
The Genome of Bifidobacterium longum subsp. infantis YLGB-1496 Provides Insights into Its Carbohydrate Utilization and Genetic Stability

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Growth Conditions
2.2. Identification of Novel YLGB-1496
2.3. Carb Trials
2.4. Continuous Subculture of YLGB-1496
2.5. Viable Microbial Counts
2.6. Scanning Electron Microscopy (SEM) Analysis of YLGB-1496
2.7. Determination of the Fermentation Viability
2.8. YLGB-1496 Genome Sequencing and Assemblies
2.9. Statistical Analyses
2.10. Sequence Analysis, Software, and Databases
3. Results
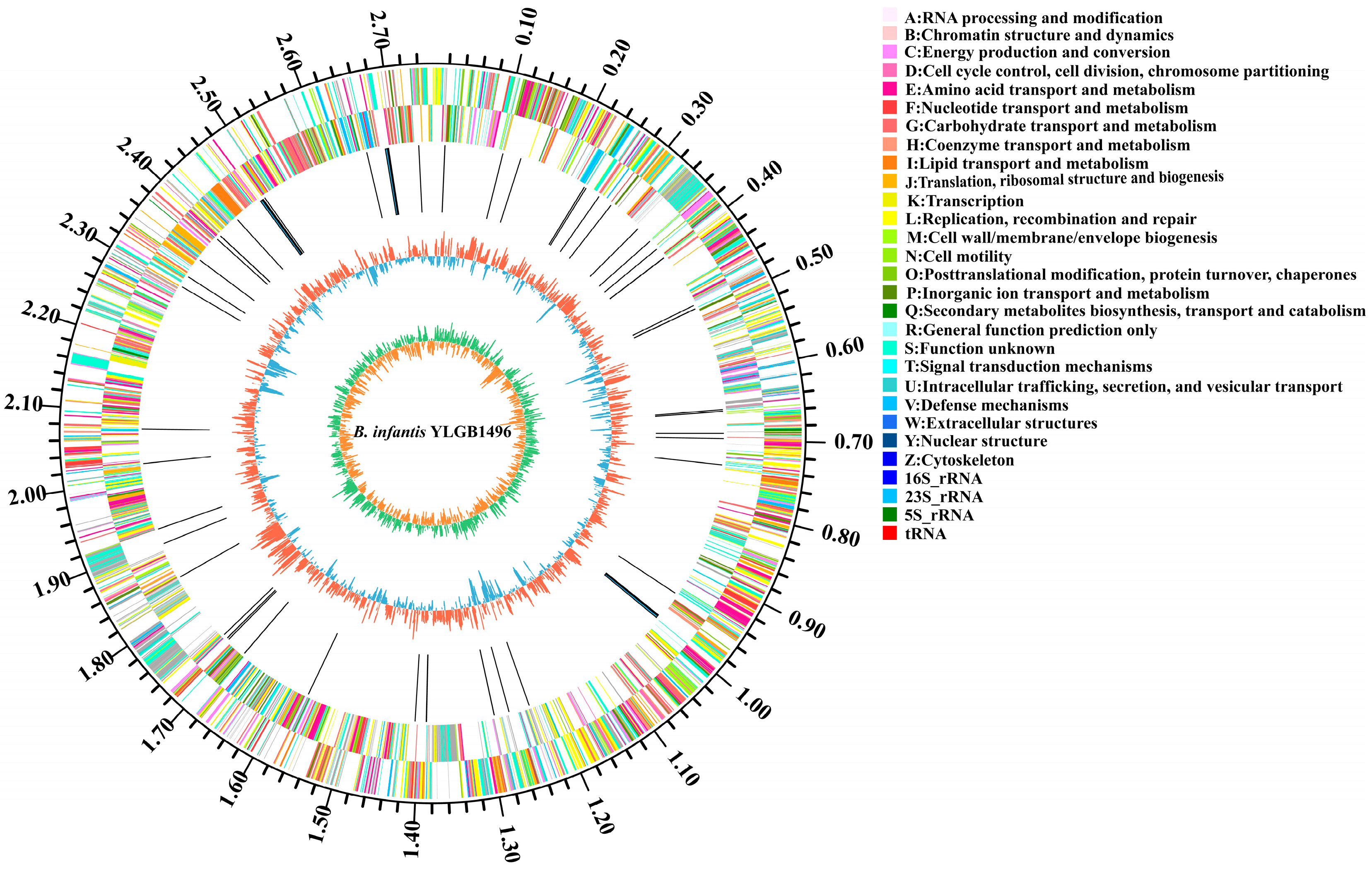
3.1. Genome Sequences of YLGB-1496
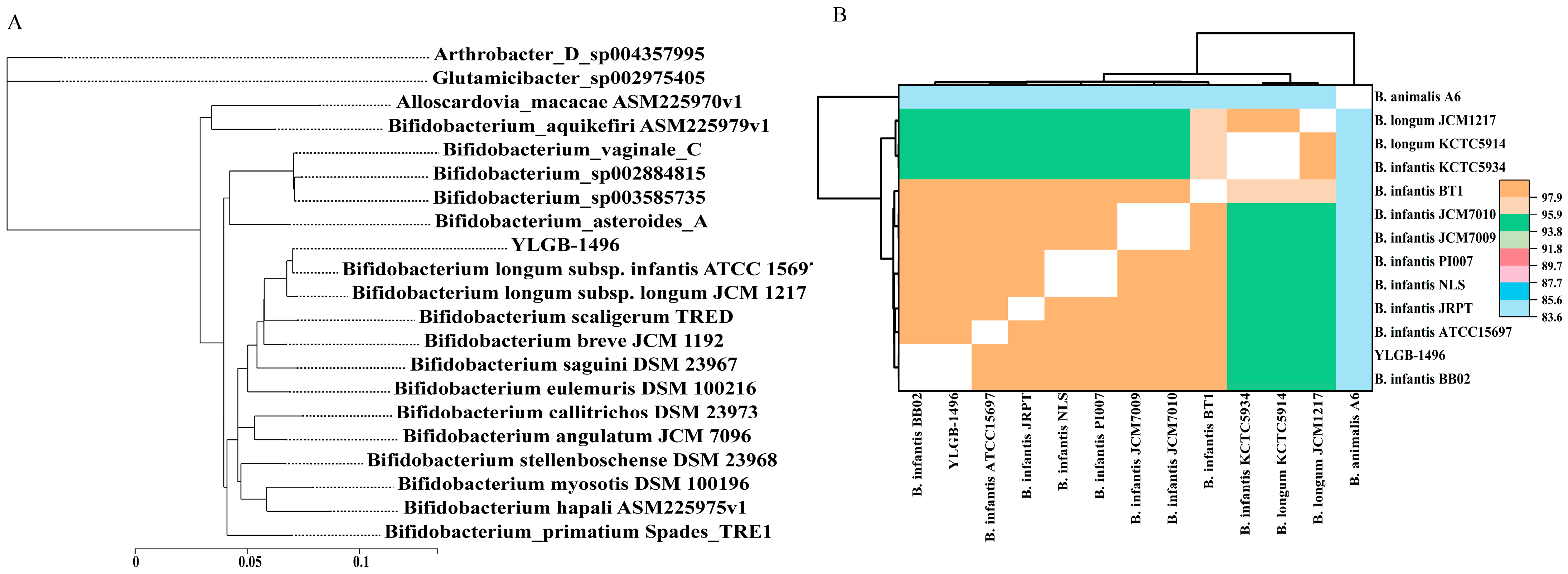
3.2. Phylogenetic Analysis of YLGB-1496
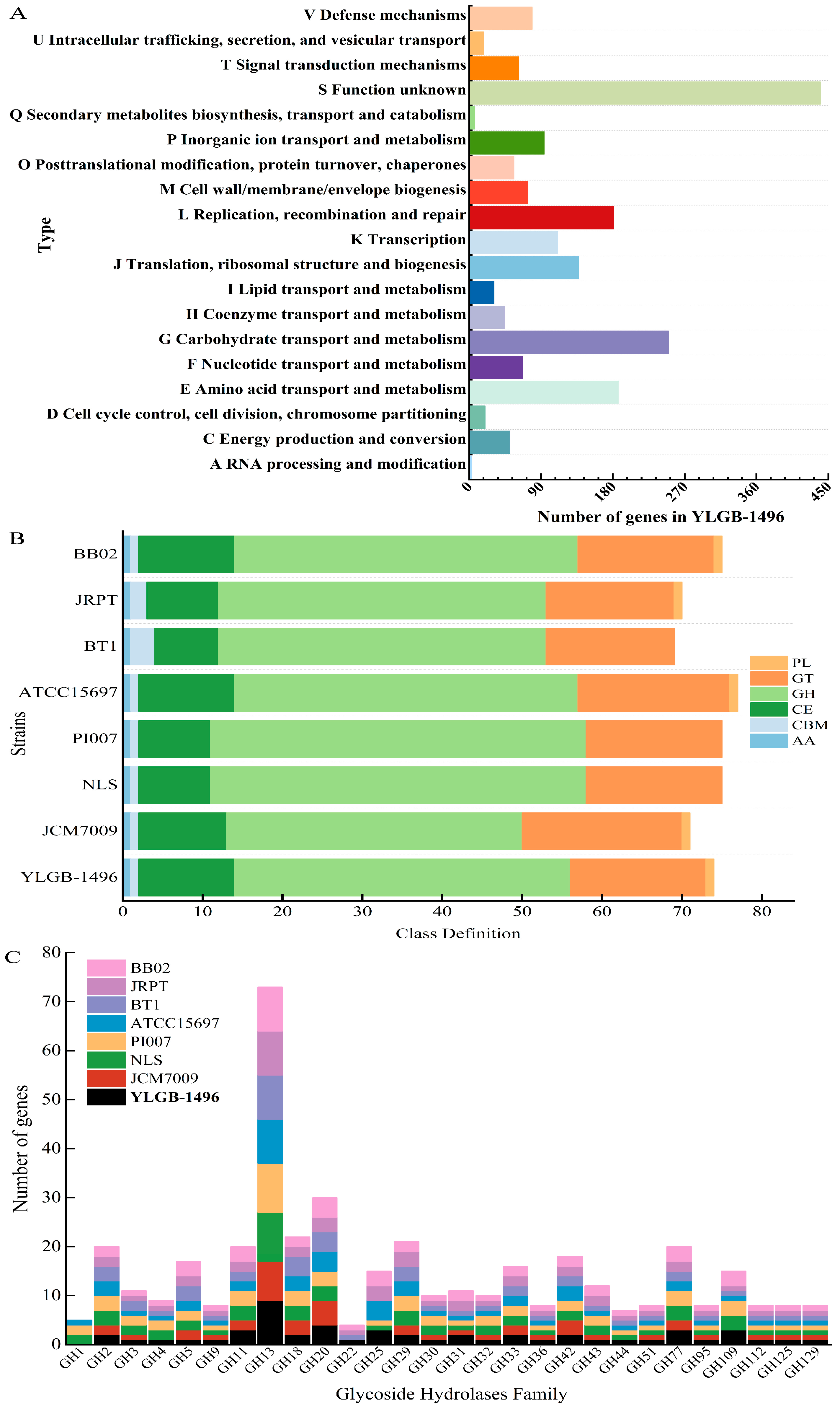
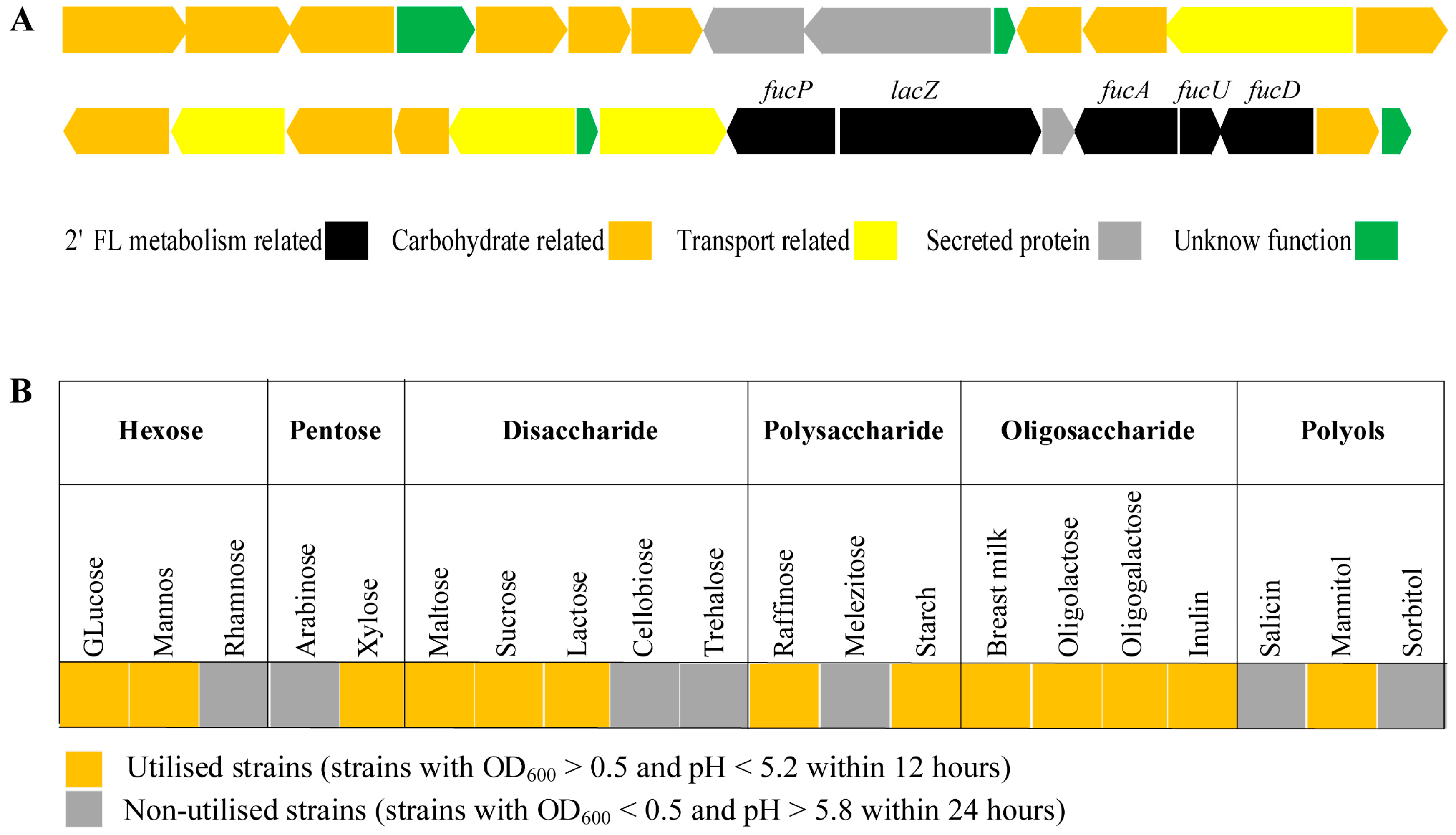
3.3. Genetic and Phenotypic Data for Carbohydrate Metabolism
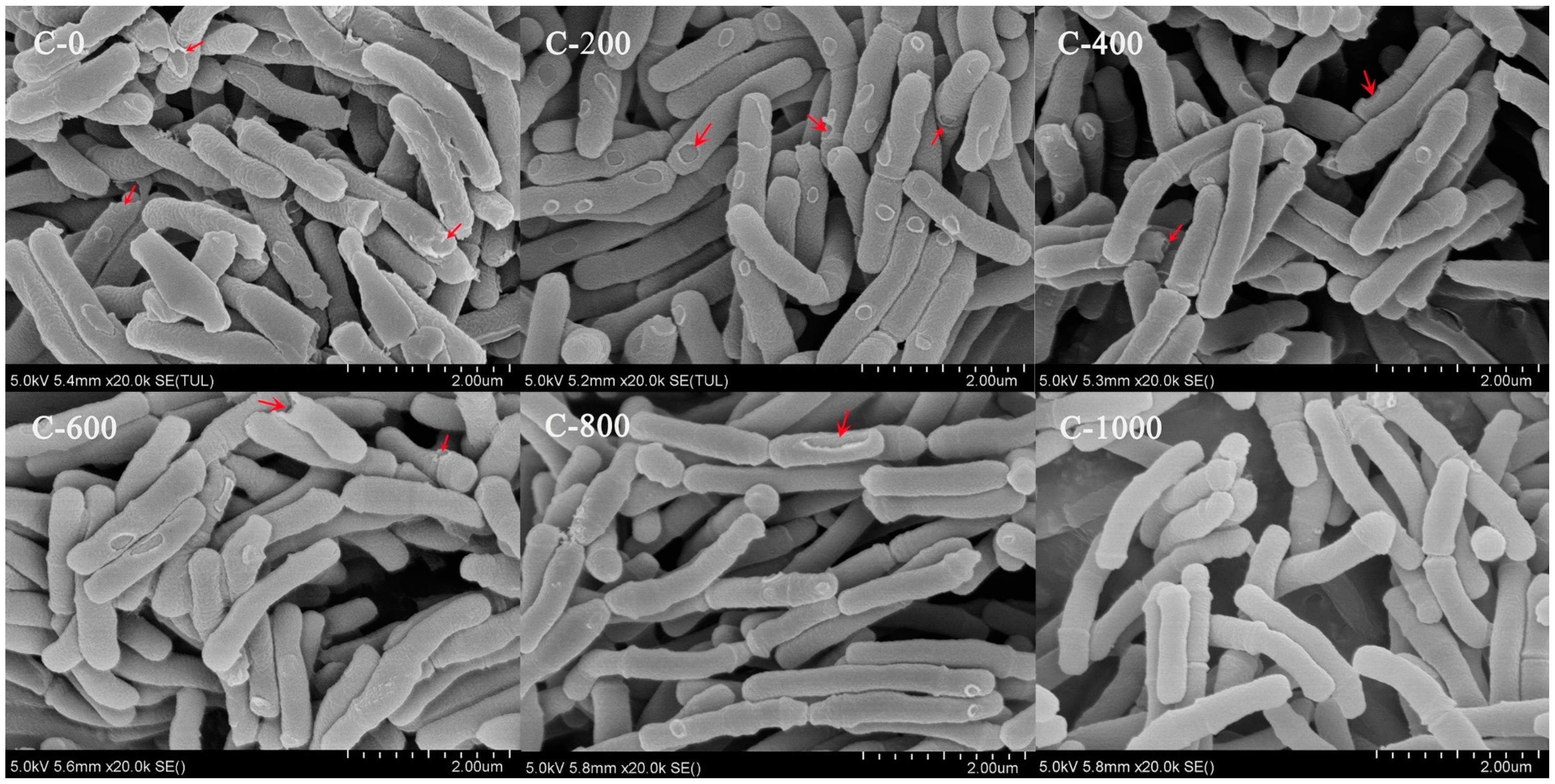
3.4. Morphology during YLGB-1496 Continuous Subculture
3.5. Growth Characteristics during YLGB-1496 Continuous Subculture
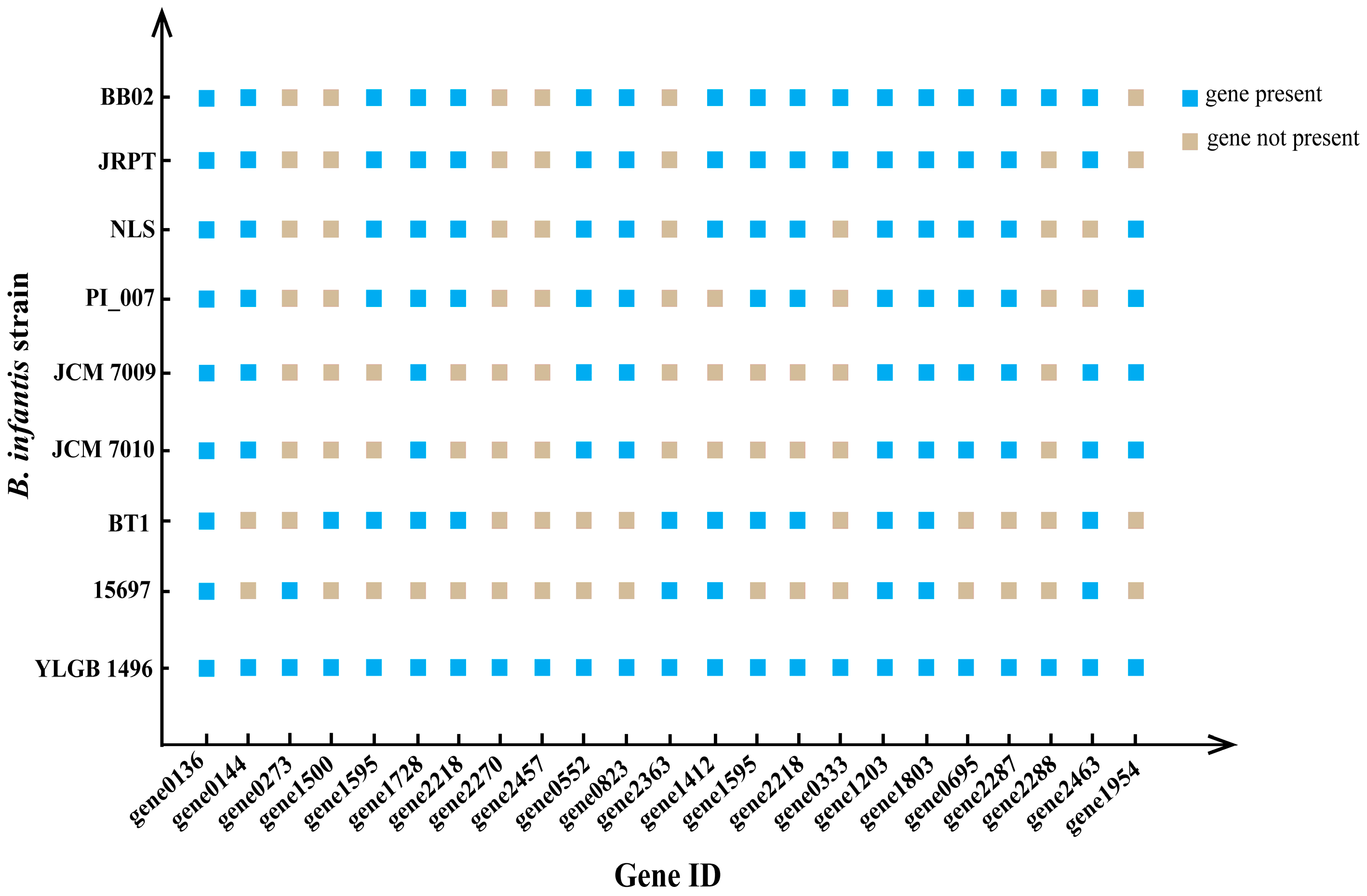
3.6. Stability of Genetic Information
3.7. Genetic Data for Extracellular Proteins Involved in Adhesion
3.8. Drug Resistance Genes in YLGB-1496
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sundararaman, A.; Bansal, K.; Sidhic, J.; Patil, P.; Halami, P.M. Genome of Bifidobacterium longum NCIM 5672 provides insights into its acid-tolerance mechanism and probiotic properties. Arch. Microbiol. 2021, 203, 6109–6118. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Pan, Y.; Zhao, W.; Sun, P.; Zhao, J.; Yan, S.; Wang, R.; Han, Y.; Liu, W.H.; Tan, S. Bifidobacterium infantis strain YLGB-1496 possesses excellent antioxidant and skin barrier-enhancing efficacy in vitro. Exp. Dermatol. 2022, 31, 1089–1094. [Google Scholar] [CrossRef]
- Berhe, T.; Ipsen, R.; Seifu, E.; Kurtu, M.Y.; Eshetu, M.; Hansen, E.B. Comparison of the acidification activities of commercial starter cultures in camel and bovine milk. LWT-Food Sci. Technol. 2018, 89, 123–127. [Google Scholar] [CrossRef]
- Zabel, B.; Yde, C.C.; Roos, P.; Marcussen, J.; Jensen, H.M.; Salli, K.; Hirvonen, J.; Ouwehand, A.C.; Morovic, W. Novel genes and metabolite trends in Bifidobacterium longum subsp. infantis Bi-26 metabolism of human milk oligosaccharide 2′-fucosyllactose. Sci. Rep. 2019, 9, 7983. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Xu, Y.; Han, D.; Teng, Q.; Jiang, D.; Liu, Q. Draft Genome Sequence of Bifidobacterium longum subsp. infantis BI-G201, a Commercialization Strain. Microbiol. Resour. Announc. 2020, 9, e00785-20. [Google Scholar] [CrossRef] [PubMed]
- Wan, M.L.Y.; Forsythe, S.J.; El-Nezami, H. Probiotics interaction with foodborne pathogens: A potential alternative to antibiotics and future challenges. Crit. Rev. Food Sci. Nutr. 2019, 59, 3320–3333. [Google Scholar] [CrossRef] [PubMed]
