR
2.9. Data Analysis
3. Results
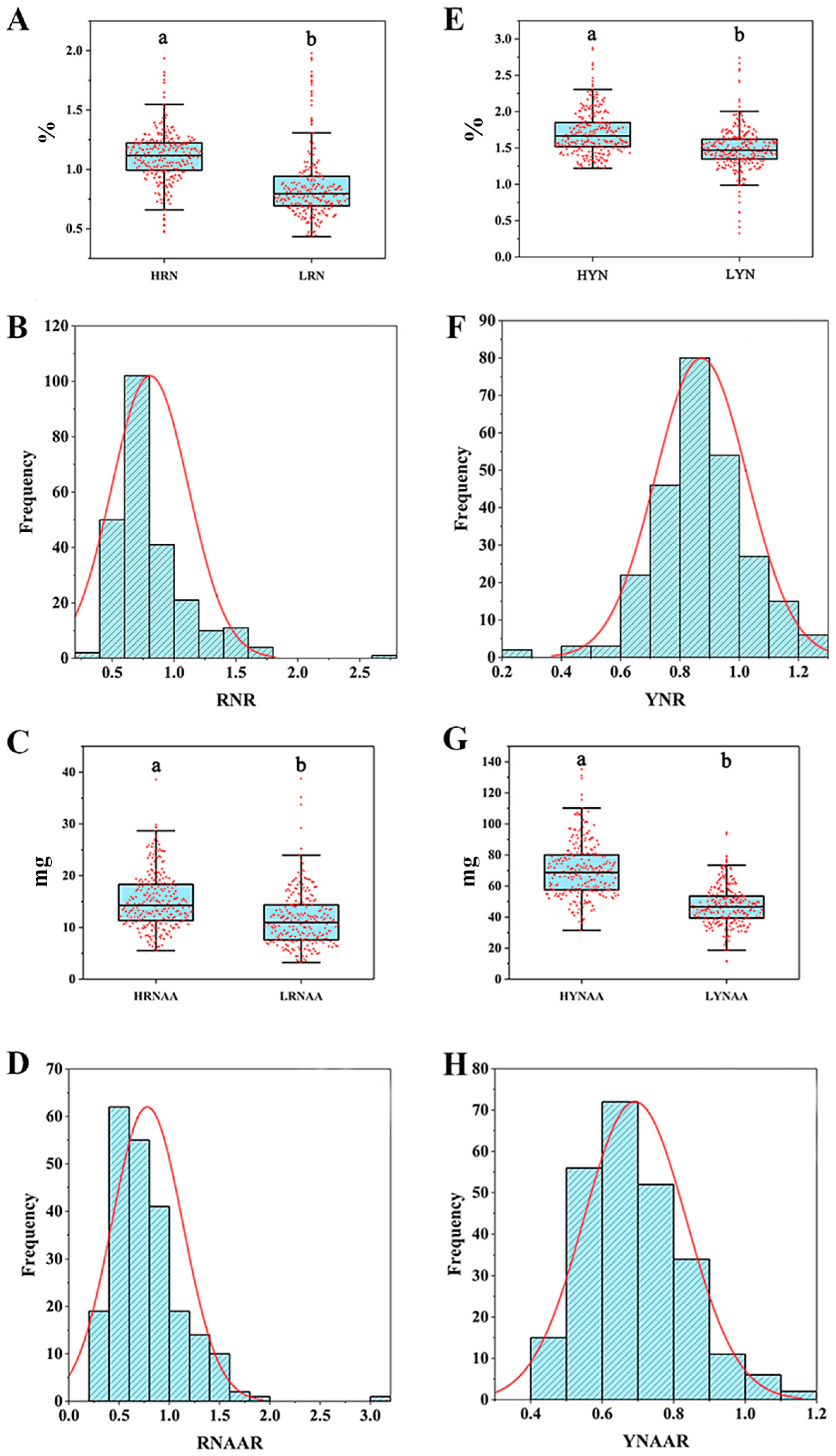
3.1. Phenotypic Data Analysis
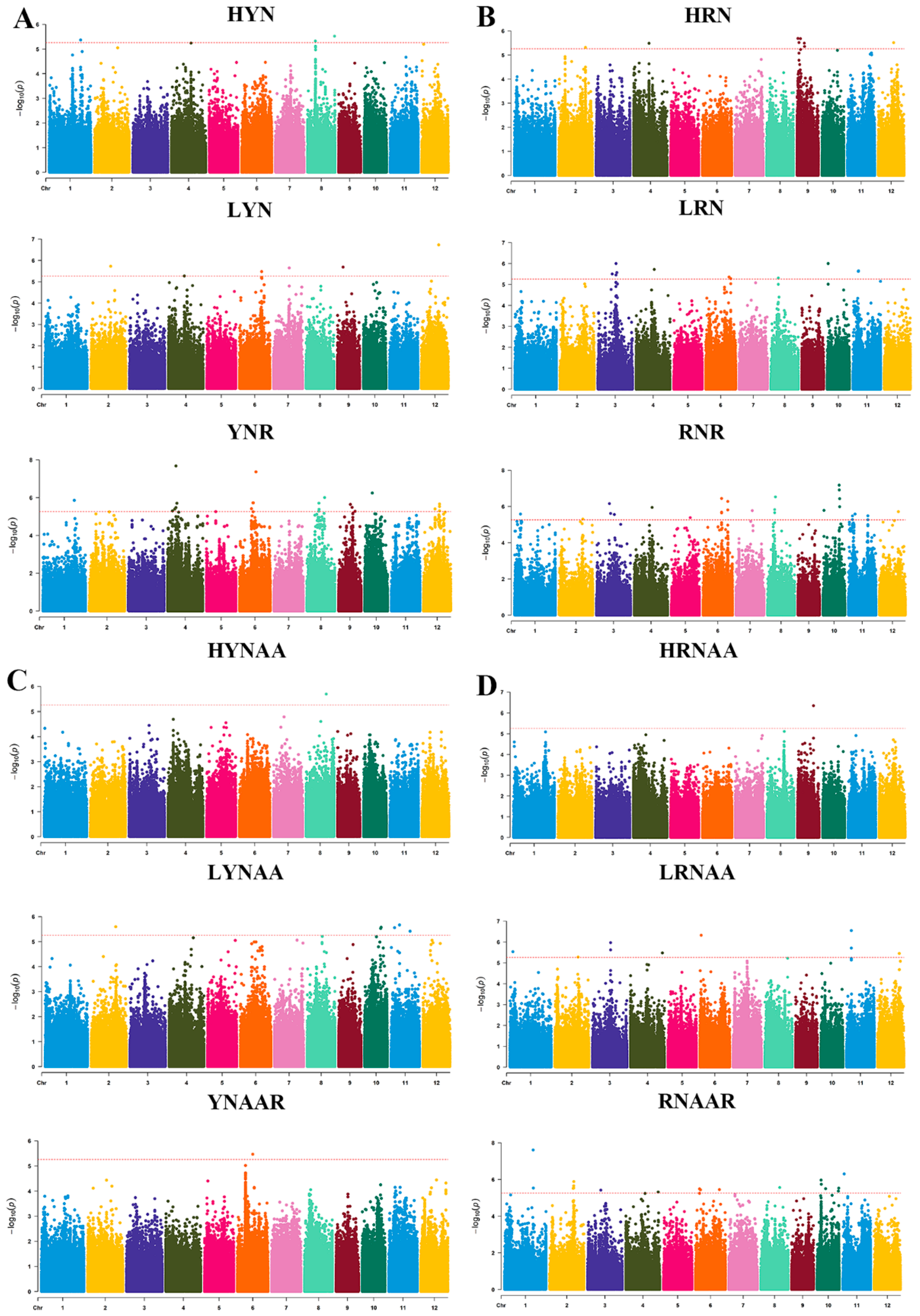
3.2. QTL Localization
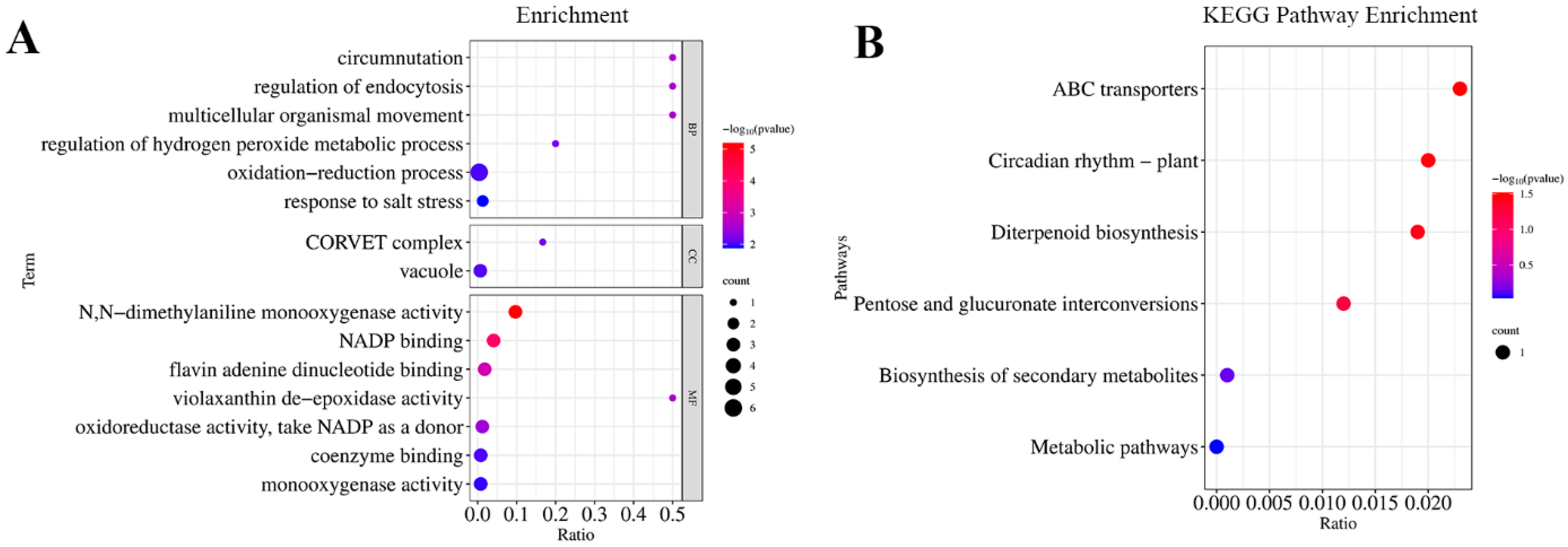
3.3. Candidate Gene Mining
3.4. Expression Analysis of Candidate Genes
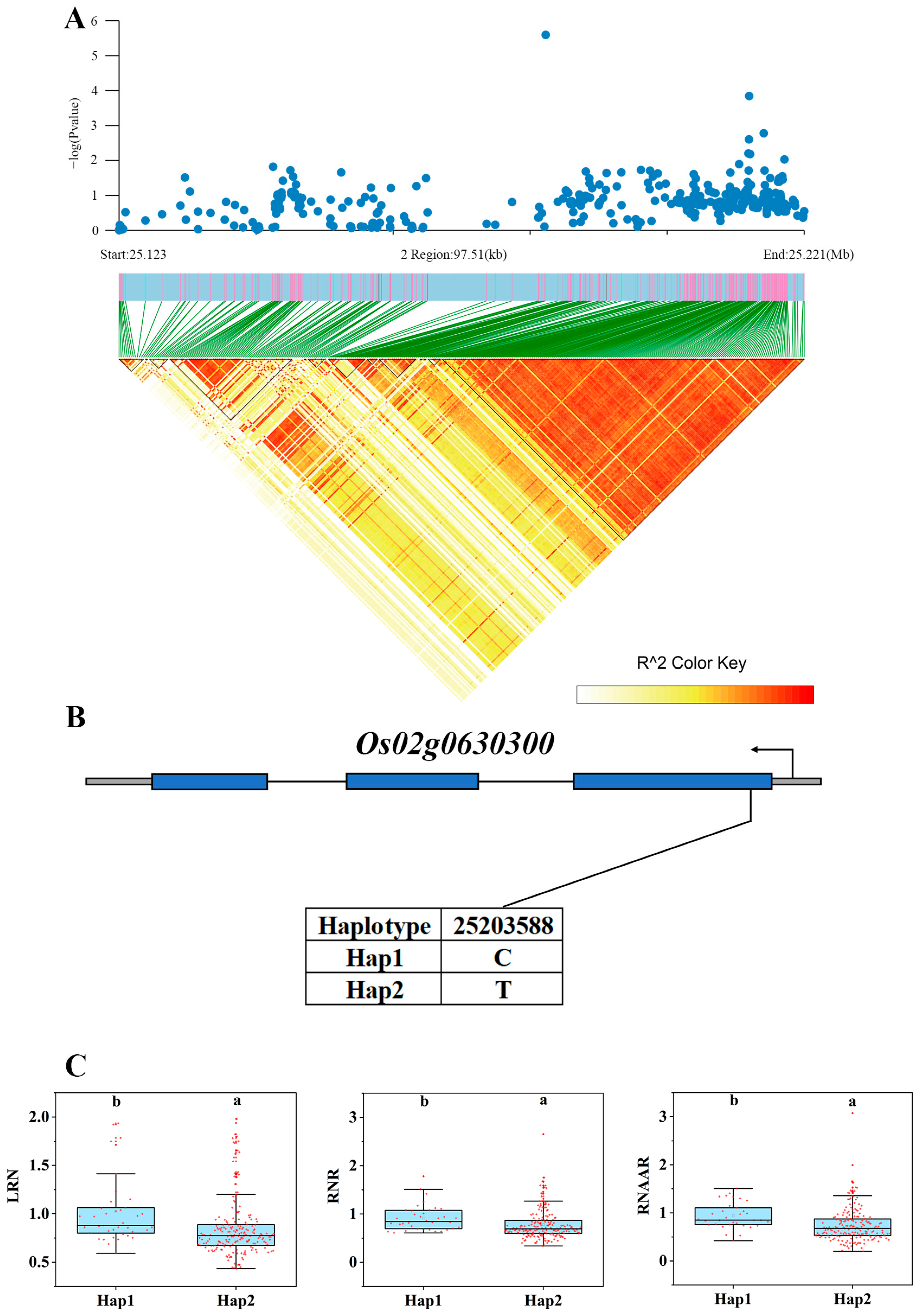
3.5. Haplotype Analysis of Candidate Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Khush, G.S. Origin, dispersal, cultivation and variation of rice. Plant Mol. Biol. 1997, 35, 25–34. [Google Scholar] [CrossRef]
- Peng, S.; Buresh, R.J.; Huang, J.; Zhong, X.; Zou, Y.; Yang, J.; Wang, G.; Liu, Y.; Hu, R.; Tang, Q.; et al. Improving nitrogen fertilization in rice by sitespecific N management. A review. Agron. Sustain. Dev. 2010, 30, 649–656. [Google Scholar] [CrossRef]
- Li, H.; Hu, B.; Chu, C.C. Nitrogen use efficiency in crops: Lessons from Arabidopsis and rice. J. Exp. Bot. 2017, 68, 2477–2488. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.C.; Jin, S.Q.; Wu, Y.Y.; Zhang, B.; Kanter, D.; Wu, B.; Xi, X.C.; Zhang, X.; Chen, D.L.; Xu, J.M.; et al. Fertilizer overuse in Chinese smallholders due to lack of fixed inputs. J. Environ. Manag. 2021, 293, 112913. [Google Scholar] [CrossRef]
- Anas, M.; Liao, F.; Verma, K.K.; Sarwar, M.A.; Mahmood, A.; Chen, Z.L.; Li, Q.; Zeng, X.P.; Liu, Y.; Li, Y.R. Fate of nitrogen in agriculture and environment: Agronomic, eco-physiological and molecular approaches to improve nitrogen use efficiency. Biol. Res. 2020, 53, 47. [Google Scholar] [CrossRef] [PubMed]
- Shah, L.; Yahya, M.; Shah, S.M.A.; Nadeem, M.; Ali, A.; Ali, A.; Wang, J.; Riaz, M.W.; Rehman, S.; Wu, W.X.; et al. Improving Lodging Resistance: Using Wheat and Rice as Classical Examples. Int. J. Mol. Sci. 2019, 20, 4211. [Google Scholar] [CrossRef] [PubMed]
- Huber, D.; Römheld, V.; Weinmann, M. Chapter 10—Relationship between Nutrition, Plant Diseases and Pests. In Marschner’s Mineral Nutrition of Higher Plants, 3rd ed.; Marschner, P., Ed.; Academic Press: San Diego, CA, USA, 2012; pp. 283–298. [Google Scholar] [CrossRef]
