
Transcriptomic Changes and Regulatory Networks Associated with Resistance to Mastitis in Xinjiang Brown Cattle

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Background
2. Materials and Methods
2.1. Animal Selection and Sample Collection
2.2. ELISA Measurements
2.3. RNA Extraction and Testing
2.4. Library Construction and Sequencing
2.5. Data Quality Control and Comparison
2.6. Transcript Splicing and Screening
2.7. Quantitative and Differential Analysis
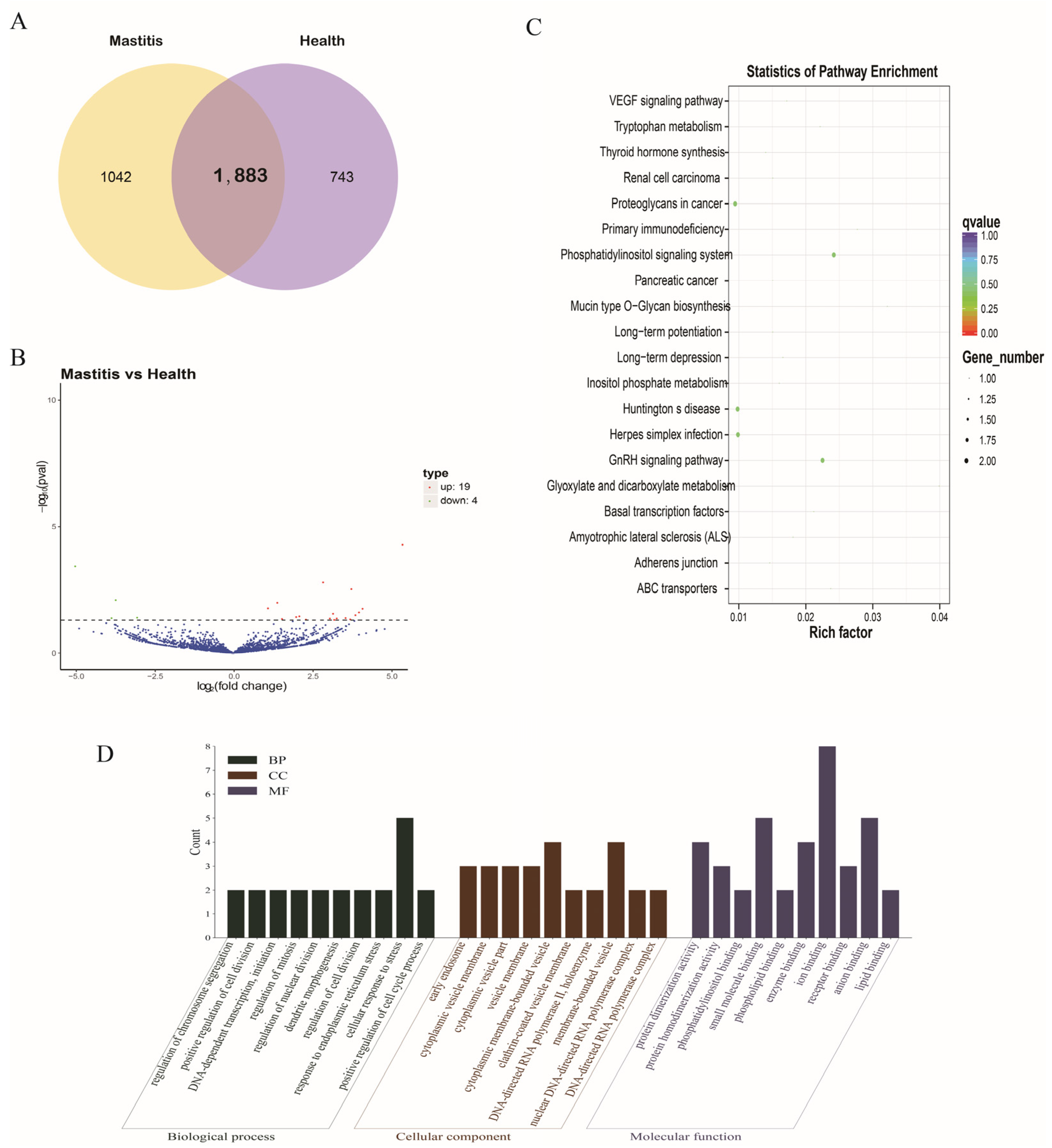
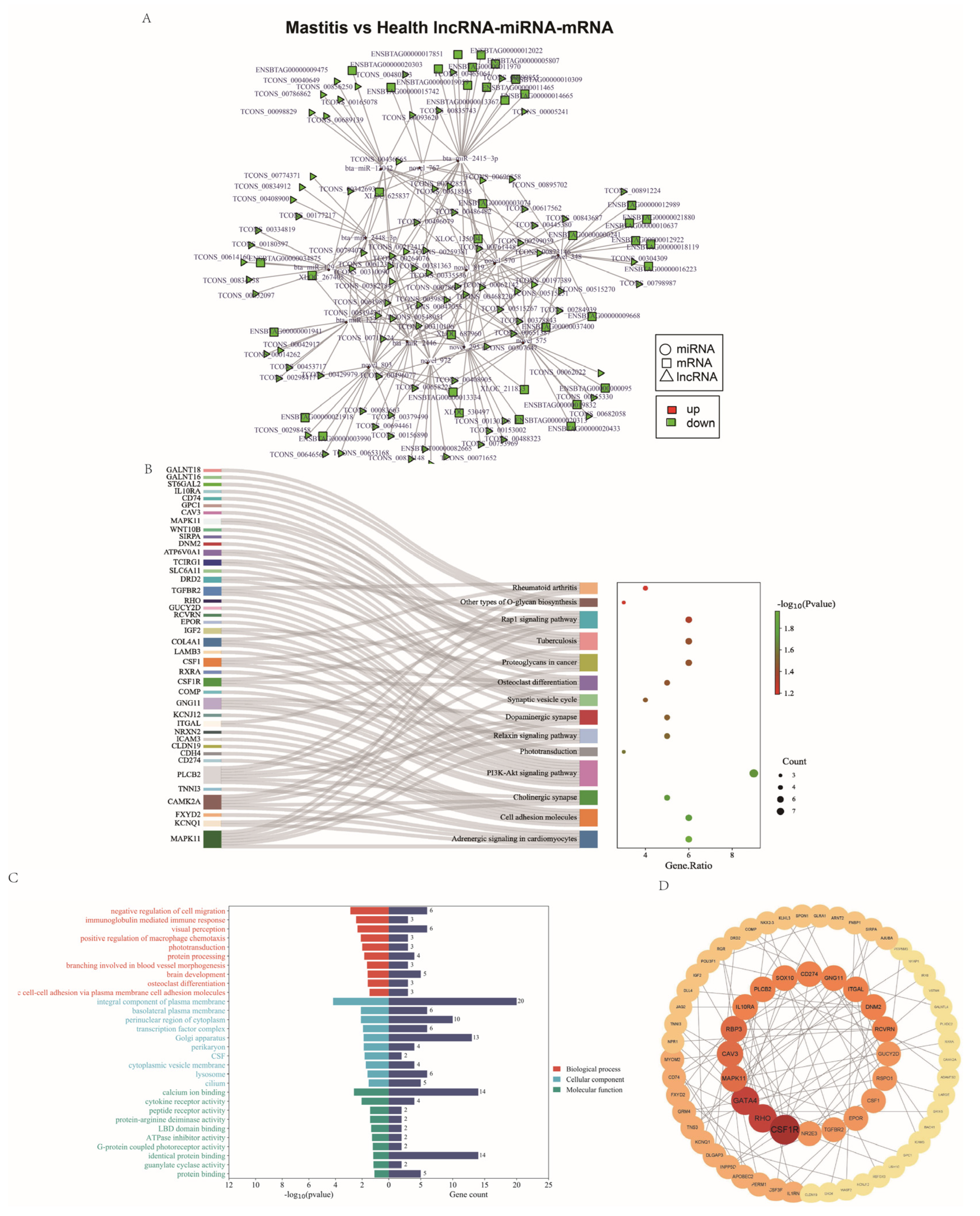
2.8. GO and KEGG Enrichment Analysis
2.9. Prediction of lncRNA Target Genes
2.10. RT-qRCR
2.11. Establishment of ceRNA Networks
3. Results
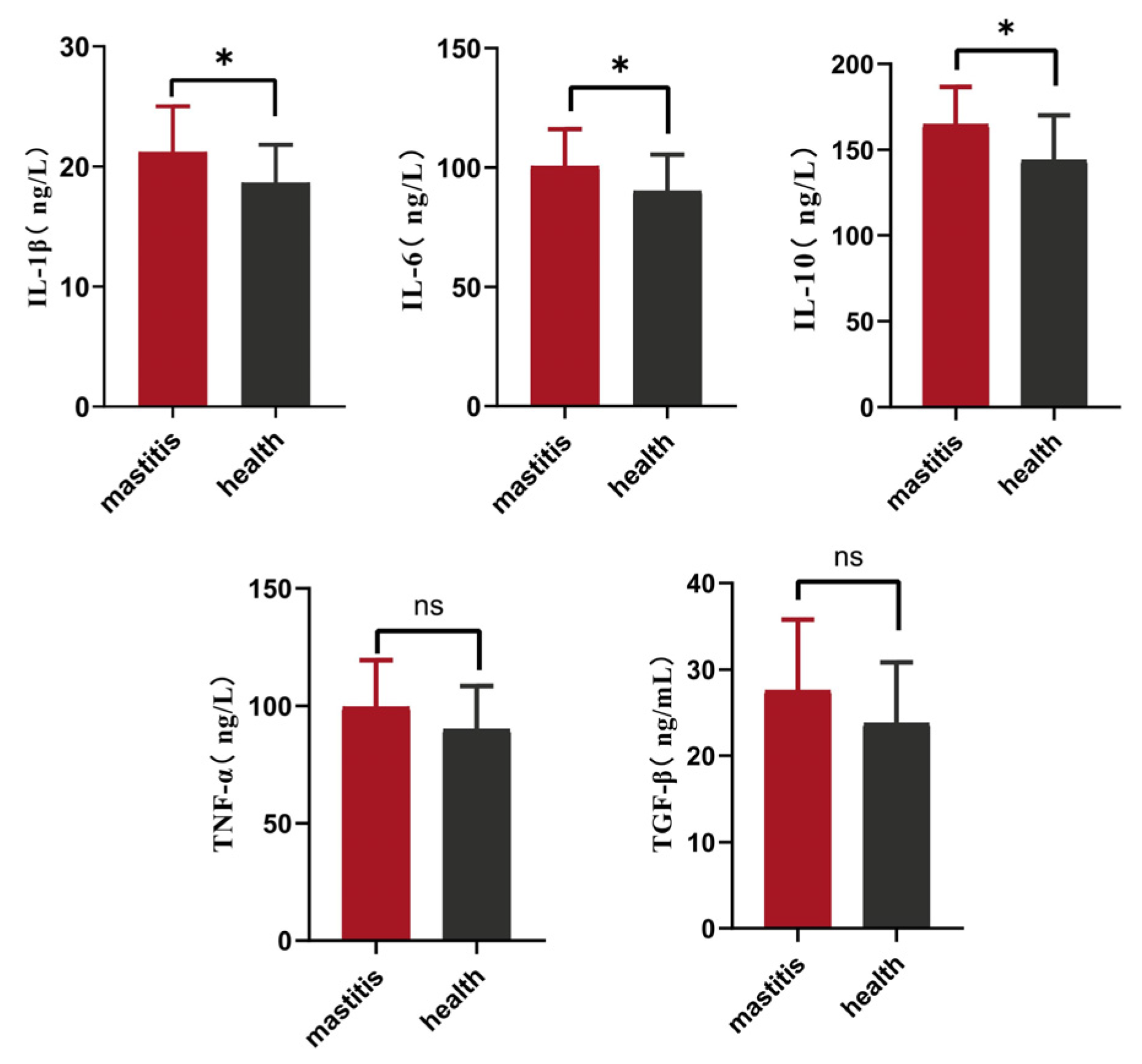
3.1. Serum Cytokine Levels in the High- and Low-SCC Xinjiang Brown Cattle
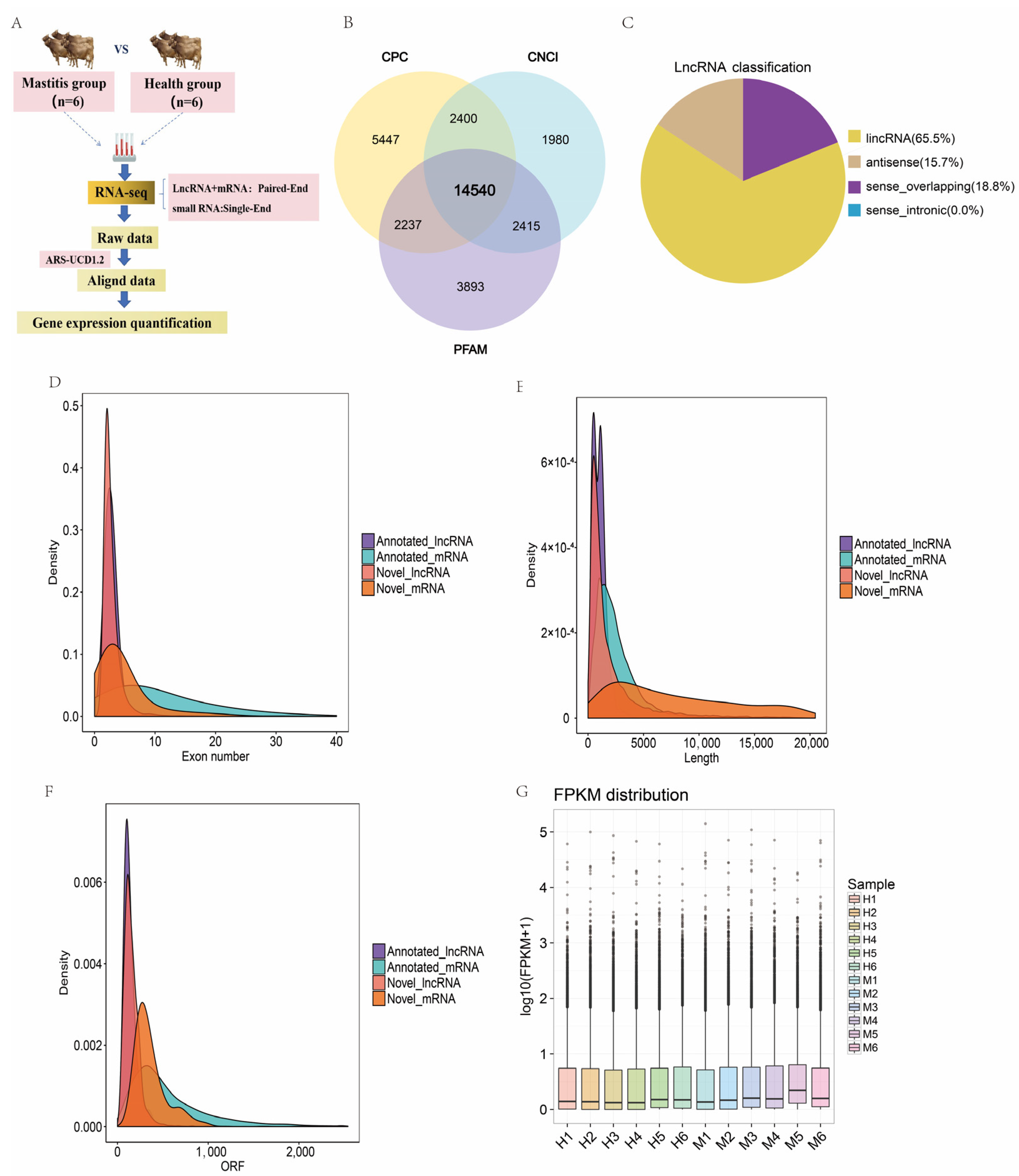
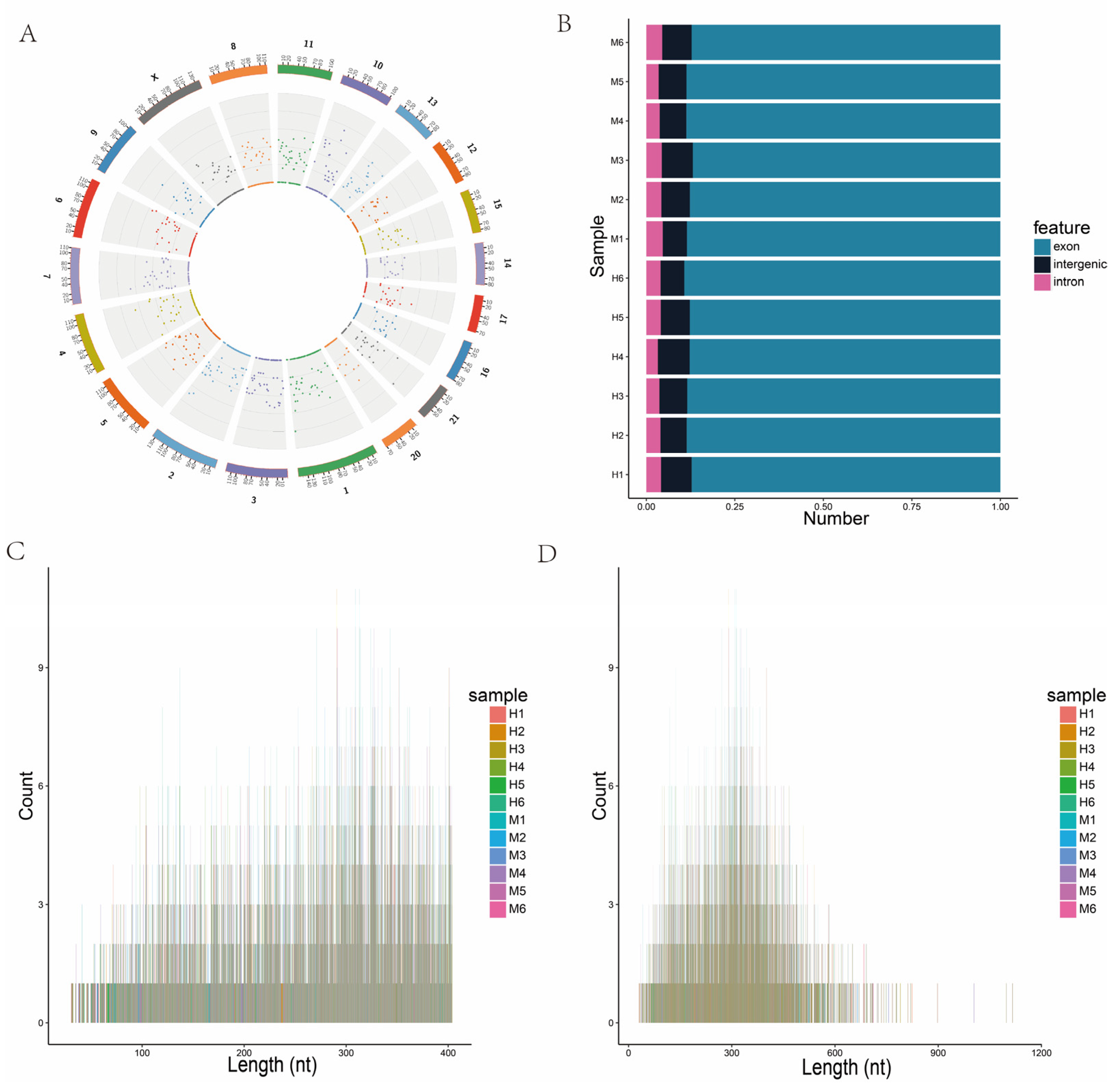
3.2. Sequencing and Characterization of lncRNAs
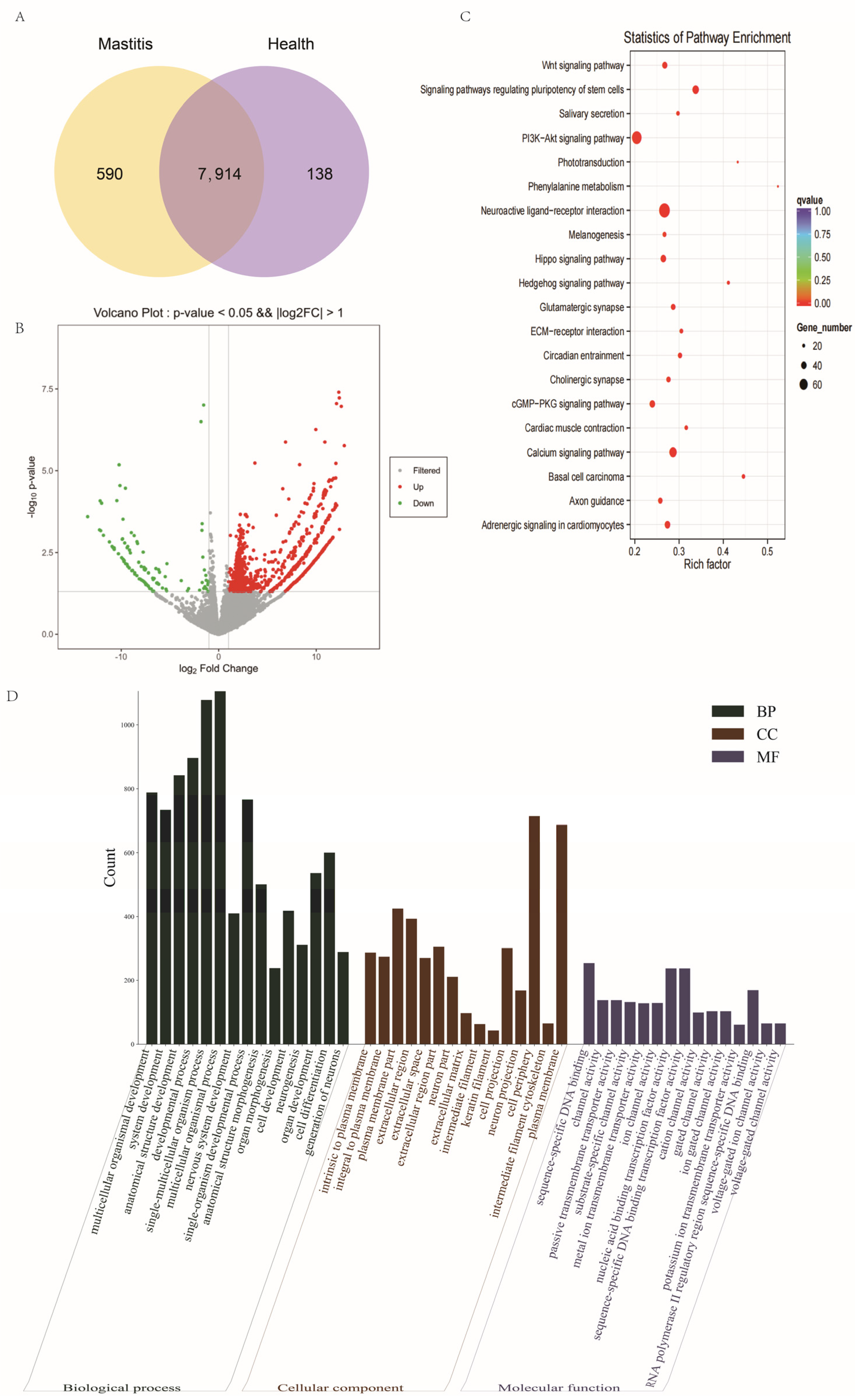
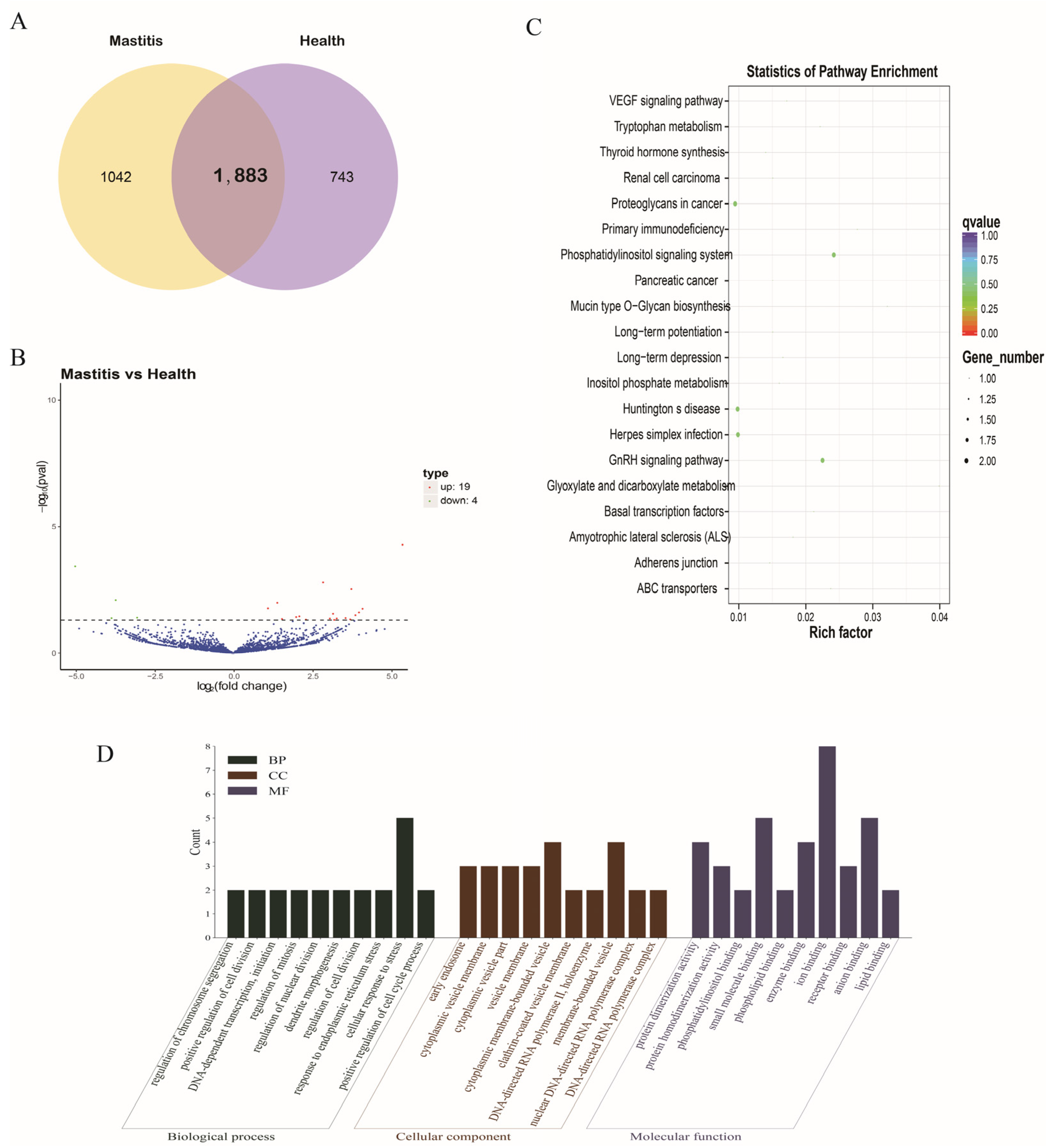
3.3. DE-lncRNAs
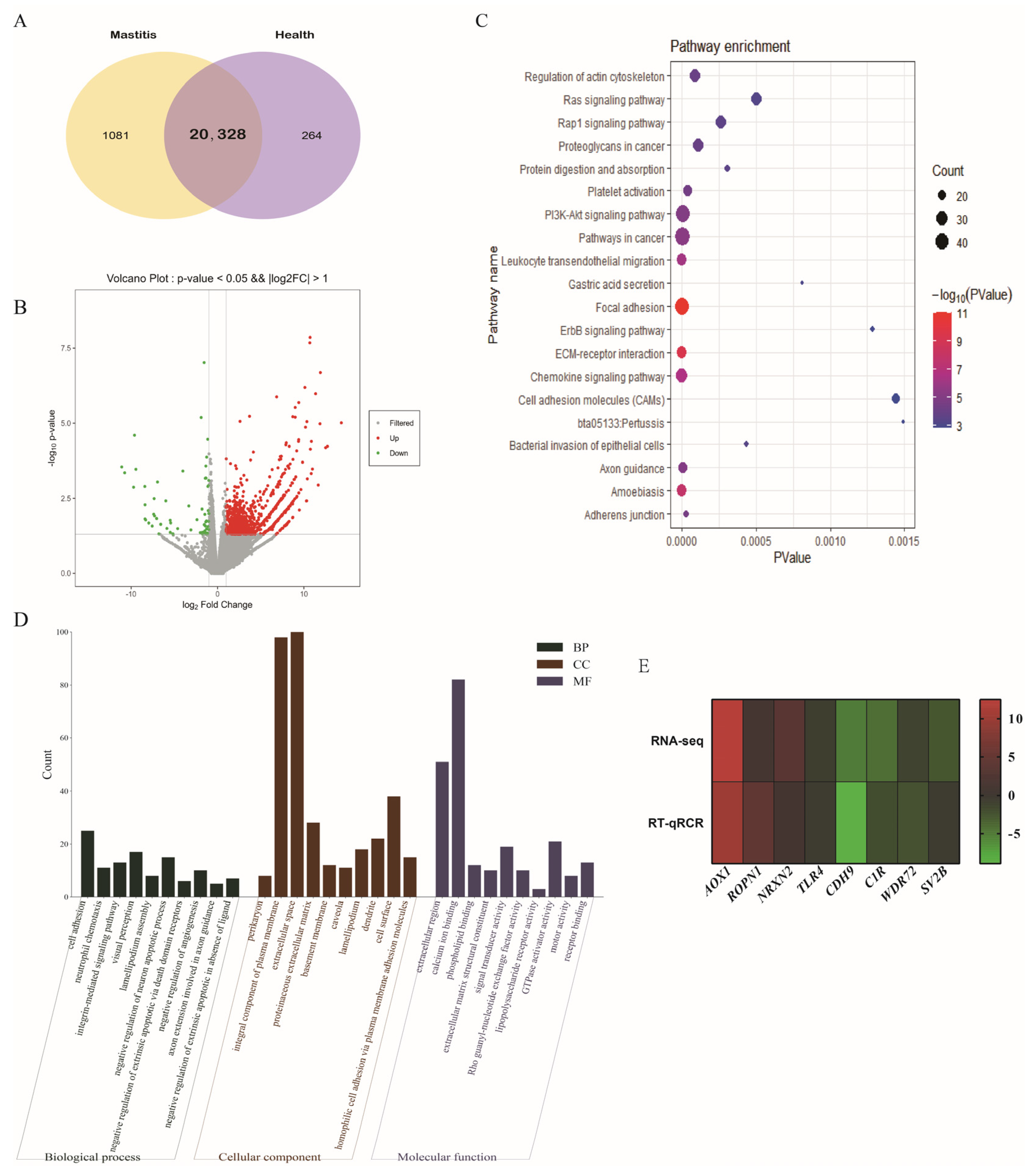
3.4. DE-mRNAs
3.5. Identification and Structural Analysis of circRNAs
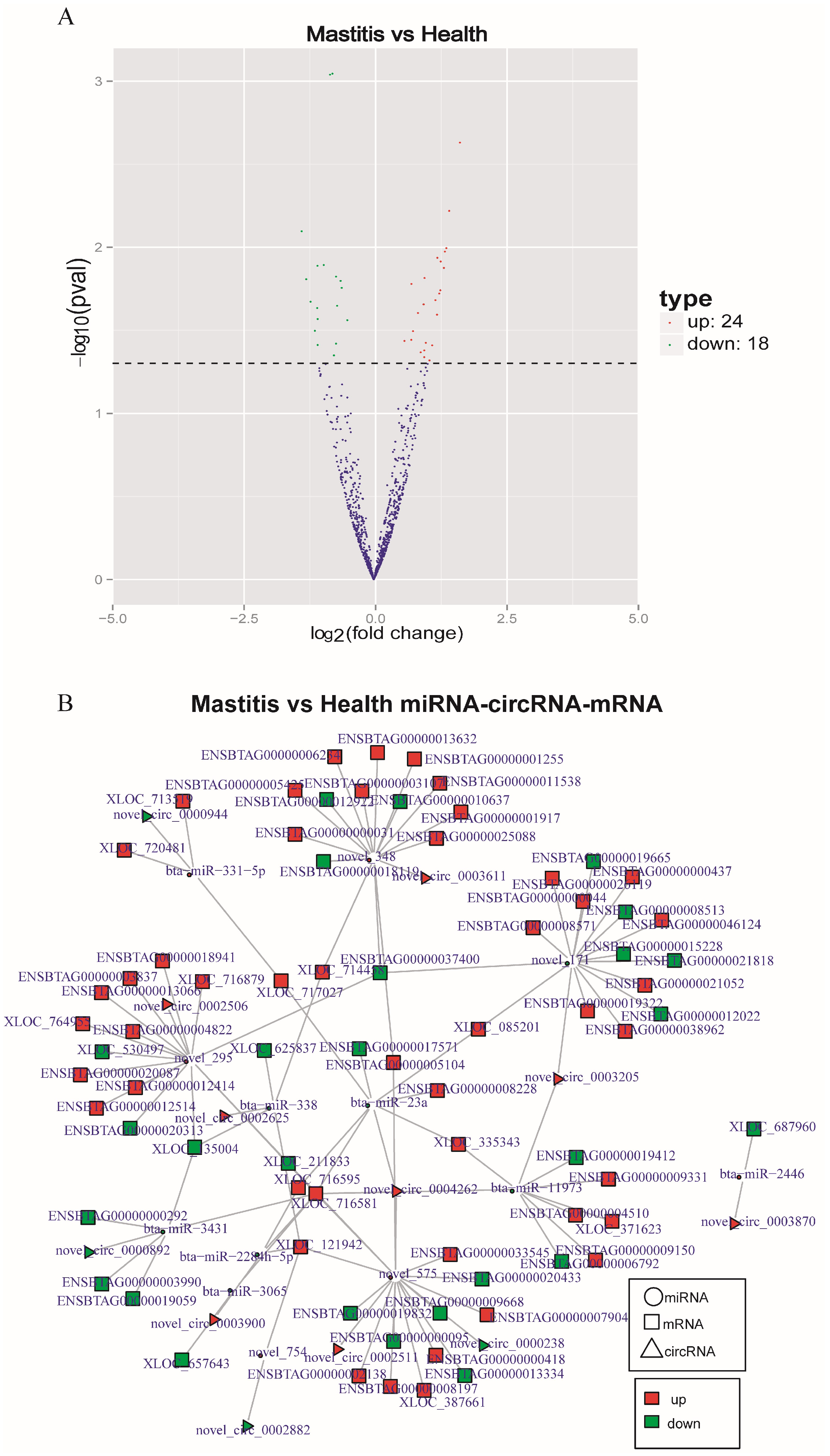
3.6. DE-circRNAs
3.7. CircRNA-miRNA-mRNA Regulatory Network
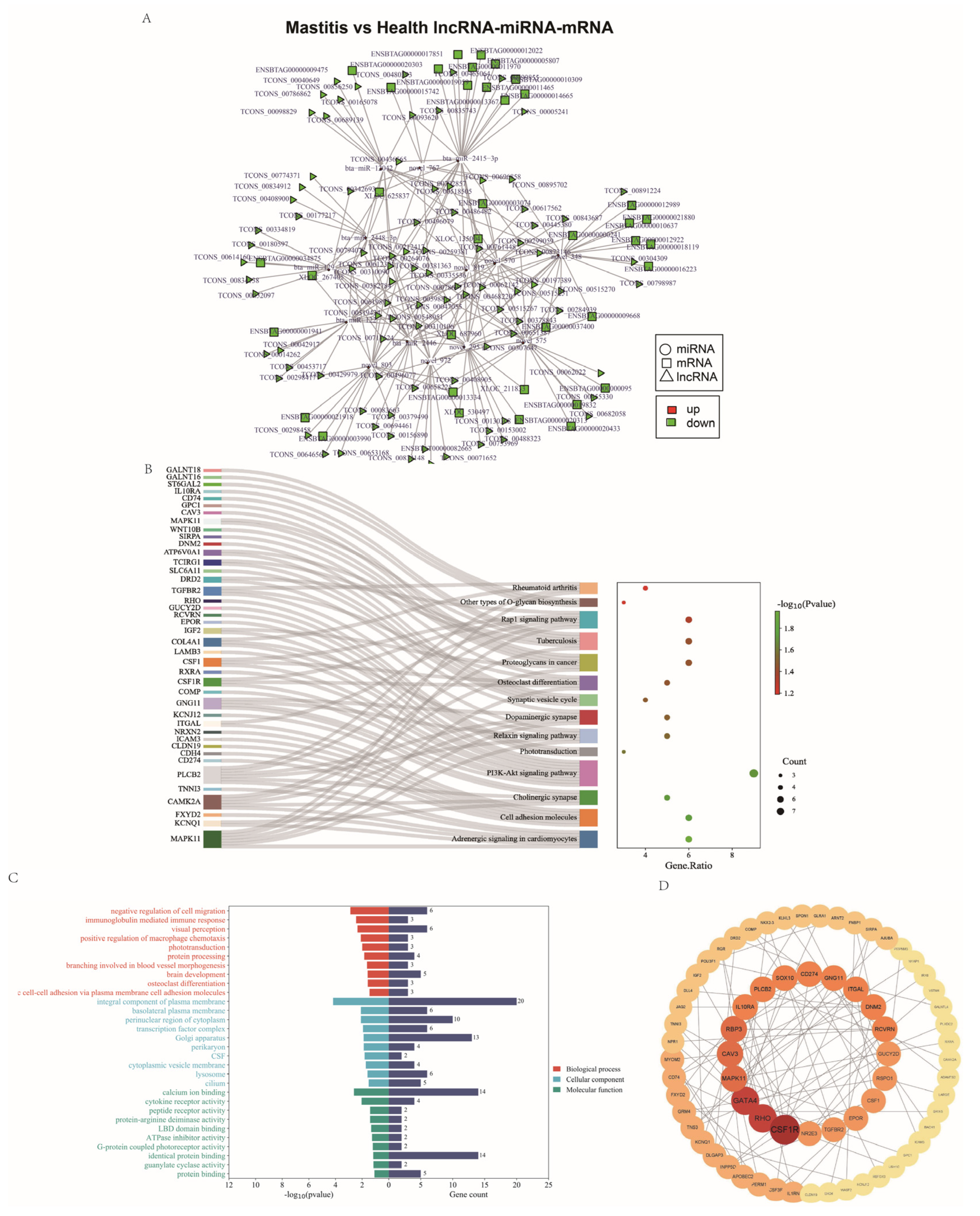
3.8. Regulatory lncRNA-miRNA-mRNA Network
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nemet-Nejat, K.R. Daily Life in Ancient Mesopotamia; Greenwood Press: Westport, CT, USA, 1998. [Google Scholar]
- Ruegg, P.L. A 100-Year Review: Mastitis detection, management, and prevention. J. Dairy Sci. 2017, 100, 10381–10397. [Google Scholar] [CrossRef]
