

Marker Density and Models to Improve the Accuracy of Genomic Selection for Growth and Slaughter Traits in Meat Rabbits
 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Phenotypes and Genotypes
2.2. Models
2.3. Marker Densities
2.4. Cross-Validation
3. Results
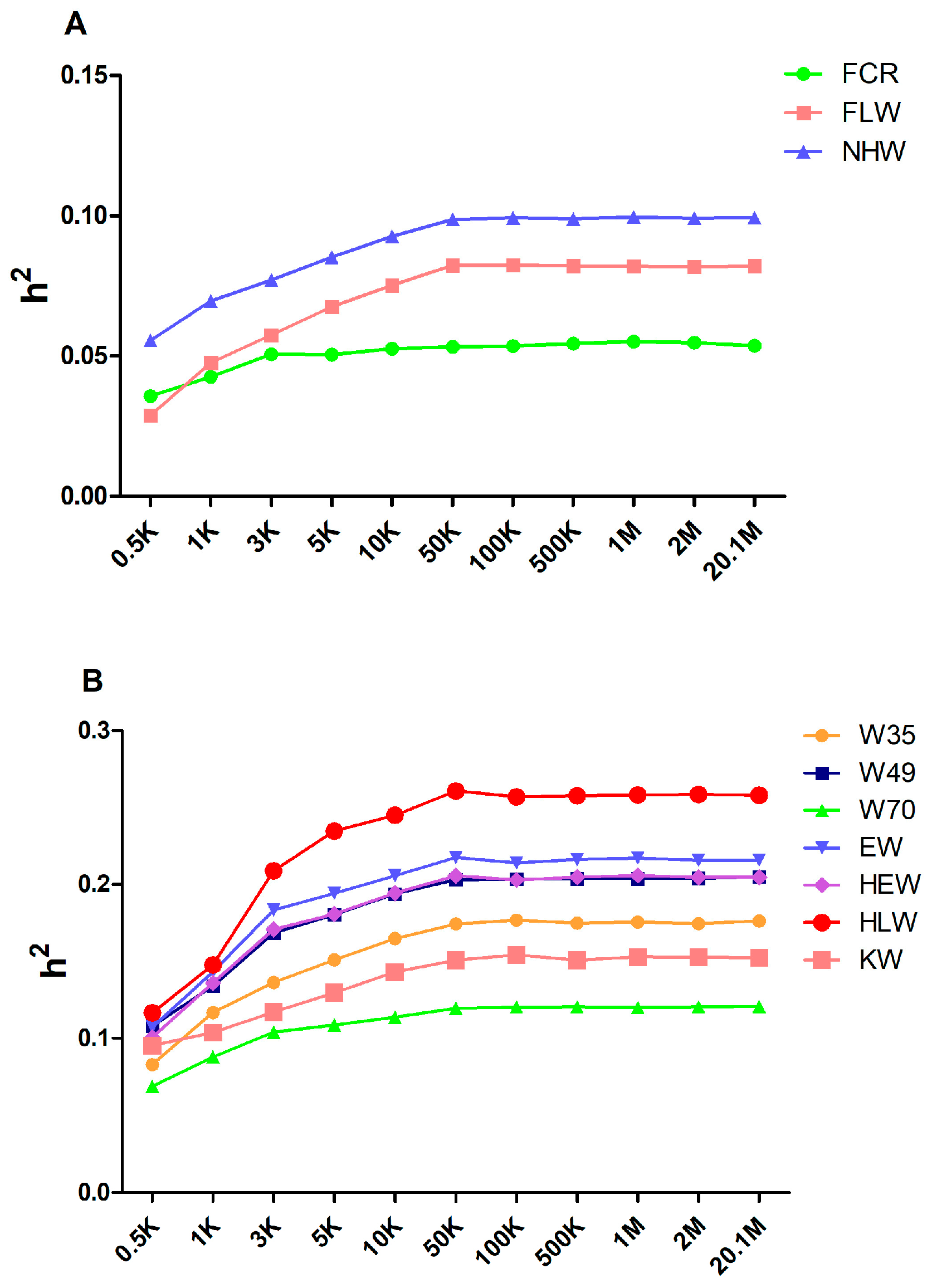
3.1. The 50 K Markers as a Baseline for Estimating Heritability for Meat Rabbits
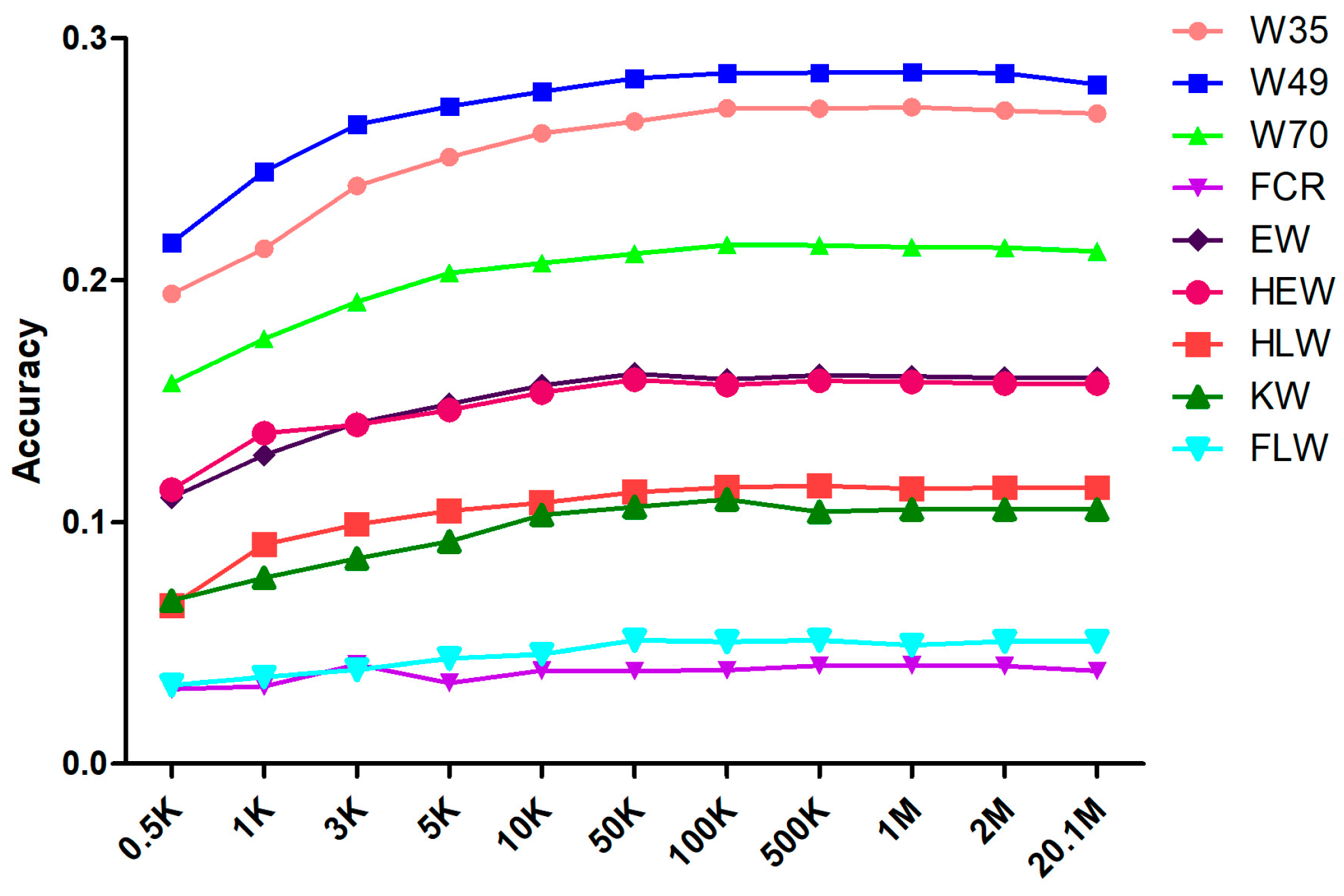
3.2. The 50 K Markers Achieve the Required Prediction Accuracy for the Meat Rabbits
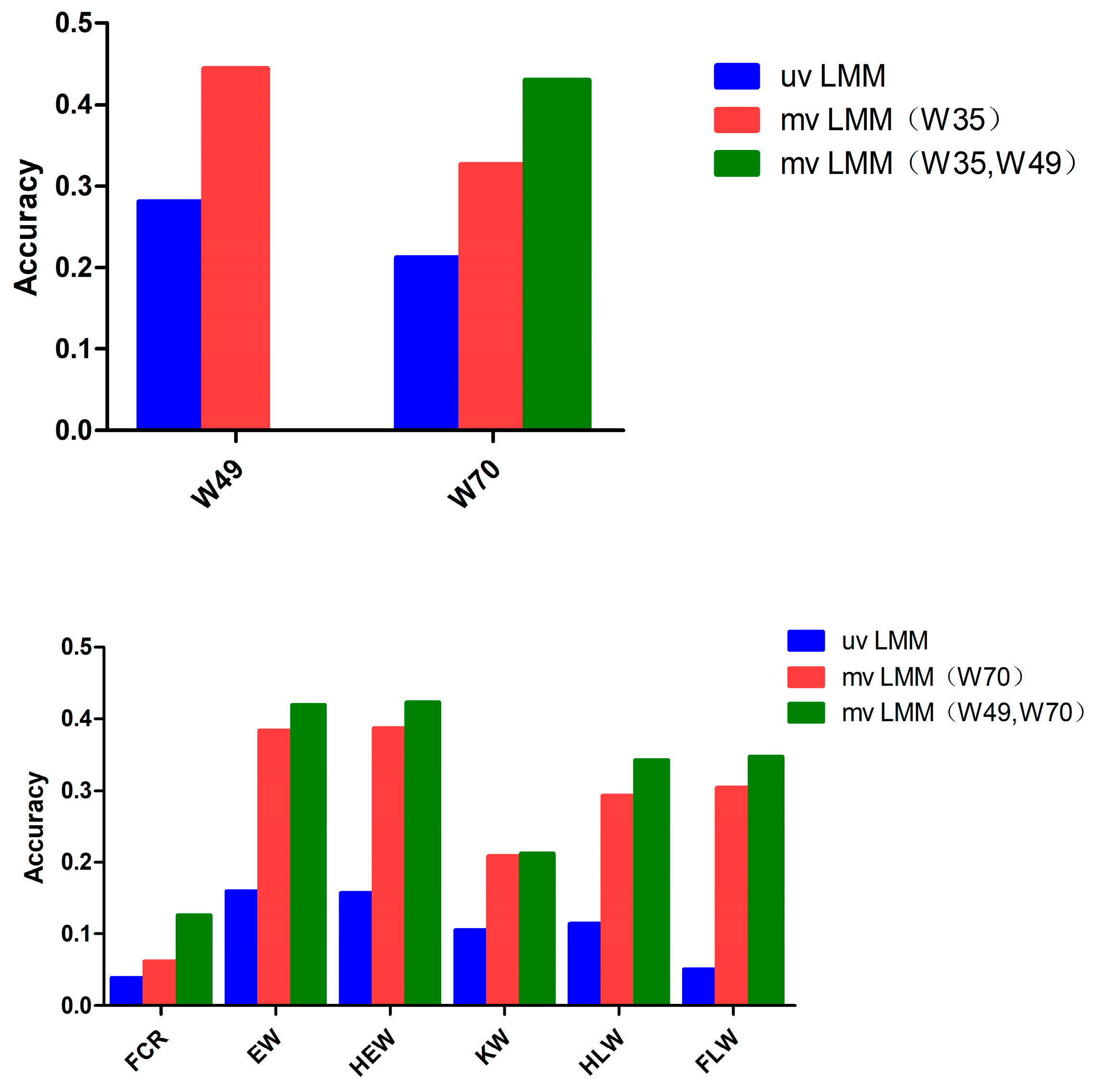
3.3. Accuracy of the Genomic Prediction
4. Discussion
4.1. Appropriate Marker Density for Heritability Estimation
4.2. Appropriate Marker Density for Genomic Prediction
4.3. Different Genome Prediction Models Using Imputation-Based Sequence Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mancini, S.; Paci, G. Probiotics in rabbit farming: Growth performance, health status, and meat quality. Animals 2021, 11, 3388. [Google Scholar] [CrossRef]
- Cullere, M.; Dalle Zotte, A. Rabbit meat production and consumption: State of knowledge and future perspectives. Meat Sci. 2018, 143, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Huang, D.P.; Xie, X.H. Progress on breeding for best-hybridized crossing of meat rabbits. Yi Chuan 2012, 34, 401–406. [Google Scholar] [CrossRef]
- Meuwissen, T.H.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Scott, B.A.; Haile-Mariam, M.; Cocks, B.G.; Pryce, J.E. How genomic selection has increased rates of genetic gain and inbreeding in the Australian national herd, genomic information nucleus, and bulls. J. Dairy Sci. 2021, 104, 11832–11849. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Iwata, H. A Bayesian method and its variational approximation for prediction of genomic breeding values in multiple traits. BMC Bioinform. 2013, 14, 34. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Wu, X.L. Methods and applications of animal genomic mating. Yi Chuan 2019, 41, 486–493. [Google Scholar] [PubMed]
- Georges, M.; Charlier, C.; Hayes, B. Harnessing genomic information for livestock improvement. Nat. Rev. Genet. 2019, 20, 135–156. [Google Scholar] [CrossRef]
