
Detailed Clinical Features of PTPRQ-Associated Hearing Loss Identified in a Large Japanese Hearing Loss Cohort


 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Clinical Evaluations
2.3. Genetic Analysis
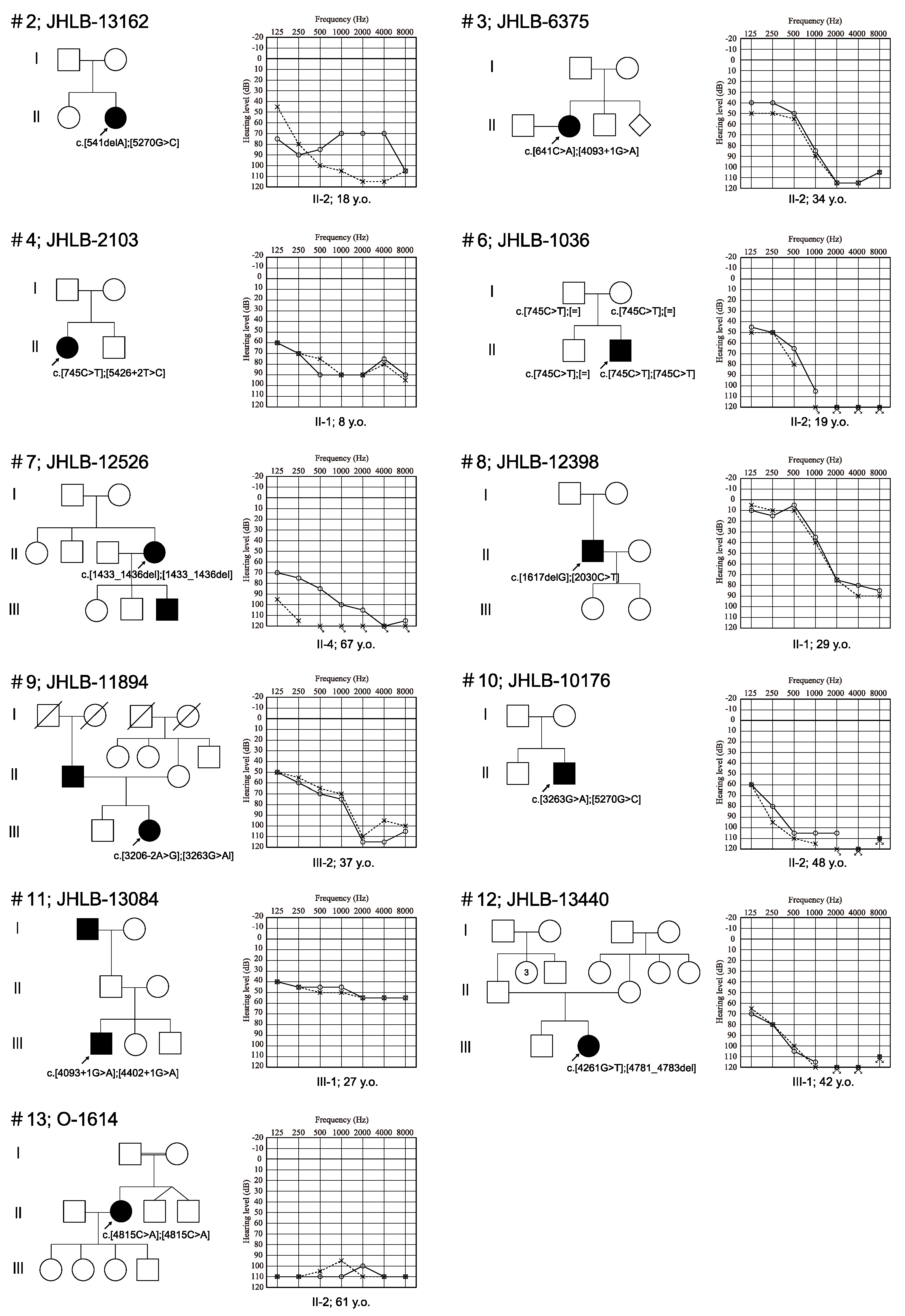
