
Determinants of Chromatin Organization in Aging and Cancer—Emerging Opportunities for Epigenetic Therapies and AI Technology

 , and
, and
Abstract
1. Introduction
2. Chromatin Organization and Its Structures
2.1. Heterochromatin
2.2. Euchromatin
3. Chromatin Imbalance in Aging and Cancer
3.1. Chromatin State in Stem Cell
3.2. Chromatin Imbalances in Cancer
3.3. Epigenetic Modifications in Aging
Progeroid Syndromes
4. Future Perspective of Epigenetic Therapies and AI Technology
4.1. Future Perspective on Aging
4.2. Future Perspective on Cancer
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Biechele, S.; Lin, C.J.; Rinaudo, P.F.; Ramalho-Santos, M. Unwind and transcribe: Chromatin reprogramming in the early mammalian embryo. Curr. Opin. Genet. Dev. 2015, 34, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Abdouh, M.; Facchino, S.; Chatoo, W.; Balasingam, V.; Ferreira, J.; Bernier, G. BMI1 sustains human glioblastoma multiforme stem cell renewal. J. Neurosci. 2009, 29, 8884–8896. [Google Scholar] [CrossRef] [PubMed]
- Morrison, O.; Thakur, J. Molecular Complexes at Euchromatin, Heterochromatin and Centromeric Chromatin. Int. J. Mol. Sci. 2021, 22, 6922. [Google Scholar] [CrossRef] [PubMed]
- Belotti, E.; Lacoste, N.; Iftikhar, A.; Simonet, T.; Papin, C.; Osseni, A.; Streichenberger, N.; Mari, P.O.; Girard, E.; Graies, M.; et al. H2A.Z is involved in premature aging and DSB repair initiation in muscle fibers. Nucleic Acids Res. 2024, 52, 3031–3049. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Zhang, L.; Ruan, H.; Li, G. Histone variant H2A.Z regulates nucleosome unwrapping and CTCF binding in mouse ES cells. Nucleic Acids Res. 2020, 48, 5939–5952. [Google Scholar] [CrossRef] [PubMed]
- Colino-Sanguino, Y.; Clark, S.J.; Valdes-Mora, F. The H2A.Z-nuclesome code in mammals: Emerging functions. Trends Genet. 2022, 38, 273–289. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Nieves-Neira, W.; Boon, C.; Pommier, Y.; Bonner, W.M. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J. Biol. Chem. 2000, 275, 9390–9395. [Google Scholar] [CrossRef] [PubMed]
- Mah, L.J.; El-Osta, A.; Karagiannis, T.C. gammaH2AX: A sensitive molecular marker of DNA damage and repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef]
- Herchenrother, A.; Wunderlich, T.M.; Lan, J.; Hake, S.B. Spotlight on histone H2A variants: From B to X to Z. Semin. Cell Dev. Biol. 2023, 135, 3–12. [Google Scholar] [CrossRef]
- Davie, J.R.; Xu, W.; Delcuve, G.P. Histone H3K4 trimethylation: Dynamic interplay with pre-mRNA splicing. Biochem. Cell Biol. 2016, 94, 1–11. [Google Scholar] [CrossRef]
- Noma, K.; Allis, C.D.; Grewal, S.I. Transitions in distinct histone H3 methylation patterns at the heterochromatin domain boundaries. Science 2001, 293, 1150–1155. [Google Scholar] [CrossRef] [PubMed]
- Poleshko, A.; Smith, C.L.; Nguyen, S.C.; Sivaramakrishnan, P.; Wong, K.G.; Murray, J.I.; Lakadamyali, M.; Joyce, E.F.; Jain, R.; Epstein, J.A. H3K9me2 orchestrates inheritance of spatial positioning of peripheral heterochromatin through mitosis. eLife 2019, 8, e49278. [Google Scholar] [CrossRef]
- Poleshko, A.; Shah, P.P.; Gupta, M.; Babu, A.; Morley, M.P.; Manderfield, L.J.; Ifkovits, J.L.; Calderon, D.; Aghajanian, H.; Sierra-Pagan, J.E.; et al. Genome-Nuclear Lamina Interactions Regulate Cardiac Stem Cell Lineage Restriction. Cell 2017, 171, 573–587.e14. [Google Scholar] [CrossRef]
- Tachibana, M.; Ueda, J.; Fukuda, M.; Takeda, N.; Ohta, T.; Iwanari, H.; Sakihama, T.; Kodama, T.; Hamakubo, T.; Shinkai, Y. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. 2005, 19, 815–826. [Google Scholar] [CrossRef]
- Cutter DiPiazza, A.R.; Taneja, N.; Dhakshnamoorthy, J.; Wheeler, D.; Holla, S.; Grewal, S.I.S. Spreading and epigenetic inheritance of heterochromatin require a critical density of histone H3 lysine 9 tri-methylation. Proc. Natl. Acad. Sci. USA 2021, 118, e2100699118. [Google Scholar] [CrossRef] [PubMed]
- Zofall, M.; Sandhu, R.; Holla, S.; Wheeler, D.; Grewal, S.I.S. Histone deacetylation primes self-propagation of heterochromatin domains to promote epigenetic inheritance. Nat. Struct. Mol. Biol. 2022, 29, 898–909. [Google Scholar] [CrossRef]
- Tachibana, M.; Sugimoto, K.; Nozaki, M.; Ueda, J.; Ohta, T.; Ohki, M.; Fukuda, M.; Takeda, N.; Niida, H.; Kato, H.; et al. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 2002, 16, 1779–1791. [Google Scholar] [CrossRef]
- Yuan, W.; Wu, T.; Fu, H.; Dai, C.; Wu, H.; Liu, N.; Li, X.; Xu, M.; Zhang, Z.; Niu, T.; et al. Dense chromatin activates Polycomb repressive complex 2 to regulate H3 lysine 27 methylation. Science 2012, 337, 971–975. [Google Scholar] [CrossRef]
- Tachibana, M.; Sugimoto, K.; Fukushima, T.; Shinkai, Y. Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J. Biol. Chem. 2001, 276, 25309–25317. [Google Scholar] [CrossRef]
- Bannister, A.J.; Schneider, R.; Myers, F.A.; Thorne, A.W.; Crane-Robinson, C.; Kouzarides, T. Spatial distribution of di- and tri-methyl lysine 36 of histone H3 at active genes. J. Biol. Chem. 2005, 280, 17732–17736. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Dang, W.; Donahue, G.; Dai, J.; Dorsey, J.; Cao, X.; Liu, W.; Cao, K.; Perry, R.; Lee, J.Y.; et al. H3K36 methylation promotes longevity by enhancing transcriptional fidelity. Genes Dev. 2015, 29, 1362–1376. [Google Scholar] [CrossRef] [PubMed]
- Pu, M.; Ni, Z.; Wang, M.; Wang, X.; Wood, J.G.; Helfand, S.L.; Yu, H.; Lee, S.S. Trimethylation of Lys36 on H3 restricts gene expression change during aging and impacts life span. Genes Dev. 2015, 29, 718–731. [Google Scholar] [CrossRef]
- Yuan, J.; Pu, M.; Zhang, Z.; Lou, Z. Histone H3-K56 acetylation is important for genomic stability in mammals. Cell Cycle 2009, 8, 1747–1753. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, Y.; Horton, J.K.; Wilson, S.H. Histone H3 Lysine 56 Acetylation Enhances AP Endonuclease 1-Mediated Repair of AP Sites in Nucleosome Core Particles. Biochemistry 2019, 58, 3646–3655. [Google Scholar] [CrossRef]
- Fang, L.; Chen, D.; Zhang, J.; Li, H.; Bradford, B.; Jin, C. Potential functions of histone H3.3 lysine 56 acetylation in mammals. Epigenetics 2022, 17, 498–517. [Google Scholar] [CrossRef]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef]
- Kuo, A.J.; Song, J.; Cheung, P.; Ishibe-Murakami, S.; Yamazoe, S.; Chen, J.K.; Patel, D.J.; Gozani, O. The BAH domain of ORC1 links H4K20me2 to DNA replication licensing and Meier-Gorlin syndrome. Nature 2012, 484, 115–119. [Google Scholar] [CrossRef]
- Moller, M.; Ridenour, J.B.; Wright, D.F.; Martin, F.A.; Freitag, M. H4K20me3 is important for Ash1-mediated H3K36me3 and transcriptional silencing in facultative heterochromatin in a fungal pathogen. PLoS Genet. 2023, 19, e1010945. [Google Scholar] [CrossRef]
- Zhao, R.; Nakamura, T.; Fu, Y.; Lazar, Z.; Spector, D.L. Gene bookmarking accelerates the kinetics of post-mitotic transcriptional re-activation. Nat. Cell Biol. 2011, 13, 1295–1304. [Google Scholar] [CrossRef]
- Park, C.S.; Rehrauer, H.; Mansuy, I.M. Genome-wide analysis of H4K5 acetylation associated with fear memory in mice. BMC Genom. 2013, 14, 539. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.P.; Zhu, L.; Tripathi, J.; Kucharski, M.; Patra, A.; Bozdech, Z. Histone 4 lysine 8 acetylation regulates proliferation and host-pathogen interaction in Plasmodium falciparum. Epigenet. Chromatin 2017, 10, 40. [Google Scholar] [CrossRef] [PubMed]