- Wolc, A.; Stricker, C.; Arango, J.; Settar, P.; Fulton, J.E.; O’Sullivan, N.P.; Preisinger, R.; Habier, D.; Fernando, R.; Garrick, D.J.; et al. Breeding value prediction for production traits in layer chickens using pedigree or genomic relationships in a reduced animal model. Genet. Sel. Evol. 2011, 43, 5. [Google Scholar] [CrossRef]
- García-Ruiz, A.; Cole, J.B.; VanRaden, P.M.; Wiggans, G.R.; Ruiz-López, F.J.; Van Tassell, C.P. Changes in genetic selection differentials and generation intervals in US Holstein dairy cattle as a result of genomic selection. Proc. Natl. Acad. Sci. USA 2016, 113, E3995–E4004. [Google Scholar] [CrossRef]
- Lillehammer, M.; Meuwissen, T.H.; Sonesson, A.K. Genomic selection for maternal traits in pigs. J. Anim. Sci. 2011, 89, 3908–3916. [Google Scholar] [CrossRef] [PubMed]
- Marina, H.; Pelayo, R.; Gutiérrez-Gil, B.; Suárez-Vega, A.; Esteban-Blanco, C.; Reverter, A.; Arranz, J.J. Low-density SNP panel for efficient imputation and genomic selection of milk production and technological traits in dairy sheep. J. Dairy Sci. 2022, 105, 8199–8217. [Google Scholar] [CrossRef] [PubMed]
- Herry, F.; Hérault, F.; Lecerf, F.; Lagoutte, L.; Doublet, M.; Picard-Druet, D.; Bardou, P.; Varenne, A.; Burlot, T.; Roy, P.L.; et al. Restriction site-associated DNA sequencing technologies as an alternative to low-density SNP chips for genomic selection: A simulation study in layer chickens. BMC Genom. 2023, 24, 271. [Google Scholar] [CrossRef] [PubMed]
- Hayes, B.J.; Bowman, P.J.; Chamberlain, A.J.; Goddard, M.E. Invited review: Genomic selection in dairy cattle: Progress and challenges. J. Dairy Sci. 2009, 92, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.M.; Crooijmans, R.P.; Affara, N.A.; Amara, A.J.; Archibald, A.L.; Beever, J.E.; Bendixen, C.; Churcher, C.; Clark, R.; Dehais, P.; et al. Design of a high density SNP genotyping assay in the pig using SNPs identified and characterized by next generation sequencing technology. PLoS ONE 2009, 4, e6524. [Google Scholar] [CrossRef] [PubMed]
- Stock, K.F.; Reents, R. Genomic selection: Status in different species and challenges for breeding. Reprod. Domest. Anim. 2013, 48 (Suppl. S1), 2–10. [Google Scholar] [CrossRef] [PubMed]
- Mancin, E.; Sosa-Madrid, B.S.; Blasco, A.; Ibáñez-Escriche, N. Genotype imputation to improve the cost-efficiency of genomic selection in rabbits. Animals 2021, 11, 803. [Google Scholar] [CrossRef]