3. Results
3.1. Identified Variations
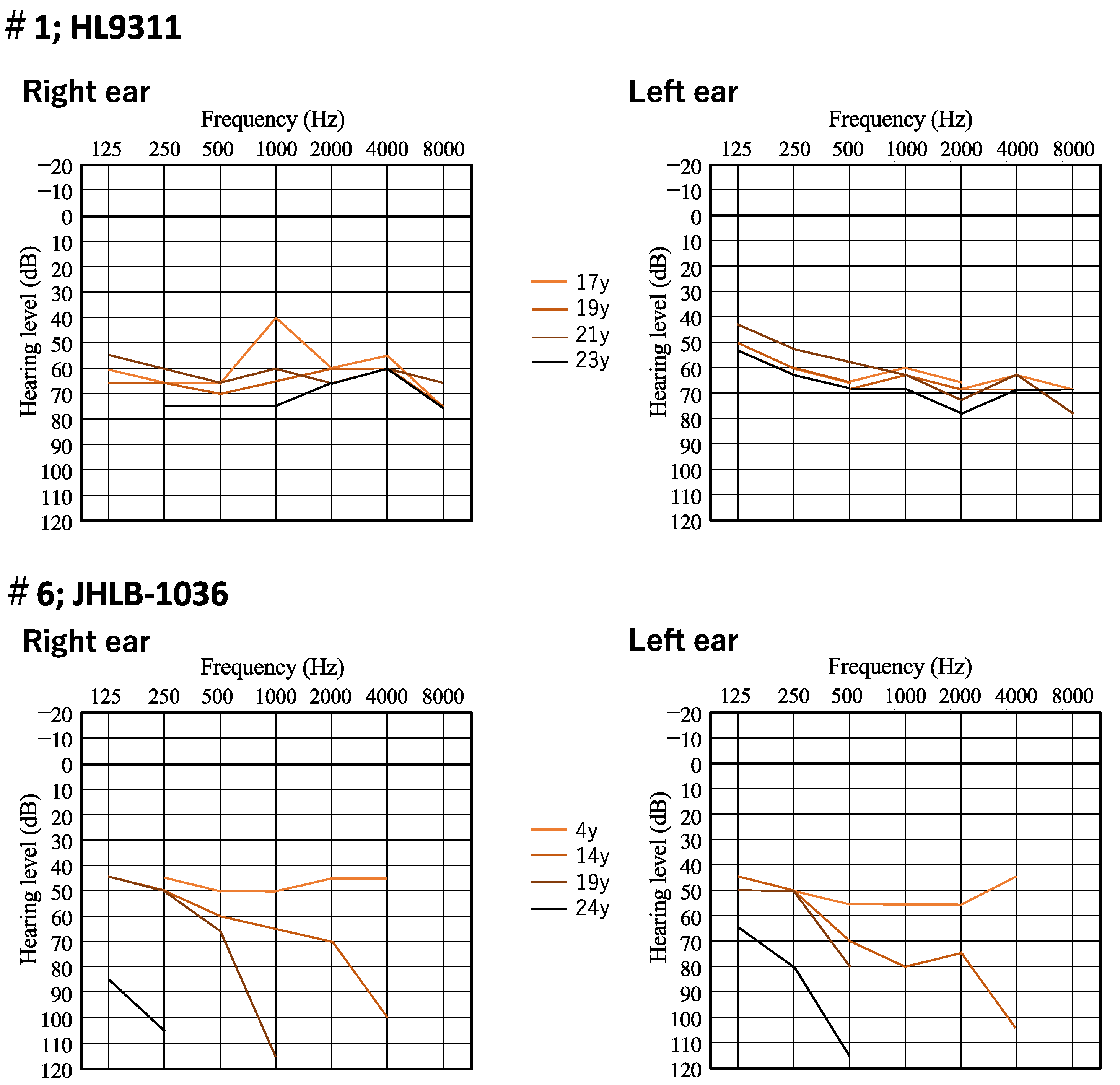
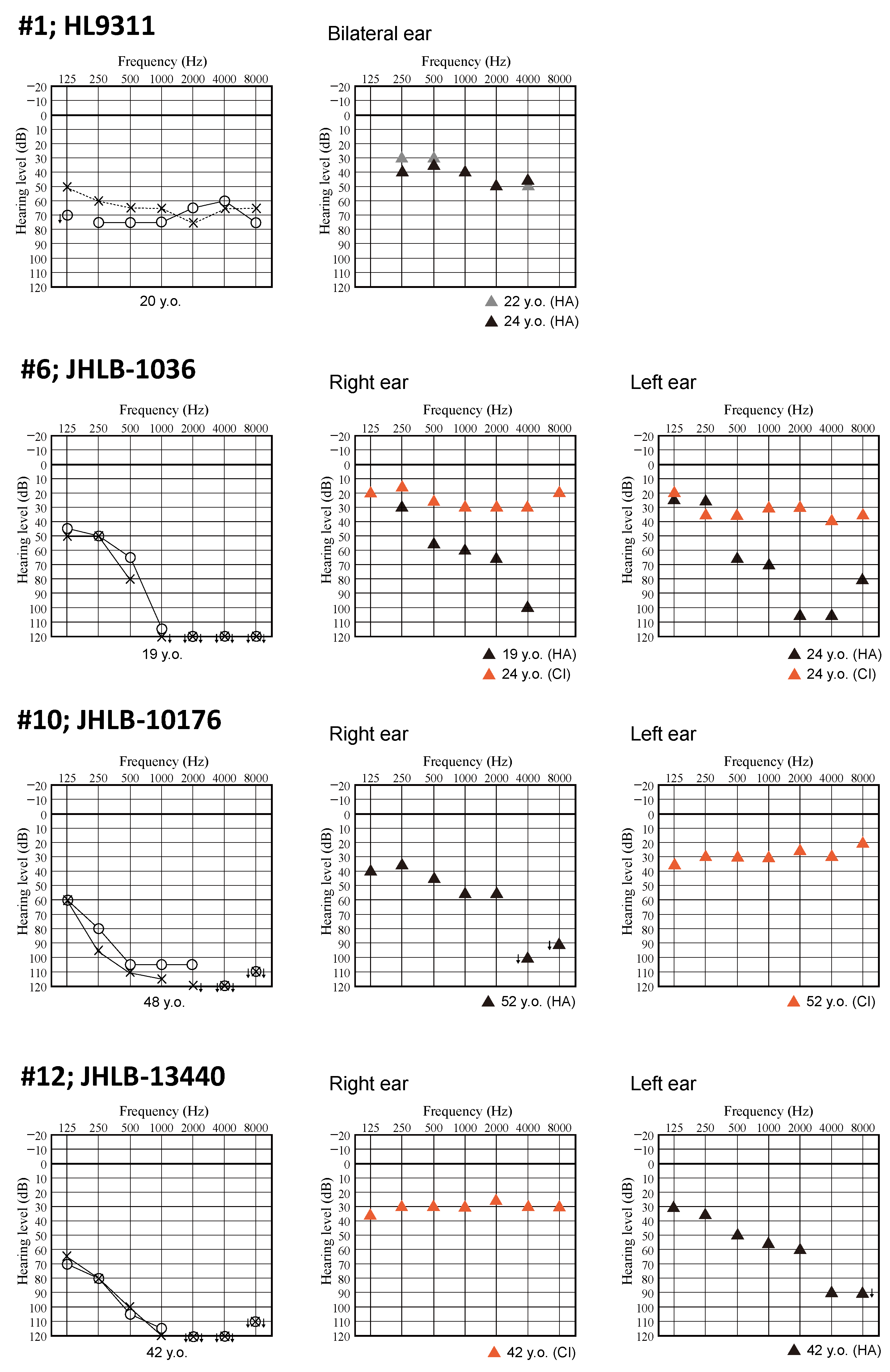
3.2. Clinical Features of Patients and Outcomes of Hearing Devices
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morton, C.C.; Nance, W.E. Newborn Hearing Screening—A Silent Revolution. N. Engl. J. Med. 2006, 354, 2151–2164. [Google Scholar] [CrossRef] [PubMed]
- Hereditary Hearing Loss Homepage. Available online: https://hereditaryhearingloss.org/ (accessed on 30 June 2023).