- Raus, A.M.; Fuller, T.D.; Nelson, N.E.; Valientes, D.A.; Bayat, A.; Ivy, A.S. Early-life exercise primes the murine neural epigenome to facilitate gene expression and hippocampal memory consolidation. Commun. Biol. 2023, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Peleg, S.; Sananbenesi, F.; Zovoilis, A.; Burkhardt, S.; Bahari-Javan, S.; Agis-Balboa, R.C.; Cota, P.; Wittnam, J.L.; Gogol-Doering, A.; Opitz, L.; et al. Altered histone acetylation is associated with age-dependent memory impairment in mice. Science 2010, 328, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, S.; Benito, E.; Fischer, A.; Johnsen, S.A. H4K12ac is regulated by estrogen receptor-alpha and is associated with BRD4 function and inducible transcription. Oncotarget 2015, 6, 7305–7317. [Google Scholar] [CrossRef] [PubMed]
- Suka, N.; Luo, K.; Grunstein, M. Sir2p and Sas2p opposingly regulate acetylation of yeast histone H4 lysine16 and spreading of heterochromatin. Nat. Genet. 2002, 32, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.G.; So, S.; Gupta, A.; Kumar, R.; Cayrou, C.; Avvakumov, N.; Bhadra, U.; Pandita, R.K.; Porteus, M.H.; Chen, D.J.; et al. MOF and histone H4 acetylation at lysine 16 are critical for DNA damage response and double-strand break repair. Mol. Cell. Biol. 2010, 30, 3582–3595. [Google Scholar] [CrossRef] [PubMed]
- Pal, D.; Patel, M.; Boulet, F.; Sundarraj, J.; Grant, O.A.; Branco, M.R.; Basu, S.; Santos, S.D.M.; Zabet, N.R.; Scaffidi, P.; et al. H4K16ac activates the transcription of transposable elements and contributes to their cis-regulatory function. Nat. Struct. Mol. Biol. 2023, 30, 935–947. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Perez, M.F.; Lehner, B. Intergenerational and transgenerational epigenetic inheritance in animals. Nat. Cell Biol. 2019, 21, 143–151. [Google Scholar] [CrossRef]
- Xavier, M.J.; Roman, S.D.; Aitken, R.J.; Nixon, B. Transgenerational inheritance: How impacts to the epigenetic and genetic information of parents affect offspring health. Hum. Reprod. Update 2019, 25, 518–540. [Google Scholar] [CrossRef] [PubMed]
- Burton, N.O.; Greer, E.L. Multigenerational epigenetic inheritance: Transmitting information across generations. Semin. Cell Dev. Biol. 2022, 127, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Ricci, M.A.; Manzo, C.; Garcia-Parajo, M.F.; Lakadamyali, M.; Cosma, M.P. Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo. Cell 2015, 160, 1145–1158. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, T.; Imai, R.; Tanbo, M.; Nagashima, R.; Tamura, S.; Tani, T.; Joti, Y.; Tomita, M.; Hibino, K.; Kanemaki, M.T.; et al. Dynamic Organization of Chromatin Domains Revealed by Super-Resolution Live-Cell Imaging. Mol. Cell 2017, 67, 282–293.e7. [Google Scholar] [CrossRef] [PubMed]
- Trojer, P.; Reinberg, D. Facultative heterochromatin: Is there a distinctive molecular signature? Mol. Cell 2007, 28, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Grewal, S.I.; Jia, S. Heterochromatin revisited. Nat. Rev. Genet. 2007, 8, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Allshire, R.C.; Madhani, H.D. Ten principles of heterochromatin formation and function. Nat. Rev. Mol. Cell Biol. 2018, 19, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, J.; Rice, J.C.; Strahl, B.D.; Allis, C.D.; Grewal, S.I. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 2001, 292, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Zegerman, P.; Partridge, J.F.; Miska, E.A.; Thomas, J.O.; Allshire, R.C.; Kouzarides, T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 2001, 410, 120–124. [Google Scholar] [CrossRef]
- Bosch-Presegue, L.; Raurell-Vila, H.; Thackray, J.K.; Gonzalez, J.; Casal, C.; Kane-Goldsmith, N.; Vizoso, M.; Brown, J.P.; Gomez, A.; Ausio, J.; et al. Mammalian HP1 Isoforms Have Specific Roles in Heterochromatin Structure and Organization. Cell Rep. 2017, 21, 2048–2057. [Google Scholar] [CrossRef]
- Horsley, D.; Hutchings, A.; Butcher, G.W.; Singh, P.B. M32, a murine homologue of Drosophila heterochromatin protein 1 (HP1), localises to euchromatin within interphase nuclei and is largely excluded from constitutive heterochromatin. Cytogenet. Cell Genet. 1996, 73, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Vakoc, C.R.; Mandat, S.A.; Olenchock, B.A.; Blobel, G.A. Histone H3 lysine 9 methylation and HP1gamma are associated with transcription elongation through mammalian chromatin. Mol. Cell 2005, 19, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Fanti, L.; Pimpinelli, S. HP1: A functionally multifaceted protein. Curr. Opin. Genet. Dev. 2008, 18, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.A.; Kingston, R.E. Mechanisms of polycomb gene silencing: Knowns and unknowns. Nat. Rev. Mol. Cell Biol. 2009, 10, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Brockdorff, N. Noncoding RNA and Polycomb recruitment. RNA 2013, 19, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, T.H. Single-Molecule Investigations on Histone H2A-H2B Dynamics in the Nucleosome. Biochemistry 2017, 56, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Elgin, S.C.; Reuter, G. Position-effect variegation, heterochromatin formation, and gene silencing in Drosophila. Cold Spring Harb. Perspect. Biol. 2013, 5, a017780. [Google Scholar] [CrossRef] [PubMed]
- Bulger, M.; Groudine, M. Functional and mechanistic diversity of distal transcription enhancers. Cell 2011, 144, 327–339. [Google Scholar] [CrossRef]
- Gaspar-Maia, A.; Alajem, A.; Polesso, F.; Sridharan, R.; Mason, M.J.; Heidersbach, A.; Ramalho-Santos, J.; McManus, M.T.; Plath, K.; Meshorer, E.; et al. Chd1 regulates open chromatin and pluripotency of embryonic stem cells. Nature 2009, 460, 863–868. [Google Scholar] [CrossRef]
- Courtot, A.M.; Magniez, A.; Oudrhiri, N.; Feraud, O.; Bacci, J.; Gobbo, E.; Proust, S.; Turhan, A.G.; Bennaceur-Griscelli, A. Morphological analysis of human induced pluripotent stem cells during induced differentiation and reverse programming. BioRes. Open Access 2014, 3, 206–216. [Google Scholar] [CrossRef]
- Klein, D.C.; Hainer, S.J. Chromatin regulation and dynamics in stem cells. Curr. Top. Dev. Biol. 2020, 138, 1–71. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.; Giuliani, A. Resolution of Complex Issues in Genome Regulation and Cancer Requires Non-Linear and Network-Based Thermodynamics. Int. J. Mol. Sci. 2019, 21, 240. [Google Scholar] [CrossRef] [PubMed]
- Ao, W.; Gaudet, J.; Kent, W.J.; Muttumu, S.; Mango, S.E. Environmentally induced foregut remodeling by PHA-4/FoxA and DAF-12/NHR. Science 2004, 305, 1743–1746. [Google Scholar] [CrossRef] [PubMed]
- Golson, M.L.; Kaestner, K.H. Fox transcription factors: From development to disease. Development 2016, 143, 4558–4570. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Kaestner, K.H. The Foxa family of transcription factors in development and metabolism. Cell. Mol. Life Sci. 2006, 63, 2317–2328. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Hayano, M.; Griffin, P.T.; Amorim, J.A.; Bonkowski, M.S.; Apostolides, J.K.; Salfati, E.L.; Blanchette, M.; Munding, E.M.; Bhakta, M.; et al. Loss of epigenetic information as a cause of mammalian aging. Cell 2023, 186, 305–326.e27. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Slattery, M.; Zhou, T.; Yang, L.; Dantas Machado, A.C.; Gordan, R.; Rohs, R. Absence of a simple code: How transcription factors read the genome. Trends Biochem. Sci. 2014, 39, 381–399. [Google Scholar] [CrossRef]
- Cirillo, L.A.; Lin, F.R.; Cuesta, I.; Friedman, D.; Jarnik, M.; Zaret, K.S. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol. Cell 2002, 9, 279–289. [Google Scholar] [CrossRef]
- Rawal, Y.; Qiu, H.; Hinnebusch, A.G. Distinct functions of three chromatin remodelers in activator binding and preinitiation complex assembly. PLoS Genet. 2022, 18, e1010277. [Google Scholar] [CrossRef]