- Jiang, Y.; Jiang, Y.; Wang, S.; Zhang, Q.; Ding, X. Optimal sequencing depth design for whole genome re-sequencing in pigs. BMC Bioinform. 2019, 20, 556. [Google Scholar] [CrossRef] [PubMed]
- Gardner, E.M.; Johnson, M.G.; Ragone, D.; Wickett, N.J.; Zerega, N.J. Low-coverage, whole-genome sequencing of Artocarpus camansi (Moraceae) for phylogenetic marker development and gene discovery. Appl. Plant Sci. 2016, 4, 1600017. [Google Scholar] [CrossRef]
- Gilly, A.; Ritchie, G.R.; Southam, L.; Farmaki, A.; Tsafantakis, E.; Dedoussis, G.; Zeggini, E. Very low-depth sequencing in a founder population identifies a cardioprotective APOC3 signal missed by genome-wide imputation. Hum. Mol. Genet. 2016, 25, 2360–2365. [Google Scholar] [CrossRef]
- Alex, B.C.; Gompert, Z. Population genomics based on low coverage sequencing: How low should we go? Mol. Ecol. 2013, 22, 3028–3035. [Google Scholar] [CrossRef] [PubMed]
- Nicod, J.; Davies, R.W.; Cai, N.; Hassett, C.; Goodstadt, L.; Cosgrove, C.; Yee, B.K.; Lionikaite, V.; McIntyre, R.E.; Remme, C.A.; et al. Genome-wide association of multiple complex traits in outbred mice by ultra-low-coverage sequencing. Nat. Genet. 2016, 48, 912–918. [Google Scholar] [CrossRef] [PubMed]
- Rubinacci, S.; Ribeiro, D.M.; Hofmeister, R.J.; Delaneau, O. Efficient phasing and imputation of low-coverage sequencing data using large reference panels. Nat. Genet. 2021, 53, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Guo, X.; Zhu, D.; Tan, C.; Bian, C.; Ren, J.; Huang, Z.; Zhao, Y.; Cai, G.; Liu, D.; et al. Accelerated deciphering of the genetic architecture of agricultural economic traits in pigs using a low-coverage whole-genome sequencing strategy. Gigascience 2021, 10, giab048. [Google Scholar] [CrossRef] [PubMed]
- Lorenzana, R.E.; Bernardo, R. Accuracy of genotypic value predictions for marker-based selection in biparental plant populations. Theor. Appl. Genet. 2009, 120, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Habier, D.; Fernando, R.L.; Garrick, D.J. Genomic BLUP decoded: A look into the black box of genomic prediction. Genetics 2013, 194, 597–607. [Google Scholar] [CrossRef] [PubMed]
- VanRaden, P.M.; Tooker, M.E.; O’Connell, J.R.; Cole, J.B.; Bickhart, D.M. Selecting sequence variants to improve genomic predictions for dairy cattle. Genet. Sel. Evol. 2017, 49, 32. [Google Scholar] [CrossRef] [PubMed]