- The PTPRQ Gene Homepage—Global Variome Shared LOVD. Available online: https://databases.lovd.nl/shared/genes/PTPRQ (accessed on 30 June 2023).
- Salles, F.T.; Andrade, L.R.; Tanda, S.; Grati, M.; Plona, K.L.; Gagnon, L.H.; Johnson, K.R.; Kachar, B.; Berryman, M.A. CLIC5 stabilizes membrane-actin filament linkages at the base of hair cell stereocilia in a molecular complex with radixin, taperin, and myosin VI. Cytoskeleton 2014, 71, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Goodyear, R.J.; Legan, P.K.; Wright, M.B.; Marcotti, W.; Oganesian, A.; Coats, S.A.; Booth, C.J.; Kros, C.J.; Seifert, R.A.; Bowen-Pope, D.F.; et al. A receptor-like inositol lipid phosphatase is required for the maturation of developing cochlear hair bundles. J. Neurosci. 2003, 23, 9208–9219. [Google Scholar] [CrossRef] [PubMed]
- Hirono, M.; Denis, C.S.; Richardson, G.P.; Gillespie, P.G. Hair cells require phosphatidylinositol 4,5-bisphosphate for mechanical transduction and adaptation. Neuron 2004, 44, 309–320. [Google Scholar] [CrossRef]
- Qin, Y.; Ma, Y.; Zeng, Z.; Zhong, Z.; Qi, Y.; Liu, Y. Delayed progressive sensorineural hearing loss due to a novel compound heterozygous PTPRQ mutation in a Chinese patient. J. Clin. Lab. Anal. 2023, 37, e24886. [Google Scholar] [CrossRef] [PubMed]
- Nishio, S.Y.; Usami, S.I. Frequency of the STRC-CATSPER2 deletion in STRC-associated hearing loss patients. Sci. Rep. 2022, 12, 634. [Google Scholar] [CrossRef] [PubMed]
- Mazzoli, M.; Camp, G.V.; Newton, V.; Giarbini, N.; Declau, F.; Parving, A. Recommendations for the Description of Genetic and Audiological Data for Families with Nonsyndromic Hereditary Hearing Impairment. Audiol. Med. 2003, 1, 148–150. [Google Scholar]
- Maekawa, K.; Nishio, S.; Abe, S.; Goto, S.I.; Honkura, Y.; Iwasaki, S.; Kanda, Y.; Kobayashi, Y.; Oka, S.; Okami, M.; et al. Mutational Spectrum and Clinical Features of Patients with LOXHD1 Variants Identified in an 8074 Hearing Loss Patient Cohort. Genes 2019, 10, 735. [Google Scholar] [CrossRef] [PubMed]
- 1000 Genome Project. Available online: https://www.internationalgenome.org/1000-genomes-summary/ (accessed on 7 October 2021).
- Genome Aggregation Database. Available online: https://gnomad.broadinstitute.org (accessed on 6 December 2019).
- The Human Genetic Variation Database. Available online: https://www.hgvd.genome.med.kyoto-u.ac.jp (accessed on 15 November 2015).
- ToMMo 38KJPN-Integrative Japanese Genome Variation Database. Available online: https://jmorp.megabank.tohoku.ac.jp/ (accessed on 29 September 2022).
