
Complete Organelle Genome of the Desiccation-Tolerant (DT) Moss Tortula atrovirens and Comparative Analysis of the Pottiaceae Family
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling and DNA/RNA Extraction
2.2. Library Construction and Sequencing
2.3. Assembly and Annotation
2.4. IR Boundaries Analysis and Selection of Candidate DNA Barcodes
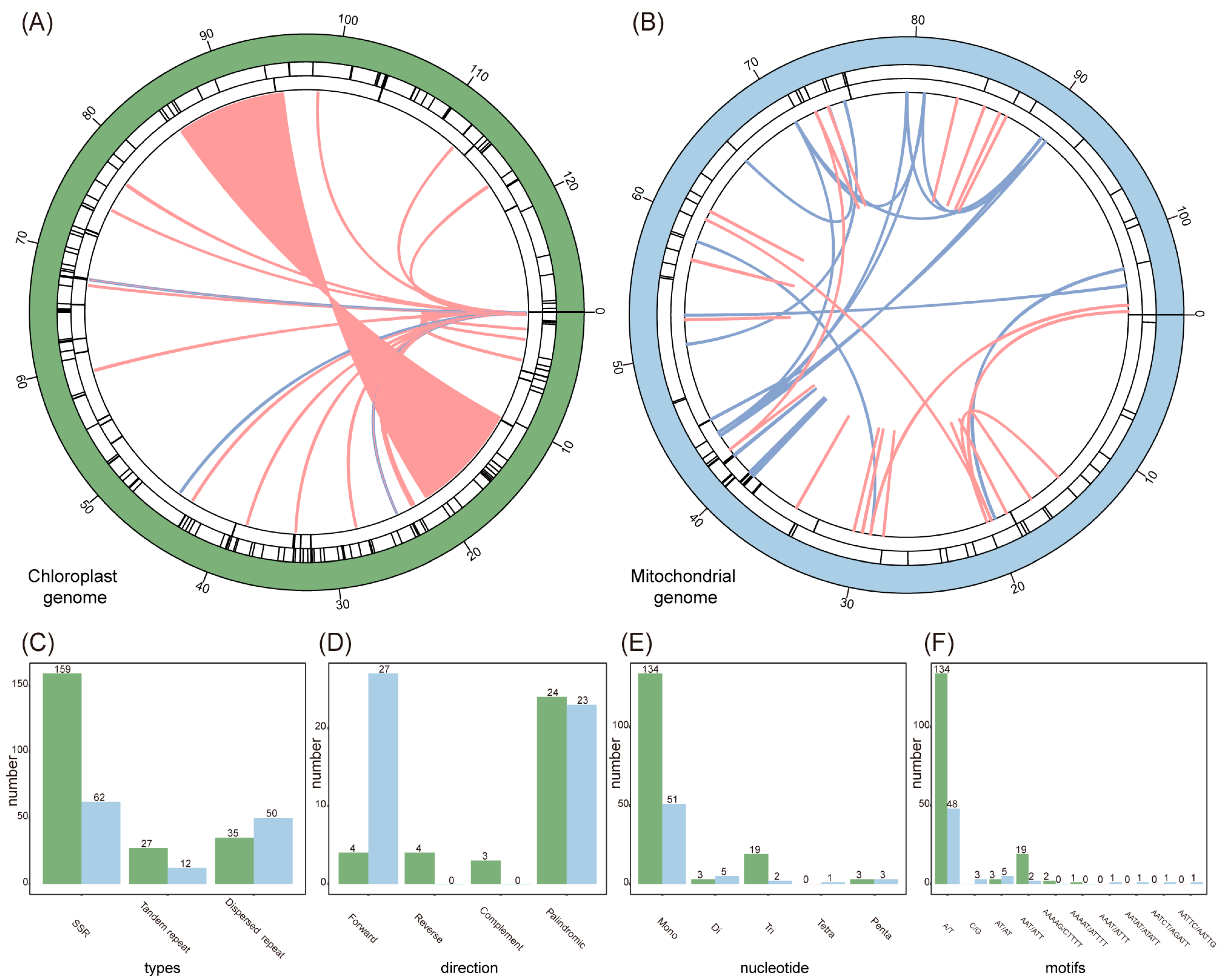