- Javasky, E.; Shamir, I.; Gandhi, S.; Egri, S.; Sandler, O.; Rothbart, S.B.; Kaplan, N.; Jaffe, J.D.; Goren, A.; Simon, I. Study of mitotic chromatin supports a model of bookmarking by histone modifications and reveals nucleosome deposition patterns. Genome Res. 2018, 28, 1455–1466. [Google Scholar] [CrossRef] [PubMed]
- Rando, O.J.; Chang, H.Y. Genome-wide views of chromatin structure. Annu. Rev. Biochem. 2009, 78, 245–271. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Talbert, P.B.; Henikoff, S. Histone variants—Ancient wrap artists of the epigenome. Nat. Rev. Mol. Cell Biol. 2010, 11, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Maze, I.; Noh, K.M.; Allis, C.D. Histone regulation in the CNS: Basic principles of epigenetic plasticity. Neuropsychopharmacology 2013, 38, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, B.; Wapinski, O.L.; Tsai, M.C.; Qu, K.; Zhang, J.; Carlson, J.C.; Lin, M.; Fang, F.; Gupta, R.A.; et al. Targeted disruption of Hotair leads to homeotic transformation and gene derepression. Cell Rep. 2013, 5, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Giadrossi, S.; Dvorkina, M.; Fisher, A.G. Chromatin organization and differentiation in embryonic stem cell models. Curr. Opin. Genet. Dev. 2007, 17, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Liu, X. Review: Chromatin organization in plant and animal stem cell maintenance. Plant Sci. 2019, 281, 173–179. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, D.; Dowell, R.; Yi, R. Single cell analysis of transcriptome and open chromatin reveals the dynamics of hair follicle stem cell aging. Front. Aging 2023, 4, 1192149. [Google Scholar] [CrossRef]
- Kucia, M.; Zuba-Surma, E.K.; Wysoczynski, M.; Wu, W.; Ratajczak, J.; Machalinski, B.; Ratajczak, M.Z. Adult marrow-derived very small embryonic-like stem cells and tissue engineering. Expert. Opin. Biol. Ther. 2007, 7, 1499–1514. [Google Scholar] [CrossRef]
- Tada, T.; Tada, M. Toti-/pluripotential stem cells and epigenetic modifications. Cell Struct. Funct. 2001, 26, 149–160. [Google Scholar] [CrossRef]
- Bayarsaihan, D.; Makeyev, A.V.; Enkhmandakh, B. Epigenetic modulation by TFII-I during embryonic stem cell differentiation. J. Cell. Biochem. 2012, 113, 3056–3060. [Google Scholar] [CrossRef]
- Ramlee, M.K.; Zhang, Q.; Idris, M.; Peng, X.; Sim, C.K.; Han, W.; Xu, F. Histone H3 K27 acetylation marks a potent enhancer element on the adipogenic master regulator gene Pparg2. Cell Cycle 2014, 13, 3414–3422. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.I.; Jenner, R.G.; Boyer, L.A.; Guenther, M.G.; Levine, S.S.; Kumar, R.M.; Chevalier, B.; Johnstone, S.E.; Cole, M.F.; Isono, K.; et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 2006, 125, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Boyer, L.A.; Plath, K.; Zeitlinger, J.; Brambrink, T.; Medeiros, L.A.; Lee, T.I.; Levine, S.S.; Wernig, M.; Tajonar, A.; Ray, M.K.; et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 2006, 441, 349–353. [Google Scholar] [CrossRef]
- Melcer, S.; Meshorer, E. Chromatin plasticity in pluripotent cells. Essays Biochem. 2010, 48, 245–262. [Google Scholar] [CrossRef]
- Farthing, C.R.; Ficz, G.; Ng, R.K.; Chan, C.F.; Andrews, S.; Dean, W.; Hemberger, M.; Reik, W. Global mapping of DNA methylation in mouse promoters reveals epigenetic reprogramming of pluripotency genes. PLoS Genet. 2008, 4, e1000116. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Huang, D.; Li, R.; Wang, W.; Zhou, C. Control of mesenchymal stem cell biology by histone modifications. Cell Biosci. 2020, 10, 11. [Google Scholar] [CrossRef]
- Sachs, M.; Onodera, C.; Blaschke, K.; Ebata, K.T.; Song, J.S.; Ramalho-Santos, M. Bivalent chromatin marks developmental regulatory genes in the mouse embryonic germline in vivo. Cell Rep. 2013, 3, 1777–1784. [Google Scholar] [CrossRef]
- Matsumura, Y.; Nakaki, R.; Inagaki, T.; Yoshida, A.; Kano, Y.; Kimura, H.; Tanaka, T.; Tsutsumi, S.; Nakao, M.; Doi, T.; et al. H3K4/H3K9me3 Bivalent Chromatin Domains Targeted by Lineage-Specific DNA Methylation Pauses Adipocyte Differentiation. Mol. Cell 2015, 60, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Khromov, T.; Pantakani, D.V.; Nolte, J.; Wolf, M.; Dressel, R.; Engel, W.; Zechner, U. Global and gene-specific histone modification profiles of mouse multipotent adult germline stem cells. Mol. Hum. Reprod. 2011, 17, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Efroni, S.; Duttagupta, R.; Cheng, J.; Dehghani, H.; Hoeppner, D.J.; Dash, C.; Bazett-Jones, D.P.; Le Grice, S.; McKay, R.D.; Buetow, K.H.; et al. Global transcription in pluripotent embryonic stem cells. Cell Stem Cell 2008, 2, 437–447. [Google Scholar] [CrossRef]
- Rodriguez-Madoz, J.R.; San Jose-Eneriz, E.; Rabal, O.; Zapata-Linares, N.; Miranda, E.; Rodriguez, S.; Porciuncula, A.; Vilas-Zornoza, A.; Garate, L.; Segura, V.; et al. Reversible dual inhibitor against G9a and DNMT1 improves human iPSC derivation enhancing MET and facilitating transcription factor engagement to the genome. PLoS ONE 2017, 12, e0190275. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Gordon, J.A.; Whitfield, T.W.; Tai, P.W.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; Lian, J.B. Chromatin dynamics regulate mesenchymal stem cell lineage specification and differentiation to osteogenesis. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xie, S.; Zhou, Y.; Xie, Y.; Liu, P.; Sun, M.; Xiao, H.; Jin, Y.; Sun, X.; Chen, Z.; et al. H3K36 histone methyltransferase Setd2 is required for murine embryonic stem cell differentiation toward endoderm. Cell Rep. 2014, 8, 1989–2002. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Song, C.; Young, N.L.; Sperling, A.S.; Xu, F.; Sridharan, R.; Conway, A.E.; Garcia, B.A.; Plath, K.; Clark, A.T.; et al. Histone h3 lysine 56 acetylation is linked to the core transcriptional network in human embryonic stem cells. Mol. Cell 2009, 33, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Evertts, A.G.; Manning, A.L.; Wang, X.; Dyson, N.J.; Garcia, B.A.; Coller, H.A. H4K20 methylation regulates quiescence and chromatin compaction. Mol. Biol. Cell 2013, 24, 3025–3037. [Google Scholar] [CrossRef] [PubMed]
- Keegan, S.E.; Haskins, J.; Simmonds, A.J.; Hughes, S.C. A chromatin remodelling SWI/SNF subunit, Snr1, regulates neural stem cell determination and differentiation. Development 2023, 150, dev201484. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, J.; Tang, Y. Advances in the role of SWI/SNF complexes in tumours. J. Cell. Mol. Med. 2023, 27, 1023–1031. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.; Dambacher, S.; Schotta, G. Heterochromatin dysregulation in human diseases. J. Appl. Physiol. 2010, 109, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.J.; Shin, Y.K. Epigenetic regulation of cancer-associated genes in ovarian cancer. Int. J. Mol. Sci. 2011, 12, 983–1008. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.Y.; Aromolaran, K.A.; Zukin, R.S. The emerging field of epigenetics in neurodegeneration and neuroprotection. Nat. Rev. Neurosci. 2017, 18, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Berson, A.; Nativio, R.; Berger, S.L.; Bonini, N.M. Epigenetic Regulation in Neurodegenerative Diseases. Trends Neurosci. 2018, 41, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Bracken, A.P.; Kleine-Kohlbrecher, D.; Dietrich, N.; Pasini, D.; Gargiulo, G.; Beekman, C.; Theilgaard-Monch, K.; Minucci, S.; Porse, B.T.; Marine, J.C.; et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007, 21, 525–530. [Google Scholar] [CrossRef]