- Wang, S.; Chen, A.; Xie, K.; Yang, X.; Luo, Z.; Chen, J.; Zeng, D.; Ren, Y.; Yang, C.; Wang, L.; et al. Functional analysis of the OsNPF4.5 nitrate transporter reveals a conserved mycorrhizal pathway of nitrogen acquisition in plants. Proc. Natl. Acad. Sci. USA 2020, 117, 16649–16659. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chu, C. Nitrogen-Use Divergence between Indica and Japonica Rice: Variation at Nitrate Assimilation. Mol. Plant 2020, 13, 6–7. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Marmagne, A.; Park, J.; Fabien, C.; Yim, Y.; Kim, S.-j.; Kim, T.-H.; Lim, P.O.; Masclaux-Daubresse, C.; Nam, H.G. Concurrent activation of OsAMT1;2 and OsGOGAT1 in rice leads to enhanced nitrogen use efficiency under nitrogen limitation. Plant J. 2020, 103, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Xu, Z.; Zhang, L.; Li, S.; Wang, S.; Yang, H.; Liu, X.; Zeng, D.; Liu, Q.; Qian, Q.; et al. MYB61 is regulated by GRF4 and promotes nitrogen utilization and biomass production in rice. Nat. Commun. 2020, 11, 5219. [Google Scholar] [CrossRef]
- Iqbal, A.; Dong, Q.; Wang, Z.; Wang, X.R.; Gui, H.P.; Zhang, H.H.; Pang, N.C.; Zhang, X.L.; Song, M.Z. Growth and nitrogen metabolism are associated with nitrogen-use efficiency in cotton genotypes. Plant Physiol. Biochem. 2020, 149, 61–74. [Google Scholar] [CrossRef]
- Yang, J.B.; Wang, M.Y.; Li, W.J.; He, X.; Teng, W.; Ma, W.Y.; Zhao, X.Q.; Hu, M.Y.; Li, H.; Zhang, Y.J.; et al. Reducing expression of a nitrate-responsive bZIP transcription factor increases grain yield and N use in wheat. Plant Biotechnol. J. 2019, 17, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Guo, N.; Gu, M.J.; Hu, J.Q.; Qu, H.Y.; Xu, G.H. Rice OsLHT1 Functions in Leaf-to-Panicle Nitrogen Allocation for Grain Yield and Quality. Front. Plant Sci. 2020, 11, 1150. [Google Scholar] [CrossRef]
- Alfatih, A.; Wu, J.; Zhang, Z.S.; Xia, J.Q.; Jan, S.U.; Yu, L.H.; Xiang, C.B. Rice NIN-LIKE PROTEIN 1 rapidly responds to nitrogen deficiency and improves yield and nitrogen use efficiency. J. Exp. Bot. 2020, 71, 6032–6042. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhu, L.; Shen, C.; Ji, Z.; Zhang, H.; Zhang, T.; Li, Y.; Yu, J.; Yang, N.; He, Y.; et al. Natural allelic variation in a modulator of auxin homeostasis improves grain yield and nitrogen use efficiency in rice. Plant Cell 2021, 33, 566–580. [Google Scholar] [CrossRef]
- Schläppi, M.R.; Jackson, A.K.; Eizenga, G.C.; Wang, A.J.; Chu, C.C.; Shi, Y.; Shimoyama, N.; Boykin, D.L. Assessment of Five Chilling Tolerance Traits and GWAS Mapping in Rice Using the USDA Mini-Core Collection. Front. Plant Sci. 2017, 8, 957. [Google Scholar] [CrossRef]