- Arboleya, S.; Stanton, C.; Ryan, C.A.; Dempsey, E.; Ross, P.R. Bosom buddies: The symbiotic relationship between infants and Bifidobacterium longum ssp. longum and ssp. infantis. Genetic and Probiotic features. Annu. Rev. Food Sci. Technol. 2016, 7, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Albert, K.; Rani, A.; Sela, D.A. Comparative pangenomics of the mammalian gut commensal Bifidobacterium longum. Microorganisms 2020, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Sela, D.A.; Chapman, J.; Adeuya, A.; Kim, J.; Chen, F.; Whitehead, T.; Lapidus, A.; Rokhsar, D.; Lebrilla, C.B.; German, J. The genome sequence of Bifidobacterium longum subsp. infantis reveals adaptations for milk utilization within the infant microbiome. Proc. Natl. Acad. Sci. USA 2008, 105, 18964–18969. [Google Scholar] [CrossRef]
- Wei, Y.-X.; Zhang, Z.-Y.; Liu, C.; Malakar, P.K.; Guo, X.-K. Safety assessment of Bifidobacterium longum JDM301 based on complete genome sequences. World J. Gastroenterol. 2012, 18, 479–488. [Google Scholar] [CrossRef]
- LoCascio, R.G.; Desai, P.; Sela, D.A.; Weimer, B.; Mills, D.A. Broad conservation of milk utilization genes in Bifidobacterium longum subsp. infantis as revealed by comparative genomic hybridization. Appl. Environ. Microbiol. 2010, 76, 7373–7381. [Google Scholar] [CrossRef]
- Fukuda, S.; Toh, H.; Hase, K.; Oshima, K.; Nakanishi, Y.; Yoshimura, K.; Tobe, T.; Clarke, J.M.; Topping, D.L.; Suzuki, T. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature 2011, 469, 543–547. [Google Scholar] [CrossRef]
- Nogacka, A.M.; Arboleya, S.; Nikpoor, N.; Auger, J.; Salazar, N.; Cuesta, I.; Alvarez-Buylla, J.R.; Mantecón, L.; Solís, G.; Gueimonde, M. In Vitro Probiotic Modulation of the Intestinal Microbiota and 2′ Fucosyllactose Consumption in Fecal Cultures from Infants at Two Months of Age. Microorganisms 2022, 10, 318. [Google Scholar] [CrossRef]
- Tobias, J.; Olyaei, A.; Laraway, B.; Jordan, B.K.; Dickinson, S.L.; Golzarri-Arroyo, L.; Fialkowski, E.; Owora, A.; Scottoline, B. Bifidobacterium longum subsp. infantis EVC001 administration is associated with a significant reduction in the Incidence of necrotizing enterocolitis in very low birth weight infants. J. Pediatr. 2022, 244, 64–71.e62. [Google Scholar] [CrossRef] [PubMed]
- Escribano, J.; Ferré, N.; Gispert-Llaurado, M.; Luque, V.; Rubio-Torrents, C.; Zaragoza-Jordana, M.; Polanco, I.; Codoner, F.M.; Chenoll, E.; Morera, M. Bifidobacterium longum subsp infantis CECT7210-supplemented formula reduces diarrhea in healthy infants: A randomized controlled trial. Pediatr. Res. 2018, 83, 1120–1128. [Google Scholar] [CrossRef]
- Lyseng-Williamson, K.A. Bifidobacterium infantis 35624 as a probiotic dietary supplement: A profile of its use. Drugs Ther. Perspect. 2017, 33, 368–374. [Google Scholar] [CrossRef]
- Al-Sahlany, S.T.; Niamah, A.K. Bacterial viability, antioxidant stability, antimutagenicity and sensory properties of onion types fermentation by using probiotic starter during storage. Food Sci. Nutr. 2022, 52, 901–916. [Google Scholar] [CrossRef]