- Al-Khudhair, A.; VanRaden, P.M.; Null, D.J.; Li, B. Marker selection and genomic prediction of economically important traits using imputed high-density genotypes for 5 breeds of dairy cattle. J. Dairy Sci. 2021, 104, 4478–4485. [Google Scholar] [CrossRef]
- Lillehammer, M.; Meuwissen, T.H.; Sonesson, A.K. A low-marker density implementation of genomic selection in aquaculture using within-family genomic breeding values. Genet. Sel. Evol. 2013, 45, 39. [Google Scholar] [CrossRef]
- Krishnappa, G.; Savadi, S.; Tyagi, B.S.; Singh, S.K.; Mamrutha, H.M.; Kumar, S.; Mishra, C.N.; Khan, H.; Gangadhara, K.; Uday, G.; et al. Integrated genomic selection for rapid improvement of crops. Genomics 2021, 113, 1070–1086. [Google Scholar] [CrossRef]
- Boison, S.A.; Utsunomiya, A.T.; Santos, D.J.; Neves, H.H.; Carvalheiro, R.; Mészáros, G.; Utsunomiya, Y.T.; Carmo, A.S.; Verneque, R.S.; Machado, M.A.; et al. Accuracy of genomic predictions in Gyr (Bos indicus) dairy cattle. J. Dairy Sci. 2017, 100, 5479–5490. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Zhao, F.; Wang, Y.; Zhang, Y.; Du, L.; Su, G. Comparison of single-trait and multiple-trait genomic prediction models. BMC Genet. 2014, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.; Cho, C.I.; Choi, T.J.; Park, B.; Choi, J.G.; Choy, Y.H.; Lee, S.S.; Cho, K.H. Estimation of Genetic Parameters for Somatic Cell Scores of Holsteins Using Multi-trait Lactation Models in Korea. Asian-Australas. J. Anim. Sci. 2015, 28, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Davies, R.W.; Flint, J.; Myers, S.; Mott, R. Rapid genotype imputation from sequence without reference panels. Nat. Genet. 2016, 48, 965–969. [Google Scholar] [CrossRef] [PubMed]
- Browning, B.L.; Zhou, Y.; Browning, S.R. One-Penny Imputed Genome from Next-Generation Reference Panels. Am. J. Hum. Genet. 2018, 103, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Xie, K.; Wang, Y.; Hu, J.; Li, W.; Yang, A.; Zhang, Q.; Ning, C.; Fan, X. Cost-effectively dissecting the genetic architecture of complex wool traits in rabbits by low-coverage sequencing. Genet. Sel. Evol. 2022, 54, 75. [Google Scholar] [CrossRef] [PubMed]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef]