- De Vries, A.; Marcondes, M.I. Review: Overview of factors affecting productive lifespan of dairy cows. Animal 2020, 14, s155–s164. [Google Scholar] [CrossRef] [PubMed]
- Bar, D.; Gröhn, Y.T.; Bennett, G.; González, R.N.; Hertl, J.A.; Schulte, H.F.; Tauer, L.W.; Welcome, F.L.; Schukken, Y.H. Effect of repeated episodes of generic clinical mastitis on milk yield in dairy cows. J. Dairy Sci. 2007, 90, 4643–4653. [Google Scholar] [CrossRef] [PubMed]
- Fernández, L.; Langa, S.; Martín, V.; Maldonado, A.; Jiménez, E.; Martín, R.; Rodríguez, J.M. The human milk microbiota: Origin and potential roles in health and disease. Pharmacol. Res. 2013, 69, 1–10. [Google Scholar] [CrossRef]
- Lyons, K.E.; Ryan, C.A.; Dempsey, E.M.; Ross, R.P.; Stanton, C. Breast Milk, a Source of Beneficial Microbes and Associated Benefits for Infant Health. Nutrients 2020, 12, 1039. [Google Scholar] [CrossRef]
- Sender, G.; Korwin-Kossakowska, A.; Pawlik, A.; Hameed, K.G.A.; Oprządek, J. Genetic basis of mastitis resistance in dairy cattle—a review/podstawy genetyczne odporności krów mlecznych Na zapalenie wymienia—Artykuł przeglądowy. Ann. Anim. Sci. 2013, 13, 663–673. [Google Scholar] [CrossRef]
- Martin, P.; Barkema, H.W.; Brito, L.F.; Narayana, S.G.; Miglior, F. Symposium review: Novel strategies to genetically improve mastitis resistance in dairy cattle. J. Dairy Sci. 2018, 101, 2724–2736. [Google Scholar] [CrossRef]
- Wang, A.F.; Huang, X.X.; Junjulieke, Z.; Liu, L.Y.; Tan, S.X.; Zhu, B.S.; Wang, Y.Q.; Ainiwa, G.; Mutielifu, P.; Liu, Z.Q. Analysis of DHI data in Holstein and Xinjiang brown cattle. China Dairy Cattle 2014, 22, 9–12. (In Chinese) [Google Scholar]