- Grześkowiak, Ł.; Isolauri, E.; Salminen, S.; Gueimonde, M. Manufacturing process influences properties of probiotic bacteria. Br. J. Nutr. 2011, 105, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Salminen, S. Handbook of Probiotics and Prebiotics; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Saarela, M.; Virkajärvi, I.; Alakomi, H.-L.; Sigvart-Mattila, P.; Mättö, J. Stability and functionality of freeze-dried probiotic Bifidobacterium cells during storage in juice and milk. Int. Dairy J. 2006, 16, 1477–1482. [Google Scholar] [CrossRef]
- Tarracchini, C.; Milani, C.; Lugli, G.A.; Mancabelli, L.; Fontana, F.; Alessandri, G.; Longhi, G.; Anzalone, R.; Viappiani, A.; Turroni, F.; et al. Phylogenomic disentangling of the Bifidobacterium longum subsp. infantis taxon. Microb. Genom. 2021, 7, 000609. [Google Scholar] [CrossRef]
- Li, X.; Zhai, Z.; Hao, Y.; Zhang, M.; Hou, C.; He, J.; Shi, S.; Zhao, Z.; Sang, Y.; Ren, F. The plasmid-encoded lactose operon plays a vital role on the acid production rate of Lacticaseibacillus casei during milk beverage fermentation. Front. Microbiol. 2022, 13, 1016904. [Google Scholar] [CrossRef] [PubMed]
- Lawley, B.; Centanni, M.; Watanabe, J.; Sims, I.; Carnachan, S.; Broadbent, R.; Lee, P.S.; Wong, K.H.; Tannock, G.W. Tuf gene sequence variation in Bifidobacterium longum subsp. infantis detected in the fecal microbiota of chinese infants. Appl. Environ. Microbiol. 2018, 84, e00336. [Google Scholar] [CrossRef] [PubMed]
- Srutkova, D.; Spanova, A.; Spano, M.; Drab, V.; Schwarzer, M.; Kozakova, H.; Rittich, B. Efficiency of PCR-based methods in discriminating Bifidobacterium longum ssp. longum and Bifidobacterium longum ssp. infantis strains of human origin. J. Microbiol. Methods 2011, 87, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Ren, L.; Song, Z.; Wang, C.; Sun, B. Purification and characteristics of bifidocin A, a novel bacteriocin produced by Bifidobacterium animals BB04 from centenarians’ intestine. Food Control 2015, 50, 889–895. [Google Scholar] [CrossRef]
- Rosche, W.A.; Foster, P.L. Determining mutation rates in bacterial populations. Methods 2000, 20, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef]
- Borodovsky, M.; Mcininch, J. GENMARK: Parallel gene recognition for both DNA strands. Comput. Chem. 1993, 17, 123–133. [Google Scholar] [CrossRef]
- Fukao, M.; Oshima, K.; Morita, H.; Toh, H.; Suda, W.; Kim, S.W.; Suzuki, S.; Yakabe, T.; Hattori, M.; Yajima, N. Genomic analysis by deep sequencing of the probiotic Lactobacillus brevis KB290 harboring nine plasmids reveals genomic stability. PLoS ONE 2013, 8, e60521. [Google Scholar] [CrossRef]
- Motorin, Y.; Marchand, V. Analysis of RNA modifications by second- and third-generation deep sequencing: 2020 update. Genes 2021, 12, 278. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The carbohydrate-active enzymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A fast phage search tool. Nucleic Acids Res. 2011, 39 (Suppl. S2), W347–W352. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Sima, V.M.; Houtgast, E.; Bertels, K.; Al-Ars, Z. Heterogeneous hardware/software acceleration of the BWA-MEM DNA alignment algorithm. In Proceedings of the 2015 IEEE/ACM International Conference on Computer-Aided Design (ICCAD), Austin, TX, USA, 2–6 November 2015; pp. 240–246. [Google Scholar]