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Rodelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [PubMed]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.; Edwards, K.J.; Day, I.N.M.; Gaunt, T.R. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Eisenberger, T.; Di Donato, N.; Decker, C.; Delle Vedove, A.; Neuhaus, C.; Nürnberg, G.; Toliat, M.; Nürnberg, P.; Mürbe, D.; Bolz, H.J. A C-terminal nonsense mutation links PTPRQ with autosomal-dominant hearing loss, DFNA73. Genet. Med. 2018, 20, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, N.; Moteki, H.; Azaiez, H.; Booth, K.T.; Takahashi, M.; Arai, Y.; Shearer, A.E.; Sloan, C.M.; Nishio, S.Y.; Kolbe, D.L.; et al. Novel PTPRQ mutations identified in three congenital hearing loss patients with various types of hearing loss. Ann. Otol. Rhinol. Laryngol. 2015, 124 (Suppl. S1), 184S–192S. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Wei, X.; Chai, Y.; Li, L.; Wu, H. Genetic etiology study of the non-syndromic deafness in Chinese Hans by targeted next-generation sequencing. Orphanet J. Rare Dis. 2013, 8, 85. [Google Scholar] [CrossRef]
- Sloan-Heggen, C.M.; Bierer, A.O.; Shearer, A.E.; Kolbe, D.L.; Nishimura, C.J.; Frees, K.L.; Ephraim, S.S.; Shibata, S.B.; Booth, K.T.; Campbell, C.A.; et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 2016, 135, 441–450. [Google Scholar] [CrossRef]
- Abu Rayyan, A.; Kamal, L.; Casadei, S.; Brownstein, Z.; Zahdeh, F.; Shahin, H.; Canavati, C.; Dweik, D.; Jaraysa, T.; Rabie, G.; et al. Genomic analysis of inherited hearing loss in the Palestinian population. Proc. Natl. Acad. Sci. USA 2020, 117, 20070–20076. [Google Scholar] [CrossRef] [PubMed]
- Vanniya, S.P.; Chandru, J.; Jeffrey, J.M.; Rabinowitz, T.; Brownstein, Z.; Krishnamoorthy, M.; Avraham, K.B.; Shomron, N.; Srikumari Srisailapathy, C.R. PNPT1, MYO15A, PTPRQ, and SLC12A2-associated genetic and phenotypic heterogeneity among hearing impaired assortative mating families in southern India. Ann. Hum. Genet. 2022, 86, 1–13. [Google Scholar] [CrossRef]
- Shahin, H.; Rahil, M.; Abu Rayan, A.; Avraham, K.B.; King, M.C.; Kanaan, M.; Walsh, T. Nonsense mutation of the stereociliar membrane protein gene PTPRQ in human hearing loss DFNB84. J. Med. Genet. 2010, 47, 643–645. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ammar-Khodja, F.; Bonnet, C.; Dahmani, M.; Ouhab, S.; Lefèvre, G.M.; Ibrahim, H.; Hardelin, J.P.; Weil, D.; Louha, M.; Petit, C. Diversity of the causal genes in hearing impaired Algerian individuals identified by whole exome sequencing. Mol. Genet. Genom. Med. 2015, 3, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Richard, E.M.; Santos-Cortez, R.L.P.; Faridi, R.; Rehman, A.U.; Lee, K.; Shahzad, M.; Acharya, A.; Khan, A.A.; Imtiaz, A.; Chakchouk, I.; et al. Global genetic insight contributed by consanguineous Pakistani families segregating hearing loss. Hum. Mutat. 2019, 40, 53–72. [Google Scholar] [CrossRef] [PubMed]