- Tong, Z.T.; Cai, M.Y.; Wang, X.G.; Kong, L.L.; Mai, S.J.; Liu, Y.H.; Zhang, H.B.; Liao, Y.J.; Zheng, F.; Zhu, W.; et al. EZH2 supports nasopharyngeal carcinoma cell aggressiveness by forming a co-repressor complex with HDAC1/HDAC2 and Snail to inhibit E-cadherin. Oncogene 2012, 31, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gonzalez, M.E.; Toy, K.; Filzen, T.; Merajver, S.D.; Kleer, C.G. Targeted overexpression of EZH2 in the mammary gland disrupts ductal morphogenesis and causes epithelial hyperplasia. Am. J. Pathol. 2009, 175, 1246–1254. [Google Scholar] [CrossRef]
- Moison, C.; Assemat, F.; Daunay, A.; Tost, J.; Guieysse-Peugeot, A.L.; Arimondo, P.B. Synergistic chromatin repression of the tumor suppressor gene RARB in human prostate cancers. Epigenetics 2014, 9, 477–482. [Google Scholar] [CrossRef]
- Kazanets, A.; Shorstova, T.; Hilmi, K.; Marques, M.; Witcher, M. Epigenetic silencing of tumor suppressor genes: Paradigms, puzzles, and potential. Biochim. Biophys. Acta 2016, 1865, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Di Croce, L.; Helin, K. Transcriptional regulation by Polycomb group proteins. Nat. Struct. Mol. Biol. 2013, 20, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.; Goode, D.L.; Iwasaki, M.; Wei, M.C.; Kuo, H.P.; Zhu, L.; Schneidawind, D.; Duque-Afonso, J.; Weng, Z.; Cleary, M.L. The H3K4-Methyl Epigenome Regulates Leukemia Stem Cell Oncogenic Potential. Cancer Cell 2015, 28, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Shilatifard, A. Epigenetic modifications of histones in cancer. Genome Biol. 2019, 20, 245. [Google Scholar] [CrossRef] [PubMed]
- Mizokami, H.; Okabe, A.; Choudhary, R.; Mima, M.; Saeda, K.; Fukuyo, M.; Rahmutulla, B.; Seki, M.; Goh, B.C.; Kondo, S.; et al. Enhancer infestation drives tumorigenic activation of inactive B compartment in Epstein-Barr virus-positive nasopharyngeal carcinoma. EBioMedicine 2024, 102, 105057. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.S.; Lee, S.; Choi, J.E.; Hong, M.J.; Do, S.K.; Lee, J.H.; Lee, W.K.; Park, J.E.; Lee, Y.H.; Choi, S.H.; et al. Promoter-Specific Variants in NeuroD1 and H3K4me3 Coincident Regions and Clinical Outcomes of Small Cell Lung Cancer. J. Korean Med. Sci. 2023, 38, e381. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.D.; Qiu, B.Q.; Xiong, D.; Pei, X.; Jie, N.; Xu, H.; Zhu, S.Q.; Long, X.; Xu, Z.; Wu, H.B.; et al. High level of H3K4 tri-methylation modification predicts poor prognosis in esophageal cancer. J. Cancer 2020, 11, 3256–3263. [Google Scholar] [CrossRef] [PubMed]
- Berger, L.; Kolben, T.; Meister, S.; Kolben, T.M.; Schmoeckel, E.; Mayr, D.; Mahner, S.; Jeschke, U.; Ditsch, N.; Beyer, S. Expression of H3K4me3 and H3K9ac in breast cancer. J. Cancer Res. Clin. Oncol. 2020, 146, 2017–2027. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.; Della Rosa, M.; Krueger, C.; Gao, Q.; Horkai, D.; King, M.; Field, L.; Houseley, J. Tri-methylation of histone H3 lysine 4 facilitates gene expression in ageing cells. eLife 2018, 7, e34081. [Google Scholar] [CrossRef]
- He, C.; Xu, J.; Zhang, J.; Xie, D.; Ye, H.; Xiao, Z.; Cai, M.; Xu, K.; Zeng, Y.; Li, H.; et al. High expression of trimethylated histone H3 lysine 4 is associated with poor prognosis in hepatocellular carcinoma. Hum. Pathol. 2012, 43, 1425–1435. [Google Scholar] [CrossRef]
- Baisya, D.R.; Lonardi, S. Prediction of histone post-translational modifications using deep learning. Bioinformatics 2021, 36, 5610–5617. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.M.; Ge, Y.; Schuster, M.B.; Pundhir, S.; Jakobsen, J.S.; Kalvisa, A.; Tapia, M.C.; Gordon, S.; Ambri, F.; Bagger, F.O.; et al. H3K9 dimethylation safeguards cancer cells against activation of the interferon pathway. Sci. Adv. 2022, 8, eabf8627. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Li, Y.; Zheng, C.; Lu, T.; Sun, R.; Mao, Y.; Yu, S.; Fan, H.; Zhang, Z. High methylation levels of histone H3 lysine 9 associated with activation of hypoxia-inducible factor 1alpha (HIF-1alpha) predict patients’ worse prognosis in human hepatocellular carcinomas. Cancer Genet. 2020, 245, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Piro, M.C.; Gasperi, V.; De Stefano, A.; Anemona, L.; Cenciarelli, C.R.; Montanaro, M.; Mauriello, A.; Catani, M.V.; Terrinoni, A.; Gambacurta, A. In Vivo Identification of H3K9me2/H3K79me3 as an Epigenetic Barrier to Carcinogenesis. Int. J. Mol. Sci. 2023, 24, 12158. [Google Scholar] [CrossRef] [PubMed]
- Yap, D.B.; Chu, J.; Berg, T.; Schapira, M.; Cheng, S.W.; Moradian, A.; Morin, R.D.; Mungall, A.J.; Meissner, B.; Boyle, M.; et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 2011, 117, 2451–2459. [Google Scholar] [CrossRef] [PubMed]
- Sasidharan Nair, V.; Toor, S.M.; Taha, R.Z.; Shaath, H.; Elkord, E. DNA methylation and repressive histones in the promoters of PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, PD-L1, and galectin-9 genes in human colorectal cancer. Clin. Epigenet. 2018, 10, 104. [Google Scholar] [CrossRef] [PubMed]
- Duan, N.; Hua, Y.; Yan, X.; He, Y.; Zeng, T.; Gong, J.; Fu, Z.; Li, W.; Yin, Y. An Imbalance in Histone Modifiers Induces tRNA-Cys-GCA Overexpression and tRF-27 Accumulation by Attenuating Promoter H3K27me3 in Primary Trastuzumab-Resistant Breast Cancer. Cancers 2024, 16, 1118. [Google Scholar] [CrossRef]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tonjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Khuong-Quang, D.A.; Buczkowicz, P.; Rakopoulos, P.; Liu, X.Y.; Fontebasso, A.M.; Bouffet, E.; Bartels, U.; Albrecht, S.; Schwartzentruber, J.; Letourneau, L.; et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012, 124, 439–447. [Google Scholar] [CrossRef]
- Lewis, P.W.; Muller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [PubMed]
- Mitchener, M.M.; Muir, T.W. Oncohistones: Exposing the nuances and vulnerabilities of epigenetic regulation. Mol. Cell 2022, 82, 2925–2938. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Jain, S.U.; Hoelper, D.; Bechet, D.; Molden, R.C.; Ran, L.; Murphy, D.; Venneti, S.; Hameed, M.; Pawel, B.R.; et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 2016, 352, 844–849. [Google Scholar] [CrossRef] [PubMed]
- Behjati, S.; Tarpey, P.S.; Presneau, N.; Scheipl, S.; Pillay, N.; Van Loo, P.; Wedge, D.C.; Cooke, S.L.; Gundem, G.; Davies, H.; et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat. Genet. 2013, 45, 1479–1482. [Google Scholar] [CrossRef]
- Bernt, K.M.; Armstrong, S.A. A role for DOT1L in MLL-rearranged leukemias. Epigenomics 2011, 3, 667–670. [Google Scholar] [CrossRef]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhou, M.; Gan, X.L.; Ren, Y.X.; Yang, Y.Z.; Weng, Z.J.; Zhang, X.F.; Guan, J.X.; Tang, L.Y.; Ren, Z.F. Combined low levels of H4K16ac and H4K20me3 predicts poor prognosis in breast cancer. Int. J. Clin. Oncol. 2023, 28, 1147–1157. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef]
- Rogenhofer, S.; Kahl, P.; Holzapfel, S.; Von Ruecker, A.; Mueller, S.C.; Ellinger, J. Decreased levels of histone H3K9me1 indicate poor prognosis in patients with renal cell carcinoma. Anticancer Res. 2012, 32, 879–886. [Google Scholar]
- Hou, Y.; Yuan, Y.; Li, Y.; Wang, L.; Hu, J.; Liu, X. The role of histone methylation in renal cell cancer: An update. Mol. Biol. Rep. 2023, 50, 2735–2742. [Google Scholar] [CrossRef]