- Wang, D.; Tang, H.; Liu, J.F.; Xu, S.; Zhang, Q.; Ning, C. Rapid epistatic mixed-model association studies by controlling multiple polygenic effects. Bioinformatics 2020, 36, 4833–4837. [Google Scholar] [CrossRef]
- Madsen, P.S.; Sørensen, P.; Su, G.; Damgaard, L.H.; Thomsen, H.; Labouriau, R.E. DMU-a package for analyzing multivariate mixed models. In Proceedings of the 8th World Congress on Genetics Applied to Livestock Production, Belo Horizonte, MG, Brazil, 13–18 August 2006. [Google Scholar]
- Garrick, D.J. The nature, scope and impact of genomic prediction in beef cattle in the United States. Genet. Sel. Evol. 2011, 43, 17. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.Q.; Chen, B.; Ran, M.L.; Yang, G.M.; Zeng, C. The application of genomic selection in pig cross breeding. Yi Chuan 2020, 42, 145–152. [Google Scholar]
- Brun, J.M.; Sanchez, A.; Ailloud, E.; Saleil, G.; Theau-Clément, M. Genetic parameters of rabbit semen traits and male fertilising ability. Anim. Reprod. Sci. 2016, 166, 15–21. [Google Scholar] [CrossRef]
- Sánchez, J.P.; Ragab, M.; Mínguez, C.; Piles, M. Genotype by feeding regimen interactions for slaughter traits in rabbit and expected responses under restricted and full feeding. J. Anim. Breed. Genet. 2022, 139, 530–539. [Google Scholar] [CrossRef]
- Montes-Vergara, D.E.; Hernndez-Herrera, D.Y.; Hurtado-Lugo, N.A. Genetic parameters of growth traits and carcass weight of New Zealand white rabbits in a tropical dry forest area. J. Adv. Vet. Anim. Res. 2021, 8, 471–478. [Google Scholar] [CrossRef]
- Larzul, C.; Gondret, F.; Combes, S.; Rochambeau, H. Divergent selection on 63-day body weight in the rabbit: Response on growth, carcass and muscle traits. Genet. Sel. Evol. 2005, 37, 105–122. [Google Scholar] [CrossRef]
- Lukefahr, S.D.; Odi, H.B.; Atakora, J.K. Mass selection for 70-day body weight in rabbits. J. Anim. Sci. 1996, 74, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Su, G.; Brøndum, R.F.; Ma, P.; Guldbrandtsen, B.; Aamand, G.P.; Lund, M.S. Comparison of genomic predictions using medium-density (∼54,000) and high-density (∼777,000) single nucleotide polymorphism marker panels in Nordic Holstein and Red Dairy Cattle populations. J. Dairy Sci. 2012, 95, 4657–4665. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Shen, L.; Guo, Z.; Gan, M.; Chen, Y.; Yang, R.; Niu, L.; Jiang, D.; Zhong, Z.; Li, W.; et al. Single nucleotide polymorphism-based analysis of the genetic structure of Liangshan pig population. Anim. Biosci. 2021, 34, 1105–1115. [Google Scholar] [CrossRef]