- Zhou, J.H.; Liu, L.Y.; Chen, C.J.; Zhang, M.H.; Lu, X.; Zhang, Z.W.; Huang, X.X.; Shi, Y.G. Genome-wide association study of milk and reproductive traits in dual-purpose Xinjiang Brown cattle. BMC Genom. 2019, 20, 827. [Google Scholar] [CrossRef]
- Ju, X.; Huang, X.X.; Zhang, M.H.; Lan, X.Y.; Wang, D.; Wei, C.; Jiang, H. Effects of eight InDel variants in FHIT on milk traits in Xinjiang brown cattle. Anim. Biotechnol. 2021, 32, 486–494. [Google Scholar] [CrossRef]
- Zhong, L.W.; Ma, S.C.; Wang, D.; Zhang, M.H.; Tian, Y.Z.; He, J.M.; Zhang, X.X.; Xu, L.; Wu, C.L.; Dong, M.M.; et al. Methylation levels in the promoter region of FHIT and PIAS1 genes associated with mastitis resistance in Xinjiang brown cattle. Genes 2023, 14, 1189. [Google Scholar] [CrossRef] [PubMed]
- Correia, C.N.; Mcloughlin, K.E.; Nalpas, N.C.; Magee, D.A.; Browne, J.A.; Rue-Albrecht, K.; Gordon, S.V.; Machugh, D.E. RNA sequencing (RNA-Seq) reveals extremely low levels of reticulocyte-derived globin gene transcripts in peripheral blood from horses (Equus caballus) and cattle (Bos taurus). Front. Genet. 2018, 9, 278. [Google Scholar] [CrossRef] [PubMed]
- Shangraw, E.M.; Rodrigues, R.O.; Choudhary, R.K.; Zhao, F.Q.; Mcfadden, T.B. Hypogalactia in mammary quarters adjacent to lipopolysaccharide-infused quarters is associated with transcriptional changes in immune genes. J. Dairy Sci. 2021, 104, 9276–9286. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.L.; Gao, Y.H.; Canela-Xandri, O.; Wang, S.; Yu, Y.; Cai, W.T.; Li, B.J.; Xiang, R.D.; Chamberlain, A.J.; Pairo-Castineira, E.; et al. A multi-tissue atlas of regulatory variants in cattle. Nat. Genet. 2022, 54, 1438–1447. [Google Scholar] [CrossRef] [PubMed]