- Boucher, S.; Tai, F.W.J.; Delmaghani, S.; Lelli, A.; Singh-Estivalet, A.; Dupont, T.; Niasme-Grare, M.; Michel, V.; Wolff, N.; Bahloul, A.; et al. Ultrarare heterozygous pathogenic variants of genes causing dominant forms of early-onset deafness underlie severe presbycusis. Proc. Natl. Acad. Sci. USA 2020, 117, 31278–31289. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Yin, Y.; Tan, Z.; Liu, J.; Deng, X.; Yang, Y. Whole-exome sequencing identified a novel heterozygous mutation of SALL1 and a new homozygous mutation of PTPRQ in a Chinese family with Townes-brocks syndrome and hearing loss. BMC Med. Genom. 2021, 14, 24. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Wang, L.; Chai, Y.; Wu, H.; Yang, T. Detection and functional verification of noncanonical splice site mutations in hereditary deafness. Front. Genet. 2021, 12, 773922. [Google Scholar] [CrossRef] [PubMed]
- Sang, S.; Ling, J.; Liu, X.; Mei, L.; Cai, X.; Li, T.; Li, W.; Li, M.; Wen, J.; Liu, X.; et al. Proband whole-exome sequencing identified genes responsible for autosomal recessive nonsyndromic hearing loss in 33 Chinese nuclear families. Front. Genet. 2019, 10, 639. [Google Scholar] [CrossRef]
- Schraders, M.; Oostrik, J.; Huygen, P.L.; Strom, T.M.; Wijk, E.; Kunst, H.P.; Hoefsloot, L.H.; Cremers, C.W.; Admiraal, R.J.; Hannie Kremer, H. Mutations in PTPRQ are a cause of autosomal-recessive non-syndromic hearing impairment DFNB84 and associated with vestibular dysfunction. Am. J. Hum. Genet. 2010, 86, 604–610. [Google Scholar] [CrossRef]
- Gao, X.; Su, Y.; Chen, Y.L.; Han, M.Y.; Yuan, Y.Y.; Xu, J.C.; Xin, F.; Zhang, M.G.; Huang, S.S.; Wang, G.J.; et al. Identification of two novel compound heterozygous PTPRQ mutations associated with autosomal recessive hearing loss in a Chinese family. PLoS ONE 2015, 10, e0124757. [Google Scholar] [CrossRef] [PubMed]
- Sang, Q.; Mei, H.; Kuermanhan, A.; Feng, R.; Guo, L.; Qu, R.; Xu, Y.; Li, H.; Jin, L.; He, L.; et al. Identification of a novel compound heterozygous mutation in PTPRQ in a DFNB84 family with prelingual sensorineural hearing impairment. Mol. Genet. Genom. 2015, 290, 1135–1139. [Google Scholar] [CrossRef] [PubMed]
- Talebi, F.; Ghanbari Mardasi, F.; Mohammadi Asl, J.; Tizno, S.; Najafvand, Z.M. Identification of novel PTPRQ and MYO1A mutations in an Iranian pedigree with autosomal recessive hearing loss. Cell J. 2018, 20, 127–131. [Google Scholar] [PubMed]
- Wu, X.; Wang, S.; Chen, S.; Wen, Y.Y.; Liu, B.; Xie, W.; Li, D.; Liu, L.; Huang, X.; Sun, Y.; et al. Autosomal recessive congenital sensorineural hearing loss due to a novel compound heterozygous PTPRQ mutation in a Chinese family. Neural Plast. 2018, 2018, 9425725. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, U.; Bukhari, S.A.; Ali, M.; Ahmed, Z.M.; Riazuddin, S.; IdentificatioGuevar, J.; Olby, N.J.; Meurs, K.M.; Yost, O.; Friedenberg, S.G. Deafness and vestibular dysfunction in a Doberman Pinscher puppy associated with a mutation in the PTPRQ gene. J. Vet. Intern. Med. 2018, 32, 665–669. [Google Scholar]
- Mahmood, U.; Bukhari, S.A.; Ali, M.; Ahmed, Z.M.; Riazuddin, S. Identification of hearing loss-associated variants of PTPRQ, MYO15A, and SERPINB6 in Pakistani families. BioMed Res. Int. 2021, 2021, 5584788. [Google Scholar]
- Jin, Y.; Liu, X.Z.; Xie, L.; Xie, W.; Chen, S.; Sun, Y. Targeted next-generation sequencing identified novel compound heterozygous variants in the PTPRQ gene causing autosomal recessive hearing loss in a Chinese family. Front. Genet. 2022, 13, 884522. [Google Scholar] [CrossRef] [PubMed]