- Yuan, J.; Wang, X.Q.; Zhao, Y.; Khan, N.U.; Zhao, Z.Q.; Zhang, Y.H.; Wen, X.R.; Tang, F.S.; Wang, F.B.; Li, Z.C. Genetic basis and identification of candidate genes for salt tolerance in rice by GWAS. Sci. Rep. 2020, 10, 9958. [Google Scholar] [CrossRef] [PubMed]
- Biselli, C.; Volante, A.; Desiderio, F.; Tondelli, A.; Gianinetti, A.; Finocchiaro, F.; Taddei, F.; Gazza, L.; Sgrulletta, D.; Cattivelli, L.; et al. GWAS for Starch-Related Parameters in Japonica Rice (Oryza sativa L.). Plants 2019, 8, 292. [Google Scholar] [CrossRef] [PubMed]
- Si, L.Z.; Chen, J.Y.; Huang, X.H.; Gong, H.; Luo, J.H.; Hou, Q.Q.; Zhou, T.Y.; Lu, T.T.; Zhu, J.J.; Shangguan, Y.Y.; et al. OsSPL13 controls grain size in cultivated rice. Nat. Genet. 2016, 48, 447–456. [Google Scholar] [CrossRef]
- Choi, B.; Yoo, J.-M.; Lee, S.-B.; Kim, G.-J.; Kim, K.-W.; Yoo, J.-H.; Oh, K.-S.; Moon, B.-C.; Kim, W.-H.; Cho, Y.-H.; et al. Genome-Wide Association Study of Rice Core Set Related to Grain Weight. Plant Breed. Biotech. 2018, 6, 293–304. [Google Scholar] [CrossRef]
- Tang, W.J.; Ye, J.; Yao, X.M.; Zhao, P.Z.; Xuan, W.; Tian, Y.L.; Zhang, Y.Y.; Xu, S.; An, H.Z.; Chen, G.M.; et al. Genome-wide associated study identifies NAC42-activated nitrate transporter conferring high nitrogen use efficiency in rice. Nat. Commun. 2019, 10, 5279. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Q.; Wang, H.R.; Jiang, Z.M.; Wang, W.; Xu, R.N.; Wang, Q.H.; Zhang, Z.H.; Li, A.F.; Liang, Y.; Ou, S.J.; et al. Genomic basis of geographical adaptation to soil nitrogen in rice (vol 590, pg 600, 2021). Nature 2022, 610, E4. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Xuan, W.; Tian, Y.L.; Fan, L.; Sun, J.; Tang, W.J.; Chen, G.M.; Wang, B.X.; Liu, Y.; Wu, W.; et al. Enhanced OsNLP4-OsNiR cascade confers nitrogen use efficiency by promoting tiller number in rice. Plant Biotechnol. J. 2021, 19, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Di, D.W.; Li, G.J.; Li, Y.L.; Kronzucker, H.J.; Shi, W.M. Transcriptome analysis of rice (Oryza sativa L.) in response to ammonium resupply reveals the involvement of phytohormone signaling and the transcription factor OsJAZ9 in reprogramming of nitrogen uptake and metabolism. J. Plant Physiol. 2020, 246, 153137. [Google Scholar] [CrossRef] [PubMed]