- Wang, B.; Zhou, M.; Shi, Y.Y.; Chen, X.L.; Ren, Y.X.; Yang, Y.Z.; Tang, L.Y.; Ren, Z.F. Survival is associated with repressive histone trimethylation markers in both HR-positive HER2-negative and triple-negative breast cancer patients. Virchows Arch. 2023, 482, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Phoyen, S.; Sanpavat, A.; Ma-On, C.; Stein, U.; Hirankarn, N.; Tangkijvanich, P.; Jindatip, D.; Whongsiri, P.; Boonla, C. H4K20me3 upregulated by reactive oxygen species is associated with tumor progression and poor prognosis in patients with hepatocellular carcinoma. Heliyon 2023, 9, e22589. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.M.; Jaber-Hijazi, F.; Cole, J.J.; Robertson, N.A.; Pawlikowski, J.S.; Norris, K.T.; Criscione, S.W.; Pchelintsev, N.A.; Piscitello, D.; Stong, N.; et al. Mapping H4K20me3 onto the chromatin landscape of senescent cells indicates a function in control of cell senescence and tumor suppression through preservation of genetic and epigenetic stability. Genome Biol. 2016, 17, 158. [Google Scholar] [CrossRef] [PubMed]
- Di Cerbo, V.; Schneider, R. Cancers with wrong HATs: The impact of acetylation. Brief. Funct. Genom. 2013, 12, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Ropero, S.; Esteller, M. The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Espin-Perez, A.; Brennan, K.; Ediriwickrema, A.S.; Gevaert, O.; Lossos, I.S.; Gentles, A.J. Peripheral blood DNA methylation profiles predict future development of B-cell Non-Hodgkin Lymphoma. NPJ Precis. Oncol. 2022, 6, 53. [Google Scholar] [CrossRef] [PubMed]
- Krivtsov, A.V.; Hoshii, T.; Armstrong, S.A. Mixed-Lineage Leukemia Fusions and Chromatin in Leukemia. Cold Spring Harb. Perspect. Med. 2017, 7, a026658. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Fong, K.W.; Mong, E.; Martin, M.C.; Schiltz, G.E.; Yu, J. Going beyond Polycomb: EZH2 functions in prostate cancer. Oncogene 2021, 40, 5788–5798. [Google Scholar] [CrossRef]
- Rossetto, D.; Avvakumov, N.; Cote, J. Histone phosphorylation: A chromatin modification involved in diverse nuclear events. Epigenetics 2012, 7, 1098–1108. [Google Scholar] [CrossRef]
- Dong, G.J.; Xu, J.L.; Qi, Y.R.; Yuan, Z.Q.; Zhao, W. Critical Roles of Polycomb Repressive Complexes in Transcription and Cancer. Int. J. Mol. Sci. 2022, 23, 9574. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, H.M.; Mohammad, H.P.; Baylin, S.B. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet. 2008, 4, e1000155. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Brommer, B.; Tian, X.; Krishnan, A.; Meer, M.; Wang, C.; Vera, D.L.; Zeng, Q.; Yu, D.; Bonkowski, M.S.; et al. Reprogramming to recover youthful epigenetic information and restore vision. Nature 2020, 588, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Ocampo, A.; Reddy, P.; Martinez-Redondo, P.; Platero-Luengo, A.; Hatanaka, F.; Hishida, T.; Li, M.; Lam, D.; Kurita, M.; Beyret, E.; et al. In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming. Cell 2016, 167, 1719–1733.e12. [Google Scholar] [CrossRef] [PubMed]
- Sidler, C.; Kovalchuk, O.; Kovalchuk, I. Epigenetic Regulation of Cellular Senescence and Aging. Front. Genet. 2017, 8, 138. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Luo, M.; Jeong, M.; Rodriguez, B.; Xia, Z.; Hannah, R.; Wang, H.; Le, T.; Faull, K.F.; Chen, R.; et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell 2014, 14, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Li, C.L.; Pu, M.; Wang, W.; Chaturbedi, A.; Emerson, F.J.; Lee, S.S. Region-specific H3K9me3 gain in aged somatic tissues in Caenorhabditis elegans. PLoS Genet. 2021, 17, e1009432. [Google Scholar] [CrossRef] [PubMed]
- Kushwaha, A.; Thakur, M.K. Increase in hippocampal histone H3K9me3 is negatively correlated with memory in old male mice. Biogerontology 2020, 21, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Sarg, B.; Koutzamani, E.; Helliger, W.; Rundquist, I.; Lindner, H.H. Postsynthetic trimethylation of histone H4 at lysine 20 in mammalian tissues is associated with aging. J. Biol. Chem. 2002, 277, 39195–39201. [Google Scholar] [CrossRef]
- O’Sullivan, R.J.; Kubicek, S.; Schreiber, S.L.; Karlseder, J. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat. Struct. Mol. Biol. 2010, 17, 1218–1225. [Google Scholar] [CrossRef]
- Ni, Z.; Ebata, A.; Alipanahiramandi, E.; Lee, S.S. Two SET domain containing genes link epigenetic changes and aging in Caenorhabditis elegans. Aging Cell 2012, 11, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kim, E.W.; Croteau, D.L.; Bohr, V.A. Heterochromatin: An epigenetic point of view in aging. Exp. Mol. Med. 2020, 52, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.P.; Donahue, G.; Otte, G.L.; Capell, B.C.; Nelson, D.M.; Cao, K.; Aggarwala, V.; Cruickshanks, H.A.; Rai, T.S.; McBryan, T.; et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.; Pawlikowski, J.; Manoharan, I.; van Tuyn, J.; Nelson, D.M.; Rai, T.S.; Shah, P.P.; Hewitt, G.; Korolchuk, V.I.; Passos, J.F.; et al. Lysosome-mediated processing of chromatin in senescence. J. Cell Biol. 2013, 202, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Santos-Rosa, H.; Schneider, R.; Bannister, A.J.; Sherriff, J.; Bernstein, B.E.; Emre, N.C.; Schreiber, S.L.; Mellor, J.; Kouzarides, T. Active genes are tri-methylated at K4 of histone H3. Nature 2002, 419, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Maures, T.J.; Hauswirth, A.G.; Green, E.M.; Leeman, D.S.; Maro, G.S.; Han, S.; Banko, M.R.; Gozani, O.; Brunet, A. Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans. Nature 2010, 466, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Jauregui-Lozano, J.; McGovern, S.E.; Bakhle, K.M.; Hagins, A.C.; Weake, V.M. Establishing the contribution of active histone methylation marks to the aging transcriptional landscape of Drosophila photoreceptors. Sci. Rep. 2023, 13, 5105. [Google Scholar] [CrossRef] [PubMed]
- Tsurumi, A.; Li, W.X. Global heterochromatin loss: A unifying theory of aging? Epigenetics 2012, 7, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Rai, K.; Jafri, I.F.; Chidester, S.; James, S.R.; Karpf, A.R.; Cairns, B.R.; Jones, D.A. Dnmt3 and G9a cooperate for tissue-specific development in zebrafish. J. Biol. Chem. 2010, 285, 4110–4121. [Google Scholar] [CrossRef]
- Sarkar, T.J.; Quarta, M.; Mukherjee, S.; Colville, A.; Paine, P.; Doan, L.; Tran, C.M.; Chu, C.R.; Horvath, S.; Qi, L.S.; et al. Transient non-integrative expression of nuclear reprogramming factors promotes multifaceted amelioration of aging in human cells. Nat. Commun. 2020, 11, 1545. [Google Scholar] [CrossRef]
- Dozmorov, M.G. Polycomb repressive complex 2 epigenomic signature defines age-associated hypermethylation and gene expression changes. Epigenetics 2015, 10, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Guillermo, A.R.R.; Chocian, K.; Gavriilidis, G.; Vandamme, J.; Salcini, A.E.; Mellor, J.; Woollard, A. H3K27 modifiers regulate lifespan in C. elegans in a context-dependent manner. BMC Biol. 2021, 19, 59. [Google Scholar] [CrossRef] [PubMed]
- Dang, W.W.; Sen, P.; Dai, J.B.; Kaeberlein, M.; Kennedy, B.; Boeke, J.D.; Berger, S. Histone Mutant Lifespan Screen Reveals That the H3k36me3 Promotes Longevity by Suppressing Intragenic Cryptic Transcription. Gerontologist 2015, 55. [Google Scholar]
- Tie, G.; Yan, J.; Khair, L.; Tutto, A.; Messina, L.M. Hypercholesterolemia Accelerates the Aging Phenotypes of Hematopoietic Stem Cells by a Tet1-Dependent Pathway. Sci. Rep. 2020, 10, 3567. [Google Scholar] [CrossRef]
- Dang, W.; Steffen, K.K.; Perry, R.; Dorsey, J.A.; Johnson, F.B.; Shilatifard, A.; Kaeberlein, M.; Kennedy, B.K.; Berger, S.L. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 2009, 459, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Dubey, S.K.; Dubey, R.; Prajapati, S.C.; Jung, K.; Mohan, K.; Liu, X.; Roney, J.; Tian, W.; Abney, J.; Giarmarco, M.M.; et al. Histone deficiency and hypoacetylation in the aging retinal pigment epithelium. Aging Cell 2024, 23, e14108. [Google Scholar] [CrossRef] [PubMed]