- Takano, S.; Matsuda, S.; Kinoshita, N.; Shimoda, N.; Sato, T.; Kato, K. Genome-wide single nucleotide polymorphisms and insertion–deletions of Oryza sativa L. subsp. japonica cultivars grown near the northern limit of rice cultivation. Mol. Breed. 2014, 34, 1007–1021. [Google Scholar] [CrossRef]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef]
- Ren, Y.; Yu, G.; Shi, C.; Liu, L.; Guo, Q.; Han, C.; Zhang, D.; Zhang, L.; Liu, B.; Gao, H. Majorbio Cloud: A one-stop, comprehensive bioinformatic platform for multiomics analyses. iMeta 2022, 1, e12. [Google Scholar] [CrossRef]
- Zabel, B.E.; Gerdes, S.; Evans, K.C.; Nedveck, D.; Singles, S.K.; Volk, B.; Budinoff, C. Strain-specific strategies of 2′-fucosyllactose, 3-fucosyllactose, and difucosyllactose assimilation by Bifidobacterium longum subsp. infantis Bi-26 and ATCC 15697. Sci. Rep. 2020, 10, 15919. [Google Scholar] [CrossRef] [PubMed]
- Smokvina, T.; Wels, M.; Polka, J.; Chervaux, C.; Brisse, S.; Boekhorst, J.; van Hylckama Vlieg, J.E.; Siezen, R.J. Lactobacillus paracasei comparative genomics: Towards species pan-genome definition and exploitation of diversity. PLoS ONE 2013, 8, e68731. [Google Scholar] [CrossRef]
- Wuyts, S.; Wittouck, S.; De Boeck, I.; Allonsius, C.N.; Pasolli, E.; Segata, N.; Lebeer, S. Large-scale phylogenomics of the Lactobacillus casei group highlights taxonomic inconsistencies and reveals novel clade-associated features. mSystems 2017, 2, e00061-17. [Google Scholar] [CrossRef]
- O’Riordan, N.; O’Callaghan, J.; Butto, L.F.; Kilcoyne, M.; Joshi, L.; Hickey, R.M. Bovine glycomacropeptide promotes the growth of Bifidobacterium longum ssp. infantis and modulates its gene expression. J. Dairy Sci. 2018, 101, 6730–6741. [Google Scholar] [CrossRef] [PubMed]
- Pightling, A.W.; Pettengill, J.B.; Luo, Y.; Baugher, J.D.; Rand, H.; Strain, E. Interpreting whole-genome sequence analyses of foodborne bacteria for regulatory applications and outbreak investigations. Front. Microbiol. 2018, 9, 1482. [Google Scholar] [CrossRef] [PubMed]
- D’Aimmo, M.R.; Modesto, M.; Biavati, B. Antibiotic resistance of lactic acid bacteria and Bifidobacterium spp. isolated from dairy and pharmaceutical products. Int. J. Food Microbiol. 2007, 115, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Serafini, F.; Bottacini, F.; Viappiani, A.; Baruffini, E.; Turroni, F.; Foroni, E.; Lodi, T.; van Sinderen, D.; Ventura, M. Insights into physiological and genetic mupirocin susceptibility in bifidobacteria. Appl. Environ. Microbiol. 2011, 77, 3141–3146. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rossello-Mora, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [PubMed]
- Jeong, D.W.; Jeong, H.M.; Shin, Y.J.; Woo, S.H.; Shim, J.H. Properties of recombinant 4-α-glucanotransferase from Bifidobacterium longum subsp. longum JCM 1217 and its application. Food Sci. Biotechnol. 2020, 29, 667–674. [Google Scholar] [CrossRef]