- Xin, W.; Zhang, L.N.; Zhang, W.Z.; Gao, J.P.; Yi, J.; Zhen, X.X.; Li, Z.; Zhao, Y.; Peng, C.C.; Zhao, C. An Integrated Analysis of the Rice Transcriptome and Metabolome Reveals Differential Regulation of Carbon and Nitrogen Metabolism in Response to Nitrogen Availability. Int. J. Mol. Sci. 2019, 20, 2349. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.R.; Yuan, Q.L.; Lin, H.; Li, X.X.; Zhang, C.; Gao, H.S.; Zhang, B.; He, H.Y.; Liu, T.J.; Jie, Z.; et al. Linkage analysis, GWAS, transcriptome analysis to identify candidate genes for rice seedlings in response to high temperature stress. BMC Plant Biol. 2021, 21, 85. [Google Scholar] [CrossRef]
- Shu, X.Y.; Wang, A.J.; Jiang, B.; Jiang, Y.Q.; Xiang, X.; Yi, X.Q.; Li, S.C.; Deng, Q.M.; Wang, S.Q.; Zhu, J.; et al. Genome-wide association study and transcriptome analysis discover new genes for bacterial leaf blight resistance in rice (Oryza sativa L.). BMC Plant Biol. 2021, 21, 255. [Google Scholar] [CrossRef]
- Yu, J.P.; Liu, C.L.; Lin, H.; Zhang, B.; Li, X.X.; Yuan, Q.L.; Liu, T.J.; He, H.Y.; Wei, Z.R.; Ding, S.L.; et al. Loci and natural alleles for cadmium-mediated growth responses revealed by a genome wide association study and transcriptome analysis in rice. BMC Plant Biol. 2021, 21, 374. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Li, N.; Zheng, H.L.; Cui, J.N.; Wang, J.G.; Liu, H.L.; Sun, J.; Liu, T.T.; Zhao, H.W.; Lai, Y.C.; Zou, D.T. Genome-wide association study and candidate gene analysis of alkalinity tolerance in japonica rice germplasm at the seedling stage. Rice 2019, 12, 24. [Google Scholar] [CrossRef]
- Han, Z.Y.; Li, F.; Qiao, W.H.; Zheng, X.M.; Cheng, Y.L.; Zhang, L.F.; Huang, J.F.; Wang, Y.Y.; Lou, D.J.; Xing, M.; et al. Global whole-genome comparison and analysis to classify subpopulations and identify resistance genes in weedy rice relevant for improving crops. Front. Plant Sci. 2023, 13, 1089445. [Google Scholar] [CrossRef]
- Li, J.; Xin, W.; Wang, W.P.; Zhao, S.J.; Xu, L.; Jiang, X.D.; Duan, Y.X.; Zheng, H.L.; Yang, L.M.; Liu, H.L.; et al. Mapping of Candidate Genes in Response to Low Nitrogen in Rice Seedlings. Rice 2022, 15, 51. [Google Scholar] [CrossRef]