- Ozieblo, D.; Sarosiak, A.; Leja, M.L.; Budde, B.S.; Tacikowska, G.; Donato, N.D.; Bolz, H.J.; Nürnberg, P.; Skarżyński, H.; Ołdak, M. First confirmatory study on PTPRQ as an autosomal dominant non-syndromic hearing loss gene. J. Transl. Med. 2019, 17, 351. [Google Scholar] [CrossRef] [PubMed]
- Nishio, S.Y.; Tono, T.; Iwaki, T.; Moteki, H.; Suzuki, K.; Tsushima, Y.; Kashio, A.; Akamatsu, Y.; Sato, H.; Yaegashi, K.; et al. Development and validation of an iPad-based Japanese language monosyllable speech perception test (iCI2004 monosyllable). Acta Otolaryngol. 2021, 141, 267–272. [Google Scholar] [CrossRef]
- Usami, S.I.; Nishio, S.Y.; Moteki, H.; Miyagawa, M.; Yoshimura, H. Cochlear Implantation From the Perspective of Genetic Background. Anat. Rec. 2020, 303, 563–593. [Google Scholar] [CrossRef]
- Nishio, S.Y.; Moteki, H.; Miyagawa, M.; Yamasoba, T.; Kashio, A.; Iwasaki, S.; Takahashi, M.; Naito, Y.; Fujiwara, K.; Sugaya, A.; et al. Etiology of hearing loss affects auditory skill development and vocabulary development in pediatric cochlear implantation cases. Acta Otolaryngol. 2022, 142, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Goodyear, R.J.; Jones, S.M.; Sharifi, L.; Forge, A.; Richardson, G.P. Hair bundle defects and loss of function in the vestibular end organs of mice lacking the receptor-like inositol lipid phosphatase. PTPRQ J. Neurosci. 2012, 32, 2762–2772. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Family Number | ID | Relationship | Base Change Allele 1 | AA Change Allele 1 | Base Change Allele 2 | AA Change Allele 2 | Hereditary | Onset | Age | Gender | Severity of HL | Type of HL | Progression | Vestibular Symptom | Hearing Device |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | HL9311 | proband | c.279T>G | p.Y93 * | c.5270G>C | p.R1757T | sporadic | 7 | 20 | F | Moderate | Flat | Y | N | HA |
| 2 | JHLB-13162 | proband | c.541delA | p.S181Afs * 12 | c.5270G>C | p.R1757T | sporadic | 0 | 18 | F | Severe | Flat | Y | N | HA |
| 3 | JHLB-6375 | proband | c.641C>A | p.S214 * | c.4093+1G>A | . | sporadic | 0 | 34 | F | Severe | HF steeply | Y | Y | CI |
| 4 | JHLB-2103 | proband | c.745C>T | p.R249 * | c.5426+2T>C | . | sporadic | 0 | 8 | F | Severe | Flat | Y | N | HA |
| 5 | HL2182 | proband | c.745C>T | p.R249 * | c.6017dupT | p.I2007Nfs * 14 | NA | NA | NA | NA | NA | NA | NA | NA | |
| 6 | JHLB-1036 | proband | c.745C>T | p.R249 * | c.745C>T | p.R249 * | sporadic | 0 | 19 | M | Profound | HF steeply | Y | N | CI |
| 7 | JHLB-12526 | proband | c.1433_1436del | p.S479Kfs * 7 | c.1433_1436del | p.S479Kfs * 7 | AD | 7 | 67 | F | Profound | Flat | Y | NA | CI |
| 8 | JHLB12398 | proband | c.1617delG | p.M539Ifs * 9 | c.2030C>T | p.T677M | sporadic | 13 | 29 | M | Moderate | HF steeply | Y | Y | |
| 9 | JHLB-11894 | proband | c.3206-2A>G | . | c.3263G>A | p.W1088 * | sporadic | 3 | 37 | F | Severe | HF steeply | Y | Y | HA |
| 10 | JHLB10176 | proband | c.3263G>A | p.W1088 * | c.5270G>C | p.R1757T | sporadic | 4 | 50 | M | Profound | Flat | Y | N | CI |
| 11 | HL13084 | proband | c.4093+1G>A | . | c.4402+1G>A | . | sporadic | 6 | 27 | M | Moderate | Flat | Y | N | |
| 12 | JHLB-13440 | proband | c.4261G>T | p.E1421 * | c.4781_4783del | p.T1596del | sporadic | NA | 42 | F | Profound | Flat | NA | N | CI |
| 13 | O-1614 | proband | c.4815C>A | p.Y1605 * | c.4815C>A | p.Y1605 * | sporadic | 0 | 66 | F | Profound | Flat | Y | NA |