- Turk, R.; Piras, C.; Kovacic, M.; Samardzija, M.; Ahmed, H.; De Canio, M.; Urbani, A.; Mestric, Z.F.; Soggiu, A.; Bonizzi, L.; et al. Proteomics of inflammatory and oxidative stress response in cows with subclinical and clinical mastitis. J. Proteom. 2012, 75, 4412–4428. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.N.; Bajic, V.B.; Zhang, Z. On the classification of long non-coding RNAs. RNA Biol. 2013, 10, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.Y.; Geng, D.H.; Li, S.Q.; Chen, Z.F.; Sun, M. LncRNA HOTAIR influences cell growth, migration, invasion, and apoptosis via the miR-20a-5p/HMGA2 axis in breast cancer. Cancer Med. 2018, 7, 842–855. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.N.; Ma, L.; Liu, N. Systematic analysis of lncRNA-miRNA-mRNA competing endogenous RNA network identifies four-lncRNA signature as a prognostic biomarker for breast cancer. J. Transl. Med. 2018, 16, 264. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambrost, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Lu, T.X.; Rothenberg, M.E. MicroRNA. J. Allergy Clin. Immun. 2018, 141, 1202–1207. [Google Scholar] [CrossRef] [PubMed]
- Recchiuti, A.; Krishnamoorthy, S.; Fredman, G.; Chiang, N.; Serhan, C.N. MicroRNAs in resolution of acute inflammation: Identification of novel resolvin Dl-miRNA circuits. FASEB J. 2010, 25, 544–560. [Google Scholar] [CrossRef] [PubMed]