- Espigolan, R.; Baldi, F.; Boligon, A.A.; Souza, F.R.; Gordo, D.G.; Tonussi, R.L.; Cardoso, D.F.; Oliveira, H.N.; Tonhati, H.; Sargolzaei, M.; et al. Study of whole genome linkage disequilibrium in Nellore cattle. BMC Genom. 2013, 14, 305. [Google Scholar] [CrossRef]
- Moghaddar, N.; Brown, D.J.; Swan, A.A.; Gurman, P.M.; Li, L.; Werf, J.H. Genomic prediction in a numerically small breed population using prioritized genetic markers from whole-genome sequence data. J. Anim. Breed. Genet. 2022, 139, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Oliveira Júnior, G.A.; Chud, T.C.; Ventura, R.V.; Garrick, D.J.; Cole, J.B.; Munari, D.P.; Ferraz, J.B.; Mullart, E.; DeNise, S.; Smith, S.; et al. Genotype imputation in a tropical crossbred dairy cattle population. J. Dairy Sci. 2017, 100, 9623–9634. [Google Scholar] [CrossRef] [PubMed]
- Ning, C.; Xie, K.; Huang, J.; Di, Y.; Wang, Y.; Yang, A.; Hu, J.; Zhang, Q.; Wang, D.; Fan, X. Marker density and statistical model designs to increase accuracy of genomic selection for wool traits in Angora rabbits. Front. Genet. 2022, 13, 968712. [Google Scholar] [CrossRef] [PubMed]
- Fayaz, H.; Tyagi, S.; Wani, A.A.; Pandey, R.; Akhtar, S.; Bhat, M.A.; Chitikineni, A.; Varshney, R.K.; Thudi, M.; Kumar, U.; et al. Genome-wide association analysis to delineate high-quality SNPs for seed micronutrient density in chickpea (Cicer arietinum L.). Sci. Rep. 2022, 12, 11357. [Google Scholar] [CrossRef] [PubMed]
- Budhlakoti, N.; Mishra, D.C.; Rai, A.; Lal, S.B.; Chaturvedi, K.K.; Kumar, R.R. A Comparative study of single-trait and multi-trait genomic selection. J. Comput. Biol. 2019, 26, 1100–1112. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Jannink, J.L. Multiple-trait genomic selection methods increase genetic value prediction accuracy. Genetics 2012, 192, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Calus, M.P.; Veerkamp, R.F. Accuracy of multi-trait genomic selection using different methods. Genet. Sel. Evol. 2011, 43, 26. [Google Scholar] [CrossRef]
- Tsuruta, S.; Misztal, I.; Aguilar, I.; Lawlor, T.J. Multiple-trait genomic evaluation of linear type traits using genomic and phenotypic data in US Holsteins. J. Dairy Sci. 2011, 94, 4198–4204. [Google Scholar] [CrossRef]