- Wang, W.; Xin, W.; Chen, N.; Yang, F.; Li, J.; Qu, G.; Jiang, X.; Xu, L.; Zhao, S.; Liu, H.; et al. Transcriptome and Co-Expression Network Analysis Reveals the Molecular Mechanism of Rice Root Systems in Response to Low-Nitrogen Conditions. Int. J. Mol. Sci. 2023, 24, 5290. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zheng, H.; Wu, W.; Liu, H.; Wang, J.; Jia, Y.; Li, J.; Yang, L.; Lei, L.; Zou, D.; et al. QTL Mapping and Candidate Gene Analysis for Alkali Tolerance in Japonica Rice at the bud Stage Based on Linkage Mapping and Genome-Wide Association Study. Rice 2020, 13, 48. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.S.; He, W.M.; Ji, J.J.; Zhang, C.; Guo, Y.; Yang, T.L. LDBlockShow: A fast and convenient tool for visualizing linkage disequilibrium and haplotype blocks based on variant call format files. Brief. Bioinf. 2021, 22, bbaa227. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Xing, M.Q.; Chen, S.H.; Zhang, X.F.; Xue, H.W. Rice OsGA2ox9 regulates seed GA metabolism and dormancy. Plant Biotechnol. J. 2023, 21, 2411–2413. [Google Scholar] [CrossRef]
- Wang, F.; Han, T.; Song, Q.; Ye, W.; Song, X.; Chu, J.; Li, J.; Chen, Z.J. The Rice Circadian Clock Regulates Tiller Growth and Panicle Development through Strigolactone Signaling and Sugar Sensing. Plant Cell 2020, 32, 3124–3138. [Google Scholar] [CrossRef]
- Schmidt, R.; Schippers, J.H.M.; Mieulet, D.; Obata, T.; Fernie, A.R.; Guiderdoni, E.; Mueller-Roeber, B. MULTIPASS, a rice R2R3-type MYB transcription factor, regulates adaptive growth by integrating multiple hormonal pathways. Plant J. 2013, 76, 258–273. [Google Scholar] [CrossRef] [PubMed]
- Bijay, S.; Craswell, E. Fertilizers and nitrate pollution of surface and ground water: An increasingly pervasive global problem. SN Appl. Sci. 2021, 3, 518. [Google Scholar] [CrossRef]
- Singh, B. Are Nitrogen Fertilizers Deleterious to Soil Health? Agronomy 2018, 8, 48. [Google Scholar] [CrossRef]
- Diaz, R.J.; Rosenberg, R. Spreading Dead Zones and Consequences for Marine Ecosystems. Science 2008, 321, 926–929. [Google Scholar] [CrossRef] [PubMed]
- Fageria, N.K.; Baligar, V.C.; Heinemann, A.B.; Carvalho, M.C.S. Nitrogen Uptake and Use Efficiency in Rice. In Nutrient Use Efficiency: From Basics to Advances; Rakshit, A., Singh, H.B., Sen, A., Eds.; Springer: New Delhi, India, 2015; pp. 285–296. [Google Scholar] [CrossRef]