- McLoughlin, K.E.; Nalpas, N.C.; Rue-Albrecht, K.V.; Browne, J.A.; Magee, D.A.; Killick, K.E.; Park, S.D.E.; Hokamp, K.; Meade, K.G.; O’Farrelly, C.; et al. RNA-seq transcriptional profiling of peripheral blood leukocytes from cattle infected with mycobacterium bovis. Front. Immunol. 2014, 5, 396. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.; Han, Y.; He, Q. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, J.; Zhao, F. CIRI: An efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015, 16, 4. [Google Scholar] [CrossRef]
- Porcherie, A.; Cunha, P.; Trotereau, A.; Roussel, P.; Gilbert, F.B.; Rainard, P.; Germon, P. Repertoire of Escherichia coli agonists sensed by innate immunity receptors of the bovine udder and mammary epithelial cells. Vet. Res. 2012, 43, 14. [Google Scholar] [CrossRef] [PubMed]
- Shuster, D.E.; Kehrli, M.E., Jr.; Rainard, P.; Paape, M. Complement fragment C5a and inflammatory cytokines in neutrophil recruitment during intramammary infection with Escherichia coli. Infect. Immun. 1997, 65, 3286–3292. [Google Scholar] [CrossRef] [PubMed]
- Alluwaimi, A.M. The cytokines of bovine mammary gland: Prospects for diagnosis and therapy. Res. Vet. Sci. 2004, 77, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Wellnitz, O.; Zbinden, C.; Huang, X.; Bruckmaier, R.M. Short communication: Differential loss of bovine mammary epithelial barrier integrity in response to lipopolysaccharide and lipoteichoic acid. J. Dairy Sci. 2016, 99, 4851–4856. [Google Scholar] [CrossRef] [PubMed]