| Nucleotide Change | AA Change | Exon | PP2 | MutTaster | REVEL | CADD | ToMMo 38KJPN | gnomAD All | Pathogenicity | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| c.279T>G | p.Y93 * | Exon 3 | . | A | . | 33 | 3.89 × 10−5 | . | Pathogenic | This study |
| c.541delA | p.S181Afs * 12 | Exon 5 | . | . | . | . | . | . | Likely pathogenic | This study |
| c.641C>A | p.S214 * | Exon 5 | . | A | . | 39 | . | . | Likely pathogenic | This study |
| c.745C>T | p.R249 * | Exon 6 | . | A | . | 37 | 9.03 × 10−5 | 1.95 × 10−5 | Pathogenic | Sakuma et al., 2015 [24] |
| c.1433_1436del | p.S479Kfs * 7 | Exon 10 | . | . | . | . | . | . | Pathogenic | This study |
| c.1617delG | p.M539Ifs * 9 | Exon 11 | . | . | . | . | . | . | Likely pathogenic | This study |
| c.2030C>T | p.T677M | Exon 13 | D | D | 0.344 | 34 | 0.0005842 | 2.70 × 10−5 | VUS | This study |
| c.3206-2A>G | . | Exon 20 | . | D | . | 22.8 | . | . | Likely pathogenic | This study |
| c.3263G>A | p.W1088 * | Exon 20 | . | A | . | 40 | 2.59 × 10−5 | 6.55 × 10−6 | Likely pathogenic | This study |
| c.4093+1G>A | . | Exon 23 | . | D | . | 25.5 | 6.46 × 10−5 | 6.58 × 10−5 | Likely pathogenic | This study |
| c.4261G>T | p.E1421 * | Exon 25 | . | A | . | 54 | . | . | Likely pathogenic | This study |
| c.4402+1G>A | . | Exon 25 | . | D | . | 27 | 0.0002614 | . | Likely pathogenic | This study |
| c.4781_4783del | p.T1596del | Exon 28 | . | . | . | . | 7.79 × 10−5 | 6.53 × 10−6 | VUS | This study |
| c.4815C>A | p.Y1605 * | Exon 28 | . | A | . | 38 | 1.30 × 10−5 | . | Pathogenic | This study |
| c.5270G>C | p.R1757T | Exon 31 | B | D | 0.153 | 24.1 | 0.0002841 | . | VUS | Yang et al., 2013 [25] |
| c.5426+2T>C | . | Exon 33 | . | D | . | 23.8 | . | . | Likely pathogenic | This study |
| c.6017dupT | p.I2007Nfs * 14 | Exon 39 | . | . | . | . | . | . | Likely pathogenic | This study |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sakuma, N.; Nishio, S.-y.; Goto, S.-i.; Honkura, Y.; Oda, K.; Takeda, H.; Kobayashi, M.; Kumakawa, K.; Iwasaki, S.; Takahashi, M.; et al. Detailed Clinical Features of PTPRQ-Associated Hearing Loss Identified in a Large Japanese Hearing Loss Cohort. Genes 2024, 15, 489. https://doi.org/10.3390/genes15040489
Sakuma N, Nishio S-y, Goto S-i, Honkura Y, Oda K, Takeda H, Kobayashi M, Kumakawa K, Iwasaki S, Takahashi M, et al. Detailed Clinical Features of PTPRQ-Associated Hearing Loss Identified in a Large Japanese Hearing Loss Cohort. Genes. 2024; 15(4):489. https://doi.org/10.3390/genes15040489
Chicago/Turabian StyleSakuma, Naoko, Shin-ya Nishio, Shin-ichi Goto, Yohei Honkura, Kiyoshi Oda, Hidehiko Takeda, Marina Kobayashi, Kozo Kumakawa, Satoshi Iwasaki, Masahiro Takahashi, and et al. 2024. "Detailed Clinical Features of PTPRQ-Associated Hearing Loss Identified in a Large Japanese Hearing Loss Cohort" Genes 15, no. 4: 489. https://doi.org/10.3390/genes15040489
APA StyleSakuma, N., Nishio, S.-y., Goto, S.-i., Honkura, Y., Oda, K., Takeda, H., Kobayashi, M., Kumakawa, K., Iwasaki, S., Takahashi, M., Ito, T., Arai, Y., Isono, Y., Obara, N., Matsunobu, T., Okubo, K., & Usami, S.-i. (2024). Detailed Clinical Features of PTPRQ-Associated Hearing Loss Identified in a Large Japanese Hearing Loss Cohort. Genes, 15(4), 489. https://doi.org/10.3390/genes15040489