- Liu, J.J.; Zhang, Q.; Meng, D.Y.; Ren, X.L.; Li, H.W.; Su, Z.Q.; Zhang, N.; Zhi, L.Y.; Ji, J.; Li, J.M.; et al. QMrl-7B Enhances Root System, Biomass, Nitrogen Accumulation and Yield in Bread Wheat. Plants 2021, 10, 764. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Fan, L.; Chen, J.N.; Jiang, L.G.; Zou, Y.B. Continuous applications of biochar to rice: Effects on nitrogen uptake and utilization. Sci. Rep. 2018, 8, 11461. [Google Scholar] [CrossRef]
- Huang, L.F.; Yu, J.; Yang, J.; Zhang, R.; Bai, Y.C.; Sun, C.M.; Zhuang, H.Y. Relationships Between Yield, Quality and Nitrogen Uptake and Utilization of Organically Grown Rice Varieties. Pedosphere 2016, 26, 85–97. [Google Scholar] [CrossRef]
- Makino, A. Photosynthesis, Grain Yield, and Nitrogen Utilization in Rice and Wheat. Plant Physiol. 2011, 155, 125–129. [Google Scholar] [CrossRef]
- Hu, B.; Wang, W.; Ou, S.J.; Tang, J.Y.; Li, H.; Che, R.H.; Zhang, Z.H.; Chai, X.Y.; Wang, H.R.; Wang, Y.Q.; et al. Variation in NRT1.1B contributes to nitrate-use divergence between rice subspecies. Nat. Genet. 2015, 47, 834–838. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, Z.S.; Xia, J.Q.; Alfatih, A.; Song, Y.; Huang, Y.J.; Wan, G.Y.; Sun, L.Q.; Tang, H.; Liu, Y.; et al. Rice NIN-LIKE PROTEIN 4 plays a pivotal role in nitrogen use efficiency. Plant Biotechnol. J. 2021, 19, 448–461. [Google Scholar] [CrossRef]
- Zhou, Y.; Tao, Y.J.; Tang, D.N.; Wang, J.; Zhong, J.; Wang, Y.; Yuan, Q.M.; Yu, X.F.; Zhang, Y.; Wang, Y.L.; et al. Identification of QTL Associated with Nitrogen Uptake and Nitrogen Use Efficiency Using High Throughput Genotyped CSSLs in Rice (Oryza sativa L.). Front. Plant Sci. 2017, 8, 1166. [Google Scholar] [CrossRef]
- Yang, X.H.; Xia, X.Z.; Zhang, Z.Q.; Nong, B.X.; Zeng, Y.; Xiong, F.Q.; Wu, Y.Y.; Gao, J.; Deng, G.F.; Li, D.T. QTL Mapping by Whole Genome Re-sequencing and Analysis of Candidate Genes for Nitrogen Use Efficiency in Rice. Front. Plant Sci. 2017, 8, 1634. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.T.; Duan, Y.Y.; Yang, J.N.; Gan, L.P.; Chen, W.J.; Yang, J.; Xiao, G.S.; Guan, L.L.; Chen, J.S. Transcriptome Analysis Reveals Genes Associated with Flooding Tolerance in Mulberry Plants. Life 2023, 13, 1087. [Google Scholar] [CrossRef]