- Ezzat Alnakip, M.; Quintela-Baluja, M.; Böhme, K.; Fernández-No, I.; Caamaño-Antelo, S.; Calo-Mata, P.; Barros-Velázquez, J. The Immunology of Mammary Gland of Dairy Ruminants between Healthy and Inflammatory Conditions. J. Vet. Med. 2014, 2014, 65980. [Google Scholar] [CrossRef] [PubMed]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.Z.; Sahana, G.; Su, G.S.; Yu, Y.; Zhang, S.L.; Lund, M.S.; Sørensen, P. Integrating sequence-based GWAS and RNA-Seq provides novel insights into the genetic basis of mastitis and milk production in dairy cattle. Sci. Rep. 2017, 7, 45560. [Google Scholar] [CrossRef] [PubMed]
- Naserkheil, M.; Ghafouri, F.; Zakizadeh, S.; Pirany, N.; Manzari, Z.; Ghorbani, S.; Banabazi, M.H.; Bakhtiarizadeh, M.R.; Huq, M.A.; Park, M.N.; et al. Multi-omics integration and network analysis reveal potential hub genes and genetic mechanisms regulating bovine mastitis. Curr. Issues Mol. Biol. 2022, 44, 309–328. [Google Scholar] [CrossRef] [PubMed]
- Zoldan, K.; Schneider, J.; Moellmer, T.; Fueldner, C.; Knauer, J.; Fuerll, M.; Starke, A.; Specht, M.; Reiche, K.; Hackermueller, J.; et al. Discovery and validation of immunological biomarkers in milk for health monitoring of dairy cows—Results from a multiomics approach. J. Adv. Dairy Res. 2017, 5, 3. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT network at the interface of oncogenic signaling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Wang, Z.J.; Yang, H.Y.; Xu, X.; Hu, H.X.; Bai, Y.X.; Hai, J.; Cheng, L.M.; Zhu, R.R. Ion elemental-optimized layered double hydroxide nanoparticles promote chondrogenic differentiation and intervertebral disc regeneration of mesenchymal stem cells through focal adhesion signaling pathway. Bioact. Mater. 