- Yoshida, E.; Sakurama, H.; Kiyohara, M.; Nakajima, M.; Kitaoka, M.; Ashida, H.; Hirose, J.; Katayama, T.; Yamamoto, K.; Kumagai, H. Bifidobacterium longum subsp. infantis uses two different β-galactosidases for selectively degrading type-1 and type-2 human milk oligosaccharides. Glycobiology 2012, 22, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Møller, P.L.; Jørgensen, F.; Hansen, O.C.; Madsen, S.M.; Stougaard, P. Intra- and extracellular β-galactosidases from Bifidobacterium bifidum and B. infantis. Appl. Environ. Microbiol. 2001, 67, 2276–2283. [Google Scholar] [CrossRef] [PubMed]
- Hallman, M.; Bry, K.; Hoppu, K.; Lappi, M.; Pohjavuori, M. Inositol supplementation in premature infants with respiratory distress syndrome. N. Engl. J. Med. 1992, 326, 1233–1239. [Google Scholar] [CrossRef]
- Urashima, T.; Asakuma, S.; Leo, F.; Fukuda, K.; Messer, M.; Oftedal, O.T. The predominance of type I oligosaccharides is a feature specific to human breast milk. Adv. Nutr. 2012, 3, 473S–482S. [Google Scholar] [CrossRef]
- LoCascio, R.G.; Ninonuevo, M.R.; Freeman, S.L.; Sela, D.A.; Grimm, R.; Lebrilla, C.B.; Mills, D.A.; German, J.B. Glycoprofiling of bifidobacterial consumption of human milk oligosaccharides demonstrates strain specific, preferential consumption of small chain glycans secreted in early human lactation. J. Agric. Food. Chem. 2007, 55, 8914–8919. [Google Scholar] [CrossRef] [PubMed]
- Sela, D.A.; Li, Y.; Lerno, L.; Wu, S.; Marcobal, A.M.; German, J.B.; Chen, X.; Lebrilla, C.B.; Mills, D.A. An infant-associated bacterial commensal utilizes breast milk sialyloligosaccharides. J. Biol. Chem. 2011, 286, 11909–11918. [Google Scholar] [CrossRef]
- Nishiyama, K.; Yamamoto, Y.; Sugiyama, M.; Takaki, T.; Urashima, T.; Fukiya, S.; Yokota, A.; Okada, N.; Mukai, T. Bifidobacterium bifidum extracellular sialidase enhances adhesion to the mucosal surface and supports carbohydrate assimilation. mBio 2017, 8, e00928-17. [Google Scholar] [CrossRef]
- Hou, B.C.; Wang, H.; Yan, T.W.; Shan, Y.; Zhou, W.Q.; Zhang, L.D.; Man, C.X.; Deng, Y.; Jiang, Y.J. Production for high-vitality starter culture of Lactobacillus plantarum NDC 75017 by high cell-density cultivation and low-temperature vacuum drying. Food Sci. Technol. Res. 2016, 22, 519–527. [Google Scholar] [CrossRef][Green Version]
- Deshwal, G.K.; Tiwari, S.; Kumar, A.; Raman, R.K.; Kadyan, S. Review on factors affecting and control of post-acidification in yoghurt and related products. Trends Food Sci. Technol. 2021, 109, 499–512. [Google Scholar] [CrossRef]
- Mirlohi, M.; Soleimanian-Zad, S.; Dokhani, S.; Sheikh-Zeinodin, M. Microbial and physiochemical changes in yoghurts containing different Lactobacillus delbrueckii subsp. bulgaricus strains in association with Lactobacillus plantarum as an adjunct culture. Int. J. Dairy Technol. 2014, 67, 246–254. [Google Scholar] [CrossRef]
- Milani, C.; Duranti, S.; Lugli, G.A.; Bottacini, F.; Strati, F.; Arioli, S.; Foroni, E.; Turroni, F.; van Sinderen, D.; Ventura, M. Comparative genomics of Bifidobacterium animalis subsp. lactis reveals a strict monophyletic bifidobacterial taxon. Appl. Environ. Microbiol. 2013, 79, 4304–4315. [Google Scholar] [CrossRef] [PubMed]