- Meng, A.J.; Wen, D.X.; Zhang, C.Q. Maize Seed Germination Under Low-Temperature Stress Impacts Seedling Growth Under Normal Temperature by Modulating Photosynthesis and Antioxidant Metabolism. Front. Plant Sci. 2022, 13, 843033. [Google Scholar] [CrossRef]
- Sun, L.J.; Wang, J.; Song, K.; Sun, Y.F.; Qin, Q.; Xue, Y. Transcriptome analysis of rice (Oryza sativa L.) shoots responsive to cadmium stress. Sci. Rep. 2019, 9, 10177. [Google Scholar] [CrossRef] [PubMed]
- Vicente, R.; Bolger, A.M.; Martínez-Carrasco, R.; Pérez, P.; Gutiérrez, E.; Usadel, B.; Morcuende, R. De Novo Transcriptome Analysis of Durum Wheat Flag Leaves Provides New Insights Into the Regulatory Response to Elevated CO2 and High Temperature. Front. Plant Sci. 2019, 10, 1605. [Google Scholar] [CrossRef] [PubMed]
- Xuan, H.D.; Huang, Y.Z.; Zhou, L.; Deng, S.S.; Wang, C.C.; Xu, J.Y.; Wang, H.T.; Zhao, J.M.; Guo, N.; Xing, H. Key Soybean Seedlings Drought-Responsive Genes and Pathways Revealed by Comparative Transcriptome Analyses of Two Cultivars. Int. J. Mol. Sci. 2022, 23, 2893. [Google Scholar] [CrossRef]
- Tan, Q.Q.; He, H.Y.; Chen, W.; Huang, L.; Zhao, D.L.; Chen, X.J.; Li, J.Y.; Yang, X.H. Integrated genetic analysis of leaf blast resistance in upland rice: QTL mapping, bulked segregant analysis and transcriptome sequencing. Aob Plants 2022, 14, plac047. [Google Scholar] [CrossRef]
- Xin, W.; Wang, J.G.; Li, J.; Zhao, H.W.; Liu, H.L.; Zheng, H.L.; Yang, L.M.; Wang, C.; Yang, F.; Chen, J.H.; et al. Candidate Gene Analysis for Nitrogen Absorption and Utilization in Japonica Rice at the Seedling Stage Based on a Genome-Wide Association Study. Front. Plant Sci. 2021, 12, 670861. [Google Scholar] [CrossRef]
- Lei, L.; Zheng, H.L.; Bi, Y.L.; Yang, L.M.; Liu, H.L.; Wang, J.G.; Sun, J.; Zhao, H.W.; Li, X.W.; Li, J.M.; et al. Identification of a Major QTL and Candidate Gene Analysis of Salt Tolerance at the Bud Burst Stage in Rice (Oryza sativa L.) Using QTL-Seq and RNA-Seq. Rice 2020, 13, 55. [Google Scholar] [CrossRef]
- Chandra, S.; Kazmi, A.Z.; Ahmed, Z.; Roychowdhury, G.; Kumari, V.; Kumar, M.; Mukhopadhyay, K. Genome-wide identification and characterization of NB-ARC resistant genes in wheat (Triticum aestivum L.) and their expression during leaf rust infection. Plant Cell Rep. 2017, 36, 1097–1112. [Google Scholar] [CrossRef] [PubMed]