- Dube, C.T.; Jahan, F.R.S.; Lim, C.Y. Key changes in chromatin mark mammalian epidermal differentiation and ageing. Epigenetics 2022, 17, 444–459. [Google Scholar] [CrossRef] [PubMed]
- Vahabikashi, A.; Adam, S.A.; Medalia, O.; Goldman, R.D. Nuclear lamins: Structure and function in mechanobiology. APL Bioeng. 2022, 6, 011503. [Google Scholar] [CrossRef]
- Prokocimer, M.; Davidovich, M.; Nissim-Rafinia, M.; Wiesel-Motiuk, N.; Bar, D.Z.; Barkan, R.; Meshorer, E.; Gruenbaum, Y. Nuclear lamins: Key regulators of nuclear structure and activities. J. Cell. Mol. Med. 2009, 13, 1059–1085. [Google Scholar] [CrossRef]
- Fragoso-Luna, A.; Askjaer, P. The Nuclear Envelope in Ageing and Progeria. Subcell. Biochem. 2023, 102, 53–75. [Google Scholar] [CrossRef]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef] [PubMed]
- De Sandre-Giovannoli, A.; Levy, N. Altered splicing in prelamin A-associated premature aging phenotypes. Prog. Mol. Subcell. Biol. 2006, 44, 199–232. [Google Scholar] [CrossRef] [PubMed]
- Capell, B.C.; Erdos, M.R.; Madigan, J.P.; Fiordalisi, J.J.; Varga, R.; Conneely, K.N.; Gordon, L.B.; Der, C.J.; Cox, A.D.; Collins, F.S. Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2005, 102, 12879–12884. [Google Scholar] [CrossRef] [PubMed]
- Burla, R.; La Torre, M.; Merigliano, C.; Verni, F.; Saggio, I. Genomic instability and DNA replication defects in progeroid syndromes. Nucleus 2018, 9, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Hitzert, M.M.; van der Crabben, S.N.; Baldewsingh, G.; van Amstel, H.K.P.; van den Wijngaard, A.; van Ravenswaaij-Arts, C.M.A.; Zijlmans, C.W.R. Mandibuloacral dysplasia type B (MADB): A cohort of eight patients from Suriname with a homozygous founder mutation in ZMPSTE24 (FACE1), clinical diagnostic criteria and management guidelines. Orphanet J. Rare Dis. 2019, 14, 294. [Google Scholar] [CrossRef] [PubMed]
- Shumaker, D.K.; Dechat, T.; Kohlmaier, A.; Adam, S.A.; Bozovsky, M.R.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Khuon, S.; Collins, F.S.; et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc. Natl. Acad. Sci. USA 2006, 103, 8703–8708. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, Z.; Zhang, L.; Ghosh, S.; Zheng, H.; Zhou, Z. Depleting the methyltransferase Suv39h1 improves DNA repair and extends lifespan in a progeria mouse model. Nat. Commun. 2013, 4, 1868. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, J.; Suzuki, K.; Qu, J.; Wang, P.; Zhou, J.; Liu, X.; Ren, R.; Xu, X.; Ocampo, A.; et al. Aging stem cells. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 2015, 348, 1160–1163. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zhang, W.; Song, M.; Wang, W.; Wei, G.; Li, W.; Lei, J.; Huang, Y.; Sang, Y.; Chan, P.; et al. Differential stem cell aging kinetics in Hutchinson-Gilford progeria syndrome and Werner syndrome. Protein Cell 2018, 9, 333–350. [Google Scholar] [CrossRef]
- Chojnowski, A.; Ong, P.F.; Foo, M.X.R.; Liebl, D.; Hor, L.P.; Stewart, C.L.; Dreesen, O. Heterochromatin loss as a determinant of progerin-induced DNA damage in Hutchinson-Gilford Progeria. Aging Cell 2020, 19, e13108. [Google Scholar] [CrossRef]
- McCord, R.P.; Nazario-Toole, A.; Zhang, H.; Chines, P.S.; Zhan, Y.; Erdos, M.R.; Collins, F.S.; Dekker, J.; Cao, K. Correlated alterations in genome organization, histone methylation, and DNA-lamin A/C interactions in Hutchinson-Gilford progeria syndrome. Genome Res. 2013, 23, 260–269. [Google Scholar] [CrossRef]
- Kohler, F.; Bormann, F.; Raddatz, G.; Gutekunst, J.; Corless, S.; Musch, T.; Lonsdorf, A.S.; Erhardt, S.; Lyko, F.; Rodriguez-Paredes, M. Epigenetic deregulation of lamina-associated domains in Hutchinson-Gilford progeria syndrome. Genome Med. 2020, 12, 46. [Google Scholar] [CrossRef] [PubMed]
- Chojnowski, A.; Ong, P.F.; Wong, E.S.; Lim, J.S.; Mutalif, R.A.; Navasankari, R.; Dutta, B.; Yang, H.; Liow, Y.Y.; Sze, S.K.; et al. Progerin reduces LAP2alpha-telomere association in Hutchinson-Gilford progeria. eLife 2015, 4, e07759. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Chow, M.Z.; Wang, Z.; Zhang, L.; Liu, B.; Liu, X.; Zhou, Z. Histone H4 lysine 16 hypoacetylation is associated with defective DNA repair and premature senescence in Zmpste24-deficient mice. Proc. Natl. Acad. Sci. USA 2011, 108, 12325–12330. [Google Scholar] [CrossRef]
- Kurup, J.T.; Han, Z.; Jin, W.; Kidder, B.L. H4K20me3 methyltransferase SUV420H2 shapes the chromatin landscape of pluripotent embryonic stem cells. Development 2020, 147. [Google Scholar] [CrossRef] [PubMed]
- González, J.; Bosch-Presegué, L.; Marazuela-Duque, A.; Guitart-Solanes, A.; Espinosa-Alcantud, M.; Fernandez, A.F.; Brown, J.P.; Ausió, J.; Vazquez, B.N.; Singh, P.B.; et al. A complex interplay between H2A.Z and HP1 isoforms regulates pericentric heterochromatin. Front. Cell Dev. Biol. 2023, 11, 1293122. [Google Scholar] [CrossRef]
- Kim, B.H.; Chung, Y.H.; Woo, T.G.; Kang, S.M.; Park, S.; Park, B.J. Progerin, an Aberrant Spliced Form of Lamin A, Is a Potential Therapeutic Target for HGPS. Cells 2023, 12, 2299. [Google Scholar] [CrossRef]
- Kychygina, A.; Dall’Osto, M.; Allen, J.A.M.; Cadoret, J.C.; Piras, V.; Pickett, H.A.; Crabbe, L. Progerin impairs 3D genome organization and induces fragile telomeres by limiting the dNTP pools. Sci. Rep. 2021, 11, 13195. [Google Scholar] [CrossRef]
- Yamagishi, M.; Kuze, Y.; Kobayashi, S.; Nakashima, M.; Morishima, S.; Kawamata, T.; Makiyama, J.; Suzuki, K.; Seki, M.; Abe, K.; et al. Mechanisms of action and resistance in histone methylation-targeted therapy. Nature 2024, 627, 221–228. [Google Scholar] [CrossRef]
- Ragnauth, C.D.; Warren, D.T.; Liu, Y.; McNair, R.; Tajsic, T.; Figg, N.; Shroff, R.; Skepper, J.; Shanahan, C.M. Prelamin A acts to accelerate smooth muscle cell senescence and is a novel biomarker of human vascular aging. Circulation 2010, 121, 2200–2210. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T. Lamin A-dependent nuclear defects in human aging. Science 2006, 312, 1059–1063. [Google Scholar] [CrossRef] [PubMed]
- Olive, M.; Harten, I.; Mitchell, R.; Beers, J.K.; Djabali, K.; Cao, K.; Erdos, M.R.; Blair, C.; Funke, B.; Smoot, L.; et al. Cardiovascular pathology in Hutchinson-Gilford progeria: Correlation with the vascular pathology of aging. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2301–2309. [Google Scholar] [CrossRef] [PubMed]
- McClintock, D.; Ratner, D.; Lokuge, M.; Owens, D.M.; Gordon, L.B.; Collins, F.S.; Djabali, K. The mutant form of lamin A that causes Hutchinson-Gilford progeria is a biomarker of cellular aging in human skin. PLoS ONE 2007, 2, e1269. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, R.; Irfan, M.; Gondal, T.M.; Khan, S.; Wu, J.; Hadi, M.U.; Heymach, J.; Le, X.; Yan, H.; Alam, T. AI in drug discovery and its clinical relevance. Heliyon 2023, 9, e17575. [Google Scholar] [CrossRef] [PubMed]
- Rauschert, S.; Raubenheimer, K.; Melton, P.E.; Huang, R.C. Machine learning and clinical epigenetics: A review of challenges for diagnosis and classification. Clin. Epigenet. 2020, 12, 51. [Google Scholar] [CrossRef]
- Xia, B.; Zhao, D.; Wang, G.; Zhang, M.; Lv, J.; Tomoiaga, A.S.; Li, Y.; Wang, X.; Meng, S.; Cooke, J.P.; et al. Machine learning uncovers cell identity regulator by histone code. Nat. Commun. 2020, 11, 2696. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, R.L.; Daniels, E.G.; Molenaars, M.; Houtkooper, R.H.; Janssens, G.E. From molecular promise to preclinical results: HDAC inhibitors in the race for healthy aging drugs. EMBO Mol. Med. 2019, 11, e9854. [Google Scholar] [CrossRef]