- Mekonnen, M.M.; Neale, C.M.; Ray, C.; Erickson, G.E.; Hoekstra, A.Y. Water productivity in meat and milk production in the US from 1960 to 2016. Environ. Int. 2019, 132, 105084. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Trait | Heritability |
|---|---|
| W35 | 0.176 ± 0.044 |
| W49 | 0.205 ± 0.044 |
| W70 | 0.121 ± 0.037 |
| FCR | 0.054 ± 0.028 |
| EW | 0.216 ± 0.003 |
| HEW | 0.205 ± 0.057 |
| KW | 0.153 ± 0.051 |
| HLW | 0.258 ± 0.063 |
| FLW | 0.082 ± 0.048 |
| Trait/Accuracy | 0.5 K | 1 K | 3 K | 5 K | 10 K | 50 K | 100 K | 500 K | 1 M | 2 M |
|---|---|---|---|---|---|---|---|---|---|---|
| W35 | 0.194 ± 0.0097 | 0.213 ± 0.0096 | 0.239 ± 0.0112 | 0.251 ± 0.0094 | 0.261 ± 0.0092 | 0.266 ± 0.0099 | 0.271 ± 0.0094 | 0.270 ± 0.0093 | 0.271 ± 0.0090 | 0.270 ± 0.0087 |
| W49 | 0.215 ± 0.0083 | 0.245 ± 0.0081 | 0.264 ± 0.0093 | 0.272 ± 0.0087 | 0.279 ± 0.0085 | 0.283 ± 0.0089 | 0.286 ± 0.0084 | 0.286 ± 0.0069 | 0.286 ± 0.0068 | 0.286 ± 0.0065 |
| W70 | 0.157 ± 0.0091 | 0.176 ± 0.0084 | 0.191 ± 0.0125 | 0.203 ± 0.0105 | 0.207 ± 0.0104 | 0.211 ± 0.0116 | 0.215 ± 0.0113 | 0.214 ± 0.0102 | 0.214 ± 0.0099 | 0.213 ± 0.0100 |
| FCR | 0.031 ± 0.0094 | 0.032 ± 0.0071 | 0.041 ± 0.0082 | 0.033 ± 0.0080 | 0.039 ± 0.0097 | 0.038 ± 0.0087 | 0.039 ± 0.0088 | 0.041 ± 0.0079 | 0.041 ± 0.0076 | 0.041 ± 0.0072 |
| EW | 0.110 ± 0.0110 | 0.126 ± 0.0089 | 0.141 ± 0.0075 | 0.149 ± 0.0103 | 0.156 ± 0.0094 | 0.161 ± 0.0084 | 0.159 ± 0.0085 | 0.161 ± 0.0075 | 0.160 ± 0.0075 | 0.160 ± 0.0070 |
| HEW | 0.113 ± 0.0104 | 0.137 ± 0.0080 | 0.140 ± 0.0066 | 0.146 ± 0.0095 | 0.154 ± 0.0083 | 0.159 ± 0.0077 | 0.157 ± 0.0077 | 0.158 ± 0.0068 | 0.158 ± 0.0061 | 0.157 ± 0.0059 |
| KW | 0.068 ± 0.0125 | 0.077 ± 0.0137 | 0.085 ± 0.0159 | 0.092 ± 0.0150 | 0.103 ± 0.0150 | 0.106 ± 0.0154 | 0.109 ± 0.0155 | 0.104 ± 0.0152 | 0.105 ± 0.0149 | 0.105 ± 0.0138 |
| HLW | 0.065 ± 0.0043 | 0.091 ± 0.0066 | 0.099 ± 0.0061 | 0.105 ± 0.0052 | 0.108 ± 0.0054 | 0.112 ± 0.0057 | 0.114 ± 0.0054 | 0.115 ± 0.0052 | 0.114 ± 0.0051 | 0.114 ± 0.0049 |
| FLW | 0.032 ± 0.0075 | 0.036 ± 0.0105 | 0.039 ± 0.0111 | 0.044 ± 0.0110 | 0.045 ± 0.0111 | 0.051 ± 0.0110 | 0.050 ± 0.0109 | 0.051 ± 0.0107 | 0.049 ± 0.0105 | 0.051 ± 0.0098 |