- Martina, M.; Tikunov, Y.; Portis, E.; Bovy, A.G. The genetic basis of tomato aroma. Genes 2021, 12, 226. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Oh, H.S.; Park, S.C.; Chun, J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 2014, 64 Pt 2, 346–351. [Google Scholar] [CrossRef]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef]
- Ben Amor, K.; Vaughan, E.E.; de Vos, W.M. Advanced molecular tools for the identification of lactic acid bacteria. J. Nutr. 2007, 137, 741S–747S. [Google Scholar] [CrossRef] [PubMed]
- Schürch, A.C.; Arredondo-Alonso, S.; Willems, R.J.L.; Goering, R.V. Whole genome sequencing options for bacterial strain typing and epidemiologic analysis based on single nucleotide polymorphism versus gene-by-gene–based approaches. Clin. Microbiol. Infect. 2018, 24, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Chen, J.; Brandt, B.W.; Zhu, Y.; Li, J.; van Loveren, C.; Deng, D.M. Identification and functional analysis of genome mutations in a fluoride-resistant Streptococcus mutans strain. PLoS ONE 2015, 10, e0122630. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Ku, S.; Kim, S.Y.; Lee, H.H.; Jin, H.; Kang, S.; Li, R.; Johnston, T.V.; Park, M.S.; Ji, G.E. Safety evaluations of Bifidobacterium bifidum BGN4 and Bifidobacterium longum BORI. Int. J. Mol. Sci. 2018, 19, 1422. [Google Scholar] [CrossRef] [PubMed]
- Peabody, C.R.; Chung, Y.J.; Yen, M.R.; Vidal-Ingigliardi, D.; Pugsley, A.P.; Saier, M.H. Type II protein secretion and its relationship to bacterial type IV pili and archaeal flagella. Microbiology 2003, 149 Pt 11, 3051–3072. [Google Scholar] [CrossRef]
- Ligthart, K.; Belzer, C.; de Vos, W.M.; Tytgat, H.L.P. Bridging bacteria and the gut: Functional aspects of type IV pili. Trends Microbiol. 2020, 28, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Kleerebezem, M.; Boekhorst, J.; van Kranenburg, R.; Molenaar, D.; Kuipers, O.P.; Leer, R.; Tarchini, R.; Peters, S.A.; Sandbrink, H.M.; Fiers, M.W. Complete genome sequence of Lactobacillus plantarum WCFS1. Proc. Natl. Acad. Sci. USA 2003, 100, 1990–1995. [Google Scholar] [CrossRef] [PubMed]
- Call, E.K.; Klaenhammer, T.R. Relevance and application of sortase and sortase-dependent proteins in lactic acid bacteria. Front. Microbiol. 2013, 4, 73. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, Z.Y.; Dong, K.; Guo, X.K. Adhesion and immunomodulatory effects of Bifidobacterium lactis HN019 on intestinal epithelial cells INT-407. World J. Gastroenterol. 2010, 16, 2283–2290. [Google Scholar] [CrossRef]
- Tsirigotaki, A.; De Geyter, J.; Sostaric, N.; Economou, A.; Karamanou, S. Protein export through the bacterial Sec pathway. Nat. Rev. Microbiol. 2017, 15, 21–36. [Google Scholar] [CrossRef]
- Kiwaki, M.; Sato, T. Antimicrobial susceptibility of Bifidobacterium breve strains and genetic analysis of streptomycin resistance of probiotic B. breve strain Yakult. Int. J. Food Microbiol. 2009, 134, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Gueimonde, M.; Flórez, A.B.; van Hoek, A.H.A.M.; Stuer-Lauridsen, B.; Strøman, P.; de los Reyes-Gavilán, C.G.; Margolles, A. Genetic basis of tetracycline resistance in Bifidobacterium animalis subsp. lactis. Appl. Environ. Microbiol. 2010, 76, 3364–3369. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| YLGB1496 | ATCC15697 | BT1 | JRPT | BB02 | PI007 | NLS | JCM7009 | |