- Yi, S.J.; Kim, K. New Insights into the Role of Histone Changes in Aging. Int. J. Mol. Sci. 2020, 21, 8241. [Google Scholar] [CrossRef]
- Fornelli, C.; Sofia Cento, A.; Nevi, L.; Mastrocola, R.; Ferreira Alves, G.; Caretti, G.; Collino, M.; Penna, F. The BET inhibitor JQ1 targets fat metabolism and counteracts obesity. J. Adv. Res. 2024, in press. [Google Scholar] [CrossRef]
- Lopez, M.; Halby, L.; Arimondo, P.B. DNA Methyltransferase Inhibitors: Development and Applications. Adv. Exp. Med. Biol. 2016, 945, 431–473. [Google Scholar] [CrossRef]
- Soto-Palma, C.; Niedernhofer, L.J.; Faulk, C.D.; Dong, X. Epigenetics, DNA damage, and aging. J. Clin. Investig. 2022, 132, e158446. [Google Scholar] [CrossRef]
- Pallauf, K.; Rimbach, G.; Rupp, P.M.; Chin, D.; Wolf, I.M. Resveratrol and Lifespan in Model Organisms. Curr. Med. Chem. 2016, 23, 4639–4680. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Ren, Z.J.; Tang, J.H. MicroRNA-34a: A potential therapeutic target in human cancer. Cell Death Dis. 2014, 5, e1327. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Liu, S.; Xu, S. Identification of hub genes, key pathways, and therapeutic agents in Hutchinson-Gilford Progeria syndrome using bioinformatics analysis. Medicine 2020, 99, e19022. [Google Scholar] [CrossRef] [PubMed]
- Vaiserman, A.; Krasnienkov, D. Telomere Length as a Marker of Biological Age: State-of-the-Art, Open Issues, and Future Perspectives. Front. Genet. 2020, 11, 630186. [Google Scholar] [CrossRef] [PubMed]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef]
- Cole, J.H.; Ritchie, S.J.; Bastin, M.E.; Valdes Hernandez, M.C.; Munoz Maniega, S.; Royle, N.; Corley, J.; Pattie, A.; Harris, S.E.; Zhang, Q.; et al. Brain age predicts mortality. Mol. Psychiatry 2018, 23, 1385–1392. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Shi, D.; Guankai, P.; Tan, Z.; Shang, X.; Hu, W.; Liao, H.; Zhang, X.; Huang, Y.; Yu, H.; et al. Retinal age gap as a predictive biomarker for mortality risk. Br. J. Ophthalmol. 2023, 107, 547–554. [Google Scholar] [CrossRef]
- Xia, X.; Chen, X.; Wu, G.; Li, F.; Wang, Y.; Chen, Y.; Chen, M.; Wang, X.; Chen, W.; Xian, B.; et al. Three-dimensional facial-image analysis to predict heterogeneity of the human ageing rate and the impact of lifestyle. Nat. Metab. 2020, 2, 946–957. [Google Scholar] [CrossRef]
- Wang, J.; Gao, Y.; Wang, F.; Zeng, S.; Li, J.; Miao, H.; Wang, T.; Zeng, J.; Baptista-Hon, D.; Monteiro, O.; et al. Accurate estimation of biological age and its application in disease prediction using a multimodal image Transformer system. Proc. Natl. Acad. Sci. USA 2024, 121, e2308812120. [Google Scholar] [CrossRef]
- Sufyan, M.; Shokat, Z.; Ashfaq, U.A. Artificial intelligence in cancer diagnosis and therapy: Current status and future perspective. Comput. Biol. Med. 2023, 165, 107356. [Google Scholar] [CrossRef] [PubMed]
- Partin, A.; Brettin, T.S.; Zhu, Y.; Narykov, O.; Clyde, A.; Overbeek, J.; Stevens, R.L. Deep learning methods for drug response prediction in cancer: Predominant and emerging trends. Front. Med. 2023, 10, 1086097. [Google Scholar] [CrossRef] [PubMed]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Arfi, S.; Srivastava, N.; Sharma, N. Artificial Intelligence: An Emerging Intellectual Sword for Battling Carcinomas. Curr. Pharm. Biotechnol. 2023, 24, 1784–1794. [Google Scholar] [CrossRef] [PubMed]
- Tansey, W.; Li, K.; Zhang, H.; Linderman, S.W.; Rabadan, R.; Blei, D.M.; Wiggins, C.H. Dose-response modeling in high-throughput cancer drug screenings: An end-to-end approach. Biostatistics 2022, 23, 643–665. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.C.; Chen, H.H.; Gorthi, A.; Mostavi, M.; Zheng, S.; Huang, Y.; Chen, Y. Deep learning of pharmacogenomics resources: Moving towards precision oncology. Brief. Bioinform. 2020, 21, 2066–2083. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Kobayashi, K.; Wakisaka, Y.; Deng, D.; Tanaka, S.; Huang, C.J.; Lei, C.; Sun, C.W.; Liu, H.; Fujiwaki, Y.; et al. Label-free chemical imaging flow cytometry by high-speed multicolor stimulated Raman scattering. Proc. Natl. Acad. Sci. USA 2019, 116, 15842–15848. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Nguyen, G.T.T.; Nguyen, T.; Le, D.H. Graph Convolutional Networks for Drug Response Prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2022, 19, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Li, H.; Li, S.; Leung, K.S. Improving prediction of phenotypic drug response on cancer cell lines using deep convolutional network. BMC Bioinform. 2019, 20, 408. [Google Scholar] [CrossRef]
- Schneider, G.; Schrodl, W.; Wallukat, G.; Muller, J.; Nissen, E.; Ronspeck, W.; Wrede, P.; Kunze, R. Peptide design by artificial neural networks and computer-based evolutionary search. Proc. Natl. Acad. Sci. USA 1998, 95, 12179–12184. [Google Scholar] [CrossRef]
- Sarkar, C.; Das, B.; Rawat, V.S.; Wahlang, J.B.; Nongpiur, A.; Tiewsoh, I.; Lyngdoh, N.M.; Das, D.; Bidarolli, M.; Sony, H.T. Artificial Intelligence and Machine Learning Technology Driven Modern Drug Discovery and Development. Int. J. Mol. Sci. 2023, 24, 2026. [Google Scholar] [CrossRef] [PubMed]
- Arora, I.; Tollefsbol, T.O. Computational methods and next-generation sequencing approaches to analyze epigenetics data: Profiling of methods and applications. Methods 2021, 187, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Chen, S.; Cui, X.; Chen, X.; Gao, Z.; Jiang, R. SilencerDB: A comprehensive database of silencers. Nucleic Acids Res. 2021, 49, D221–D228. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Ovcharenko, I. Enhancer-silencer transitions in the human genome. Genome Res. 2022, 32, 437–448. [Google Scholar] [CrossRef]
- Ghandi, M.; Mohammad-Noori, M.; Ghareghani, N.; Lee, D.; Garraway, L.; Beer, M.A. gkmSVM: An R package for gapped-kmer SVM. Bioinformatics 2016, 32, 2205–2207. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Li, L.; Sun, H.; Xu, D.; Wang, G. DeepICSH: A complex deep learning framework for identifying cell-specific silencers and their strength from the human genome. Brief. Bioinform. 2023, 24, bbad316. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Quan, L.; Zhou, Y.; Jiang, Y.; Li, K.; Wu, T.; Lyu, Q. Identifying modifications on DNA-bound histones with joint deep learning of multiple binding sites in DNA sequence. Bioinformatics 2022, 38, 4070–4077. [Google Scholar] [CrossRef] [PubMed]
- Tavares, G.C.; Pereira, F.L.; Barony, G.M.; Rezende, C.P.; da Silva, W.M.; de Souza, G.; Verano-Braga, T.; de Carvalho Azevedo, V.A.; Leal, C.A.G.; Figueiredo, H.C.P. Delineation of the pan-proteome of fish-pathogenic Streptococcus agalactiae strains using a label-free shotgun approach. BMC Genom. 2019, 20, 11. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; He, X.; Jing, J. Overview of Artificial Intelligence in Breast Cancer Medical Imaging. J. Clin. Med. 2023, 12, 419. [Google Scholar] [CrossRef]
- Schopf, C.M.; Ramwala, O.A.; Lowry, K.P.; Hofvind, S.; Marinovich, M.L.; Houssami, N.; Elmore, J.G.; Dontchos, B.N.; Lee, J.M.; Lee, C.I. Artificial Intelligence-Driven Mammography-Based Future Breast Cancer Risk Prediction: A Systematic Review. J. Am. Coll. Radiol. 2024, 21, 319–328. [Google Scholar] [CrossRef]