| Trait/GEBV | Single-Trait | W35 | W49 | W70 |
|---|---|---|---|---|
| W49 | 0.281 ± 0.0082 | 0.444 ± 0.0087 | ||
| W70 | 0.212 ± 0.0079 | 0.326 ± 0.0106 | 0.432 ± 0.0065 | |
| FCR | 0.038 ± 0.0082 | 0.044 ± 0.0087 | 0.061 ± 0.0093 | 0.044 ± 0.0086 |
| EW | 0.160 ± 0.0084 | 0.226 ± 0.0082 | 0.352 ± 0.0098 | 0.383 ± 0.0102 |
| HEW | 0.157 ± 0.0076 | 0.221 ± 0.0077 | 0.354 ± 0.0091 | 0.386 ± 0.0096 |
| KW | 0.105 ± 0.0151 | 0.097 ± 0.0160 | 0.190 ± 0.0160 | 0.208 ± 0.0163 |
| HLW | 0.114 ± 0.0055 | 0.160 ± 0.0046 | 0.271 ± 0.0068 | 0.293 ± 0.0076 |
| FLW | 0.051 ± 0.0112 | 0.141 ± 0.0107 | 0.332 ± 0.0105 | 0.304 ± 0.0106 |
| Trait/GC | W35 | W49 | W70 |
|---|---|---|---|
| W49 | 0.810 ± 0.0243 | ||
| W70 | 0.617 ± 0.0303 | 0.881 ± 0.0227 | |
| FCR | 0.191 ± 0.0362 | −0.246 ± 0.0352 | 0.081 ± 0.0340 |
| EW | 0.201 ± 0.0390 | 0.756 ± 0.0324 | 0.899 ± 0.0115 |
| HEW | 0.189 ± 0.0390 | 0.754 ± 0.0324 | 0.914 ± 0.0104 |
| KW | 0.182 ± 0.0471 | 0.414 ± 0.0443 | 0.689 ± 0.0371 |
| HLW | 0.160 ± 0.0425 | 0.654 ± 0.0361 | 0.732 ± 0.0159 |
| FLW | 0.226 ± 0.0421 | 0.794 ± 0.0372 | 0.870 ± 0.0214 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.; Li, W.; Song, Z.; Gao, Z.; Xie, K.; Wang, Y.; Wang, B.; Hu, J.; Zhang, Q.; Ning, C.; et al. Marker Density and Models to Improve the Accuracy of Genomic Selection for Growth and Slaughter Traits in Meat Rabbits. Genes 2024, 15, 454. https://doi.org/10.3390/genes15040454
Li W, Li W, Song Z, Gao Z, Xie K, Wang Y, Wang B, Hu J, Zhang Q, Ning C, et al. Marker Density and Models to Improve the Accuracy of Genomic Selection for Growth and Slaughter Traits in Meat Rabbits. Genes. 2024; 15(4):454. https://doi.org/10.3390/genes15040454
Chicago/Turabian StyleLi, Wenjie, Wenqiang Li, Zichen Song, Zihao Gao, Kerui Xie, Yubing Wang, Bo Wang, Jiaqing Hu, Qin Zhang, Chao Ning, and et al. 2024. "Marker Density and Models to Improve the Accuracy of Genomic Selection for Growth and Slaughter Traits in Meat Rabbits" Genes 15, no. 4: 454. https://doi.org/10.3390/genes15040454
APA StyleLi, W., Li, W., Song, Z., Gao, Z., Xie, K., Wang, Y., Wang, B., Hu, J., Zhang, Q., Ning, C., Wang, D., & Fan, X. (2024). Marker Density and Models to Improve the Accuracy of Genomic Selection for Growth and Slaughter Traits in Meat Rabbits. Genes, 15(4), 454. https://doi.org/10.3390/genes15040454