|---|---|---|---|---|---|---|---|---|
| General genome feature | ||||||||
| Size | 2,758,242 | 2,832,748 | 2,578,115 | 2,776,348 | 2,757,833 | 2,604,585 | 2,598,286 | 2,673,222 |
| GC content (%) | 59.87 | 59.86 | 59.39 | 59.77 | 59.87 | 59.32 | 59.32 | 59.77 |
| CDS No. | 2442 | 2553 | 2198 | 2461 | 2441 | 2234 | 2232 | 2328 |
| Number of RNAs | 64 | 85 | 61 | 64 | 63 | 64 | 62 | 63 |
| COG gene No. | 1896 | 1836 | 1800 | 1847 | 1829 | 1806 | 1805 | 1846 |
| Percent of All Genes (%) | 77.64 | 71.92 | 81.89 | 75.05 | 74.93 | 80.84 | 80.87 | 79.3 |
| Amino acid metabolism | 117 | 115 | 117 | 115 | 114 | 113 | 113 | 112 |
| Biosynthesis of other secondary metabolites | 30 | 27 | 30 | 28 | 27 | 33 | 33 | 28 |
| Carbohydrate metabolism | 128 | 121 | 119 | 120 | 131 | 137 | 137 | 123 |
| Energy metabolism | 48 | 47 | 48 | 47 | 49 | 50 | 50 | 47 |
| Glycan biosynthesis and metabolism | 29 | 41 | 47 | 40 | 38 | 48 | 48 | 42 |
| Lipid metabolism | 24 | 25 | 29 | 27 | 26 | 28 | 28 | 29 |
| Metabolism of cofactors and vitamins | 76 | 73 | 73 | 73 | 73 | 75 | 75 | 75 |
| Metabolism of other amino acids | 27 | 27 | 32 | 27 | 26 | 27 | 27 | 28 |
| Metabolism of terpenoids and polyketides | 15 | 13 | 17 | 14 | 15 | 19 | 19 | 14 |
| Nucleotide metabolism | 59 | 60 | 60 | 62 | 58 | 59 | 59 | 63 |
| Xenobiotics biodegradation and metabolism | 11 | 14 | 12 | 12 | 13 | 13 | 13 | 12 |
| Generation Time (n) | OD600 | pH Values | Viable Bacteria Number (lg(CFU/mL)) | Fermentation Activity (U) |
|---|---|---|---|---|
| 0 | 2.18 ± 0.02 | 3.96 ± 0.01 | 9.45 ± 0.04 | 59.30 ± 0.27 |
| 200 | 2.16 ± 0.02 | 3.98 ± 0.02 | 9.38 ± 0.04 | 60.40 ± 0.38 |
| 400 | 2.20 ± 0.01 | 3.95 ± 0.01 | 9.41 ± 0.04 | 59.97 ± 0.49 |
| 600 | 2.20 ± 0.01 | 3.97 ± 0.00 | 9.47 ± 0.04 | 60.43 ± 0.35 |
| 800 | 2.22 ± 0.02 | 3.98 ± 0.01 | 9.37 ± 0.04 | 60.70 ± 0.34 |
| 1000 | 2.20 ± 0.01 | 3.96 ± 0.02 | 9.42 ± 0.04 | 60.20 ± 0.22 |
| Gene Mutation Type | Generation Time (n) | |||||
|---|---|---|---|---|---|---|
| 0 | 200 | 400 | 600 | 800 | 1000 | |
| Single-nucleotide polymorphism (SNP) | 0 | 14 | 14 | 18 | 18 | 20 |
| Multiple-nucleotide polymorphism (MNP) | 0 | 0 | 0 | 0 | 0 | 0 |
| Insertion mutation (INS) | 0 | 2 | 1 | 1 | 1 | 2 |
| Deletion mutation (DEL) | 0 | 0 | 0 | 0 | 0 | 1 |
| Inversion mutation (INV) | 0 | 0 | 0 | 0 | 0 | 0 |
| Duplicate mutation (DUP) | 0 | 0 | 0 | 0 | 0 | 0 |
| Gene ID | Gene Name | Description | Transmembrane Structural Domain | COG Type | ||
|---|---|---|---|---|---|---|
| First 60 Exp AAs No. | Total Prob of N-in | Topology | ||||
| gene0102 | urtE | High-affinity branched-chain amino acid transport ATP-binding protein LivF | - | - | - | E |
| gene0139 | ilvC | Ketol-acid reductoisomerase | - | - | - | E |
| gene0143 | ilvC | ketol-acid reductoisomerase | - | - | - | E |
| gene0230 | ftsY | fused signal recognition particle receptor | 22.67 | 0.83 | inside: 1–4; TMhelix: 5–27; outside: 28–420; | U |