- Nassif, A.B.; Talib, M.A.; Nasir, Q.; Afadar, Y.; Elgendy, O. Breast cancer detection using artificial intelligence techniques: A systematic literature review. Artif. Intell. Med. 2022, 127, 102276. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Ali, M.; Naseem, U.; Nezhadmoghadam, F.; Jatoi, M.A.; Gulliver, T.A.; Tamez-Pena, J.G. Breast cancer risk prediction using machine learning: A systematic review. Front. Oncol. 2024, 14, 1343627. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.; Zhao, Z.; Chen, X.F.; Sauer, J.; Saraf, S.A.; Jialdasani, R.; Taghipour, K.; Sathe, A.; Khor, L.Y.; Lim, K.H.; et al. A promising deep learning-assistive algorithm for histopathological screening of colorectal cancer. Sci. Rep. 2022, 12, 2222. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Tang, R.S.Y.; Lam, T.Y.T.; Zhao, G.; Lau, J.Y.W.; Liu, Y.; Wu, Q.; Rong, L.; Xu, W.; Li, X.; et al. Artificial Intelligence-Assisted Colonoscopy for Colorectal Cancer Screening: A Multicenter Randomized Controlled Trial. Clin. Gastroenterol. Hepatol. 2023, 21, 337–346.e3. [Google Scholar] [CrossRef]
- Qiu, H.; Ding, S.; Liu, J.; Wang, L.; Wang, X. Applications of Artificial Intelligence in Screening, Diagnosis, Treatment, and Prognosis of Colorectal Cancer. Curr. Oncol. 2022, 29, 1773–1795. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Histone | Function | Citations |
|---|---|---|
| H2A.Z | Active transcription, also found in premature aging. | [4,5,6] |
| H2A.X | Early response to double-strand breaks. | [7,8,9,10] |
| H3K4me3 | Involved in transcription initiation, elongation and RNA splicing. Typically located at the surrounding euchromatic regions. | [11,12] |
| H3K9me2 | Evolutionarily conserved mark of peripheral heterochromatin. Marker of heterochromatin at the nuclear periphery. Involved in the reassembly of the nuclear lamina after cellular mitosis. | [13,14] |
| H3K9me3 | Hallmark of constitutive heterochromatin, gene silencing, epigenetic inheritance, heterochromatin assembly. | [15,16,17,18] |
| H3K27me3 | Hallmark of facultative heterochromatin, transcription repression. Maintenance of transcriptional silencing throughout cell divisions. | [19,20] |
| H3K36me3 | Regulate life span and active transcription. | [21,22,23] |
| H3K56ac | Genomic stability, chromosome segregation and cell division. | [24,25,26] |
| H4K20me1 H4K20me2 | Associated with transcriptional activation. | [27,28] |
| H4K20me3 | Transcription repression. | [27,29] |
| H4K5ac | Rapid transcription and bookmarking, memory formation | [30,31] |
| H4K8ac | Marker of transcriptionally active regions. Enriched in neural cells of the brain exposed to early life exercise. | [27,32,33] |
| H4K12ac | Associated with learning and memory, active transcription on estrogen-induced genes. | [27,34,35] |
| H4K16ac | Associated with transcription, DNA repair, active chromatin landscape | [36,37,38] |
| Histone | Function | Citations |
|---|---|---|
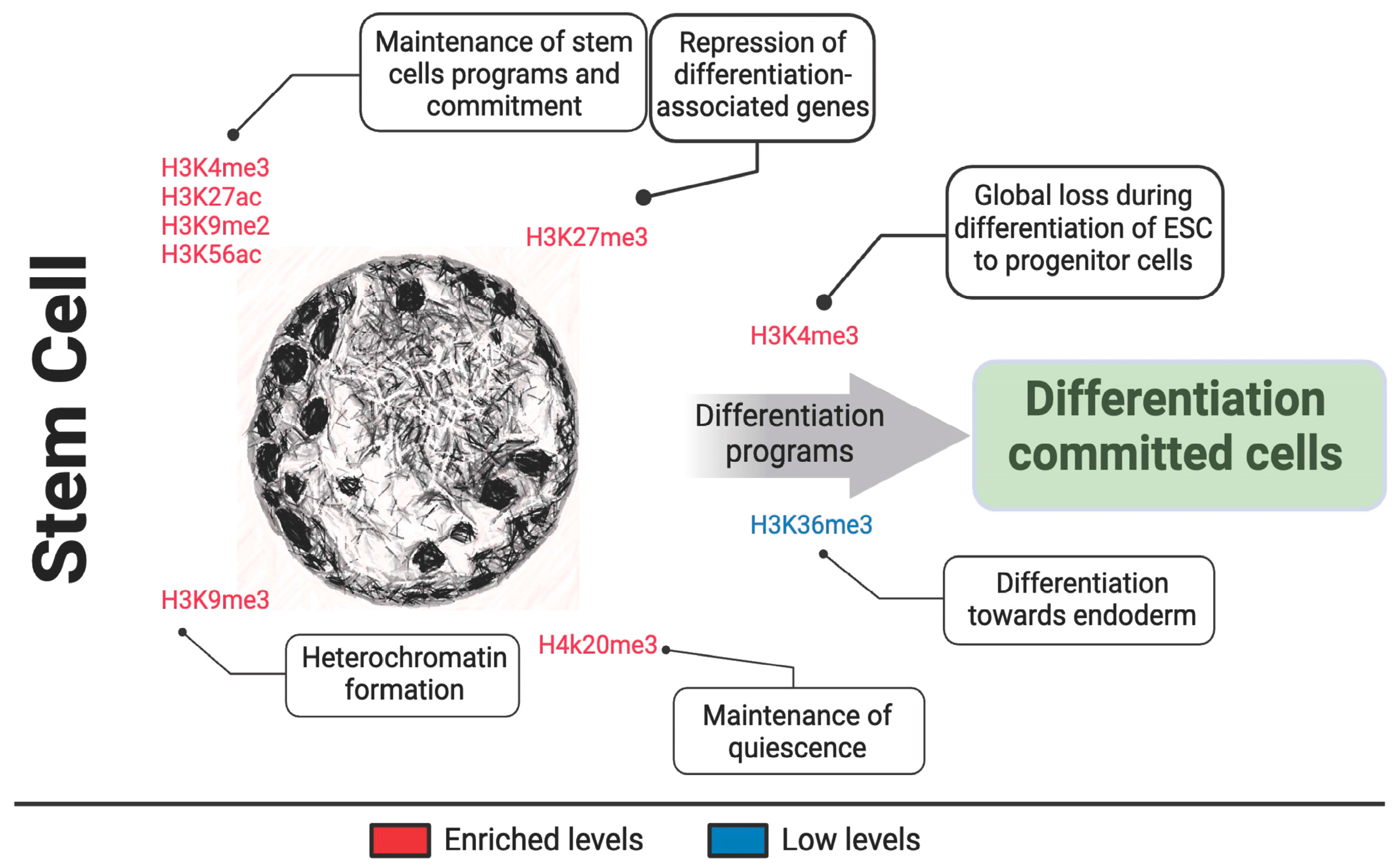
| H3K4me3 H3K27ac | Maintenance of stem cell pluripotency, activated during embryonic stem cell development, global loss during differentiation of ESC to progenitor cells | [83,84,89,90,91,92,93] |
| H3K9me2 | Maintenance of stem cell identity | [89,94] |
| K3K9me3 | Heterochromatin formation, inhibited during embryonic stem cell development | [91,92] |
| H3K27me3 | Loss during stem cell line commitment, inhibited during embryonic stem cell development, repression and silencing of genes associated with differentiation in stem cells. | [85,86,90,91,92,95] |
| H3K36me3 | Embryonic stem cell differentiation towards endoderm. | [96] |
| H3K56ac | Human core transcriptional network of pluripotency | [97] |
| H4K20me3 | Maintenance of stem cell self-renew | [98] |
| Histone | Function | Citations |
|---|---|---|
| H3K4me3 | Linked to tumorigenesis, maintenance of leukemia stem cell; associated with poor prognosis of hepatocellular carcinomas | [113,114,115,116,117,118,119,120] |
| H3K9me2 | Repressive marks; found to safeguard cancer cells from interferon pathway; found highly methylated in hepatocellular carcinomas; prevent carcinogens in normal cells. | [114,121,122,123,124] |
| H3K9me3 | High methylation is associated with poor prognosis of hepatocellular carcinomas; prevent carcinogens in normal cells. | [123,124] |
| H3K27me3 | Mixed function: Reduced in breast, colorectal and nasopharyngeal cancers; elevated in B-cell lymphoma (EZH2Y641F/N). | [108,125,126,127] |
| H3K27M | Hotspots mutations to the unstructured N-terminal tail of histone H3 in pediatric high-grade gliomas; lead to global reduction of H3K27me3. | [128,129,130,131] |
| H3K36M | Mutated in glioblastoma, induces formation of sarcomas, found prevalent in chondroblastomas, leads to global reduction of H3K36me2/3. | [131,132,133,134] |
| H3K79me | Active in Mixed Lineage Leukemia (MML-ENL) and hypomethylated in leukemia stem cells. | [113,135,136] |
| H4K16ac | Loss is a common hallmark of cancer; reduced levels in breast cancer. | [137,138] |
| H4K20me2 | Reduction levels in renal cell cancer. | [138,139,140] |
| H4K20me3 | Reduction levels in renal cell cancer; oncogene-induced senescence-associated proliferation arrest and tumor suppression function; poor prognosis of hepatocellular carcinoma; reduced levels in breast cancer. | [138,139,140,141,142,143] |
| Histone | Function | Citations |
|---|---|---|
| H3K4me3 | Facilitates gene expression in aging cells, globally decrease across all actively expressed genes with age. | [119,165,166,167] |
| H3K9me2 | Hallmark of inactive euchromatin, decrease in aging | [168,169] |
| H3K9me3 | Hallmark of constitutive heterochromatin, decrease in aging | [168,169,170] |
| H3K27me3 | Cell-specific heterochromatin regions, regulation of lifespan. | [107,171,172] |
| H3K36me3 | Promotes longevity, mutation reduces life span, globally decrease across all actively expressed genes with age, mark of transcribed regions. | [22,167,173,174] |
| H3K56ac | Mutation reduces life span | [160,170,175] |
| H4K16ac | Mutation reduces life span, hypoacetylation in human retinal pigment epithelium, hypoacetylated in aged epidermal basal cells. | [175,176,177] |
| H4K20me3 | Repression is associated with cellular senescence and is involved in the control of cell senescence. | [143,155,159,160] |
| Histone | Function | Citations |
|---|---|---|
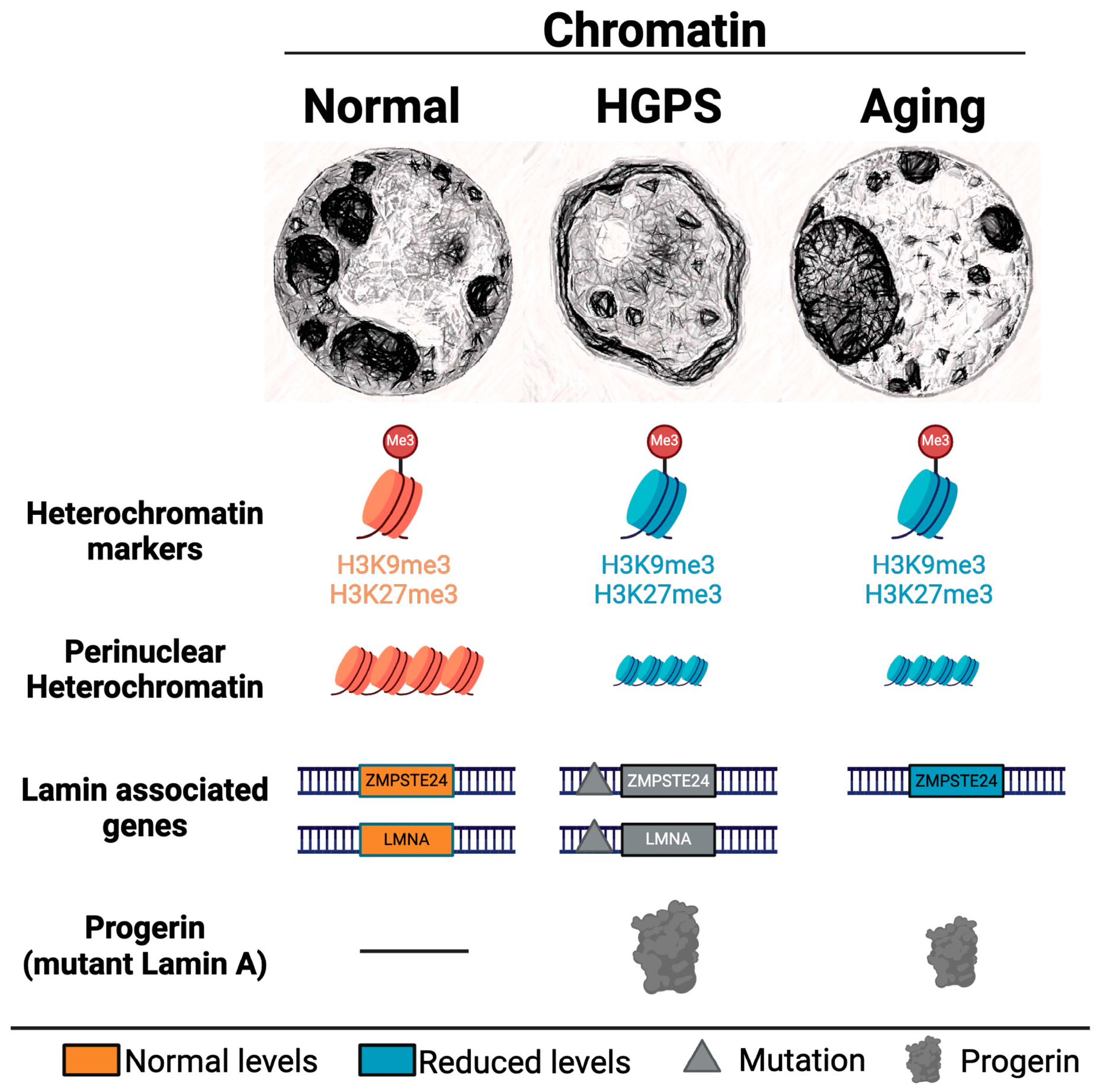
| H3K9me3 | Loss in Hutchinson-Gilford progeria syndrome and Werner syndrome. | [157,186,187,188,189,190] |
| H3K27me3 | Loss in Hutchinson-Gilford progeria syndrome and Werner syndrome. | [188,190,191,192,193] |
| H3K36me3 | Underrepresented in Hutchinson-Gilford progeria syndrome. | [192] |
| H4K16ac | Hypoacetylated in Hutchinson-Gilford progeria syndrome | [194] |
| H4K20me3 | Increase in trimethylation in Hutchinson-Gilford progeria syndrome. | [177,186,187,195,196] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castilho, R.M.; Castilho, L.S.; Palomares, B.H.; Squarize, C.H. Determinants of Chromatin Organization in Aging and Cancer—Emerging Opportunities for Epigenetic Therapies and AI Technology. Genes 2024, 15, 710. https://doi.org/10.3390/genes15060710
Castilho RM, Castilho LS, Palomares BH, Squarize CH. Determinants of Chromatin Organization in Aging and Cancer—Emerging Opportunities for Epigenetic Therapies and AI Technology. Genes. 2024; 15(6):710. https://doi.org/10.3390/genes15060710
Chicago/Turabian StyleCastilho, Rogerio M., Leonard S. Castilho, Bruna H. Palomares, and Cristiane H. Squarize. 2024. "Determinants of Chromatin Organization in Aging and Cancer—Emerging Opportunities for Epigenetic Therapies and AI Technology" Genes 15, no. 6: 710. https://doi.org/10.3390/genes15060710
APA StyleCastilho, R. M., Castilho, L. S., Palomares, B. H., & Squarize, C. H. (2024). Determinants of Chromatin Organization in Aging and Cancer—Emerging Opportunities for Epigenetic Therapies and AI Technology. Genes, 15(6), 710. https://doi.org/10.3390/genes15060710