- Herawati, R.; Herlinda, S.; Ganefianti, D.W.; Bustamam, H.; Sipriyadi. Improving Broad Spectrum Blast Resistance by Introduction of the Pita2 Gene: Encoding the NB-ARC Domain of Blast-Resistant Proteins into Upland Rice Breeding Programs. Agronomy 2022, 12, 2373. [Google Scholar] [CrossRef]
- Wang, Y.; Teng, Z.; Li, H.; Wang, W.; Xu, F.; Sun, K.; Chu, J.; Qian, Y.; Loake, G.J.; Chu, C.; et al. An activated form of NB-ARC protein RLS1 functions with cysteine-rich receptor-like protein RMC to trigger cell death in rice. Plant Commun. 2023, 4, 100459. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Candidate Genes | Regulated | QTLs | Annotation | Reference |
|---|---|---|---|---|
| Os01g0159250 | up | qLRNAA1 | Hypothetical protein | |
| Os02g0630300 | down | qLYNAA2 | 2OG-Fe (II) oxygenase domain-containing protein | Xing et al., 2023 [40] |
| Os02g0618200 | down | qLRNAA2 | Circadian-associated rice pseudo-response regulator, control of flowering time | Wang et al., 2020 [41] |
| Os02g0618400 | down | qLRNAA2 | R2R3-type MYB transcription factor, adaptive growth regulation | Schmidt et al., 2013 [42] |
| Os06g0619000 | down | qRNAAR6-2 | NB-ARC domain-containing protein |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, N.; Ma, T.; Xia, S.; Li, C.; Liu, Y.; Wang, J.; Qu, G.; Liu, H.; Zheng, H.; Yang, L.; et al. Mapping of Candidate Genes for Nitrogen Uptake and Utilization in Japonica Rice at Seedling Stage. Genes 2024, 15, 327. https://doi.org/10.3390/genes15030327
Chen N, Ma T, Xia S, Li C, Liu Y, Wang J, Qu G, Liu H, Zheng H, Yang L, et al. Mapping of Candidate Genes for Nitrogen Uptake and Utilization in Japonica Rice at Seedling Stage. Genes. 2024; 15(3):327. https://doi.org/10.3390/genes15030327
Chicago/Turabian StyleChen, Ning, Tianze Ma, Sijia Xia, Chengxin Li, Yinuo Liu, Jiaqi Wang, Guize Qu, Hualong Liu, Hongliang Zheng, Luomiao Yang, and et al. 2024. "Mapping of Candidate Genes for Nitrogen Uptake and Utilization in Japonica Rice at Seedling Stage" Genes 15, no. 3: 327. https://doi.org/10.3390/genes15030327
APA StyleChen, N., Ma, T., Xia, S., Li, C., Liu, Y., Wang, J., Qu, G., Liu, H., Zheng, H., Yang, L., Zou, D., Wang, J., & Xin, W. (2024). Mapping of Candidate Genes for Nitrogen Uptake and Utilization in Japonica Rice at Seedling Stage. Genes, 15(3), 327. https://doi.org/10.3390/genes15030327