2023, 22, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.C.; Ping, L.Y.; Hsu, S.H.; Tsai, H.Y.; Cheng, M.C. Indicators of HSV1 infection, ECM–receptor interaction, and chromatin modulation in a nuclear family with schizophrenia. J. Pers. Med. 2023, 13, 1392. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hu, Q.C.; Wang, J.P.; Ren, Q.Q.; Wang, X.P.; Luoreng, Z.M.; Wei, D.W.; Ma, Y. RNA-Seq reveals the role of miR-29c in regulating inflammation and oxidative stress of bovine mammary epithelial cells. Front. Vet. Sci. 2022, 9, 865415. [Google Scholar] [CrossRef] [PubMed]
- Ran, H.B.; Yang, Y.; Luo, M.N.; Liu, X.R.; Yue, B.L.; Chai, Z.X.; Zhong, J.C.; Wang, H. Molecular regulation of yak preadipocyte differentiation and proliferation by LncFAM200B and ceRNA regulatory network analysis. Cells 2022, 11, 2366. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Huang, K.W.; Wang, P.C.; Feng, T.; Shi, D.S.; Cui, K.Q.; Luo, C.; Shafique, L.; Qian, Q.; Ruan, J.; et al. Comparison of long non-coding RNA expression profiles of cattle and buffalo differing in muscle characteristics. Front. Genet. 2020, 11, 98. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Ge, F.; Ma, X.; Dai, R.; Dingkao, R.; Zhaxi, Z.; Burenchao, G.; Bao, P.; Wu, X.; Guo, X.; et al. Comprehensive analysis of mRNA, lncRNA, circRNA, and miRNA expression profiles and their ceRNA networks in the longissimus dorsi muscle of cattle-yak and yak. Front. Genet. 2021, 12, 772557. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.C.; Raza, S.H.A.; Naqvi, M.A.R.; Feng, Y.R.; Khan, R.; Mohammedsaleh, Z.M.; Shater, A.F.; Al-Ahmadi, B.M.; Saleh, F.M.; Bilal, M.A.; et al. Construction of adipogenic ceRNA network based on lncRNA expression profile of adipogenic differentiation of human MSC cells. Biochem. Genet. 2022, 60, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.H.; Gu, X.Y.; Lv, X.Y.; Cao, X.K.; Yuan, Z.H.; Wang, S.H.; Sun, W. Non-coding transcriptomic profiles in the sheep mammary gland during different lactation periods. Front. Vet. Sci. 2022, 9, 983562. [Google Scholar] [CrossRef]
- Jia, C.L.; Bai, Y.; Liu, J.N.; Cai, W.T.; Liu, L.; He, Y.H.; Song, J.Z. Metabolic Regulations by lncRNA, miRNA, and ceRNA under grass-fed and grain-fed regimens in angus beef cattle. Front. Genet. 2021, 12, 579393. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; Yang, H.; Ma, S.; Liu, T.; Yan, M.; Dong, M.; Zhang, M.; Zhang, T.; Zhang, X.; Xu, L.; et al. Transcriptomic Changes and Regulatory Networks Associated with Resistance to Mastitis in Xinjiang Brown Cattle. Genes 2024, 15, 465. https://doi.org/10.3390/genes15040465
Wang D, Yang H, Ma S, Liu T, Yan M, Dong M, Zhang M, Zhang T, Zhang X, Xu L, et al. Transcriptomic Changes and Regulatory Networks Associated with Resistance to Mastitis in Xinjiang Brown Cattle. Genes. 2024; 15(4):465. https://doi.org/10.3390/genes15040465
Chicago/Turabian StyleWang, Dan, Haiyan Yang, Shengchao Ma, Tingting Liu, Mengjie Yan, Mingming Dong, Menghua Zhang, Tao Zhang, Xiaoxue Zhang, Lei Xu, and et al. 2024. "Transcriptomic Changes and Regulatory Networks Associated with Resistance to Mastitis in Xinjiang Brown Cattle" Genes 15, no. 4: 465. https://doi.org/10.3390/genes15040465