| gene0352 | - | FKBP-type peptidyl-prolyl cis-trans isomerase | - | - | - | - |
| gene0430 | ffh | signal recognition particle subunit SRP54 [EC: 3.6.5.4] | - | - | - | U |
| gene0574 | - | ABC transporter substrate-binding component | 82.73 | 20.23 | inside: 1–30; TMhelix: 31–48; outside: 49–62; TMhelix: 63–82; inside: 83–144; TMhelix: 145–164; outside:165–167; TMhelix: 168–190; inside: 191–286; | G |
| gene0631 | - | ABC transporter substrate-binding component | - | - | - | G |
| gene0818 | bioM | ABC transporter substrate-binding component | - | - | - | - |
| gene0938 | - | ABC transporter substrate-binding component | - | - | - | G |
| gene1038 | secA | preprotein translocase subunit SecA [EC: 7.4.2.8] | - | - | - | E; G |
| gene1192 | secG | preprotein translocase subunit SecG | 24.69 | 0.73 | inside: 1–6; TMhelix: 7–26; outside: 27–54; TMhelix: 55–73; inside: 74–82; | E; G |
| gene1274 | yajC | preprotein translocase subunit YajC | 16.79 | 0.09 | outside: 1–4; TMhelix: 5–22; inside: 23–148; | U |
| gene1933 | - | ABC transporter substrate-binding component | - | - | - | P |
| gene2189 | secY | preprotein translocase subunit SecY | 20.87 | 1 | inside: 1–16; TMhelix: 17–38; outside: 39–74; TMhelix: 75–97; inside: 98–116; TMhelix: 117–139; outside: 140–153; TMhelix: 154–176; inside: 177–187; TMhelix: 188–210; outside: 211–219; TMhelix: 220–242; inside: 243–273; TMhelix: 274–296; outside: 297–315; TMhelix: 316–338; inside: 339–373; TMhelix: 374–396; outside: 397–399; TMhelix: 400–417; inside: 418–445; | E; G |
| gene2304 | secE | preprotein translocase subunit SecE | 19.83 | 1 | inside: 1–40; TMhelix: 41–63; outside: 64–75; | E; G |
| gene2504 | yidC | YidC/Oxa1 family membrane protein insertase | 22.58 | 0.41 | outside: 1–46; TMhelix: 47–69; inside: 70–189; TMhelix: 190–209; outside: 210–232; TMhelix: 233–255; inside: 256–335; | U |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Yang, J.; Shi, S.; Lan, H.; Zhao, W.; Hung, W.; He, J.; Wang, R. The Genome of Bifidobacterium longum subsp. infantis YLGB-1496 Provides Insights into Its Carbohydrate Utilization and Genetic Stability. Genes 2024, 15, 466. https://doi.org/10.3390/genes15040466
Li X, Yang J, Shi S, Lan H, Zhao W, Hung W, He J, Wang R. The Genome of Bifidobacterium longum subsp. infantis YLGB-1496 Provides Insights into Its Carbohydrate Utilization and Genetic Stability. Genes. 2024; 15(4):466. https://doi.org/10.3390/genes15040466
Chicago/Turabian StyleLi, Xiaoxia, Jianjun Yang, Shaoqi Shi, Hanglian Lan, Wen Zhao, Weilian Hung, Jian He, and Ran Wang. 2024. "The Genome of Bifidobacterium longum subsp. infantis YLGB-1496 Provides Insights into Its Carbohydrate Utilization and Genetic Stability" Genes 15, no. 4: 466. https://doi.org/10.3390/genes15040466
APA StyleLi, X., Yang, J., Shi, S., Lan, H., Zhao, W., Hung, W., He, J., & Wang, R. (2024). The Genome of Bifidobacterium longum subsp. infantis YLGB-1496 Provides Insights into Its Carbohydrate Utilization and Genetic Stability. Genes, 15(4), 466. https://doi.org/10.3390/genes15040466