2.5. Repetitive Sequences Analysis
2.6. Codon Usage Preference and RNA Editing Sites Analysis
2.7. Mitochondrial Plastid Sequences Analysis
2.8. Synteny and Selective Pressure Analysis
2.9. Phylogenetic Analyses
3. Results
3.1. Feature of Organelle Genome
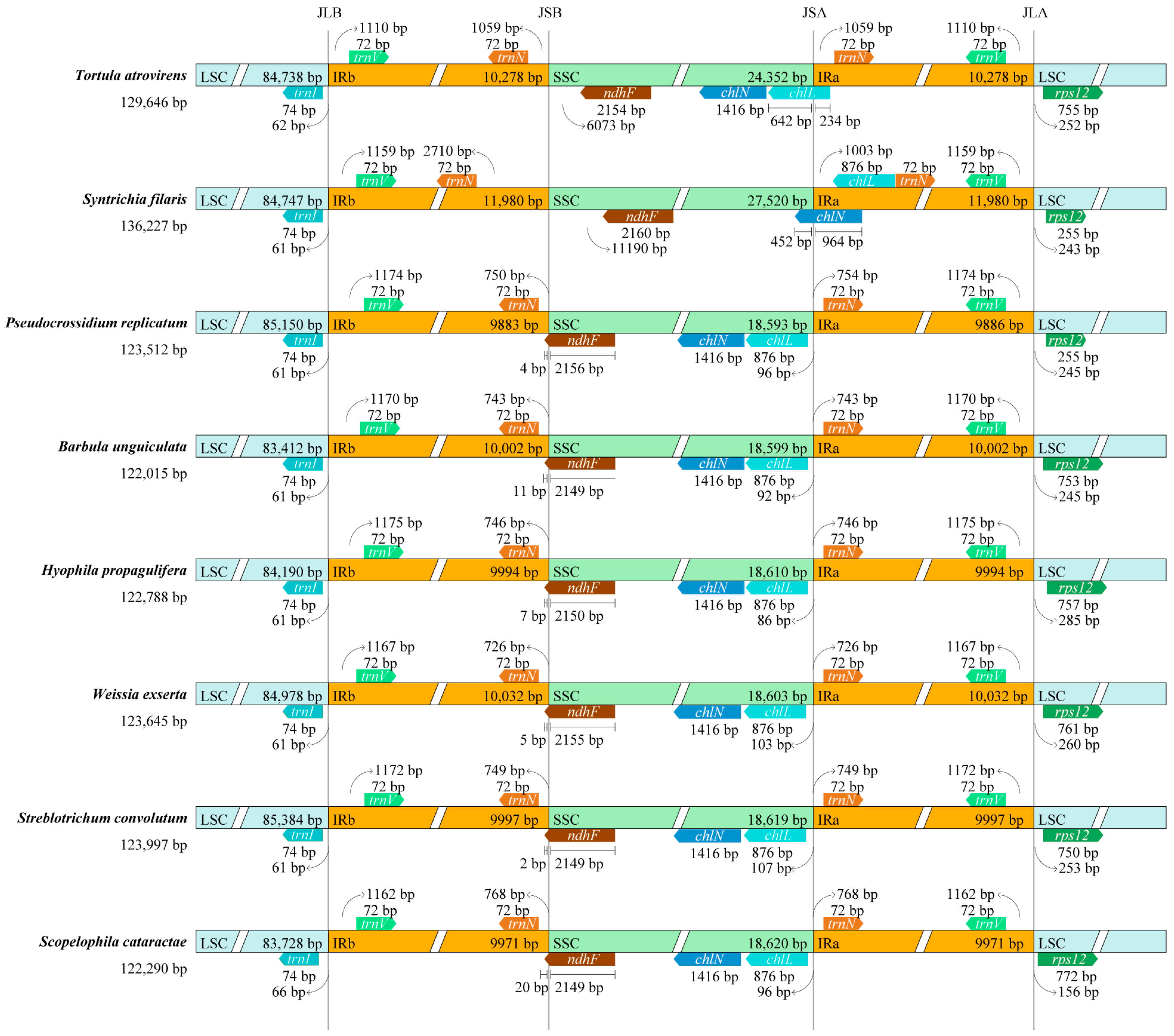
3.2. IR Boundaries Analysis
3.3. Selection of Candidate DNA Barcodes
3.4. Comparative Analysis of Organelle Genomes
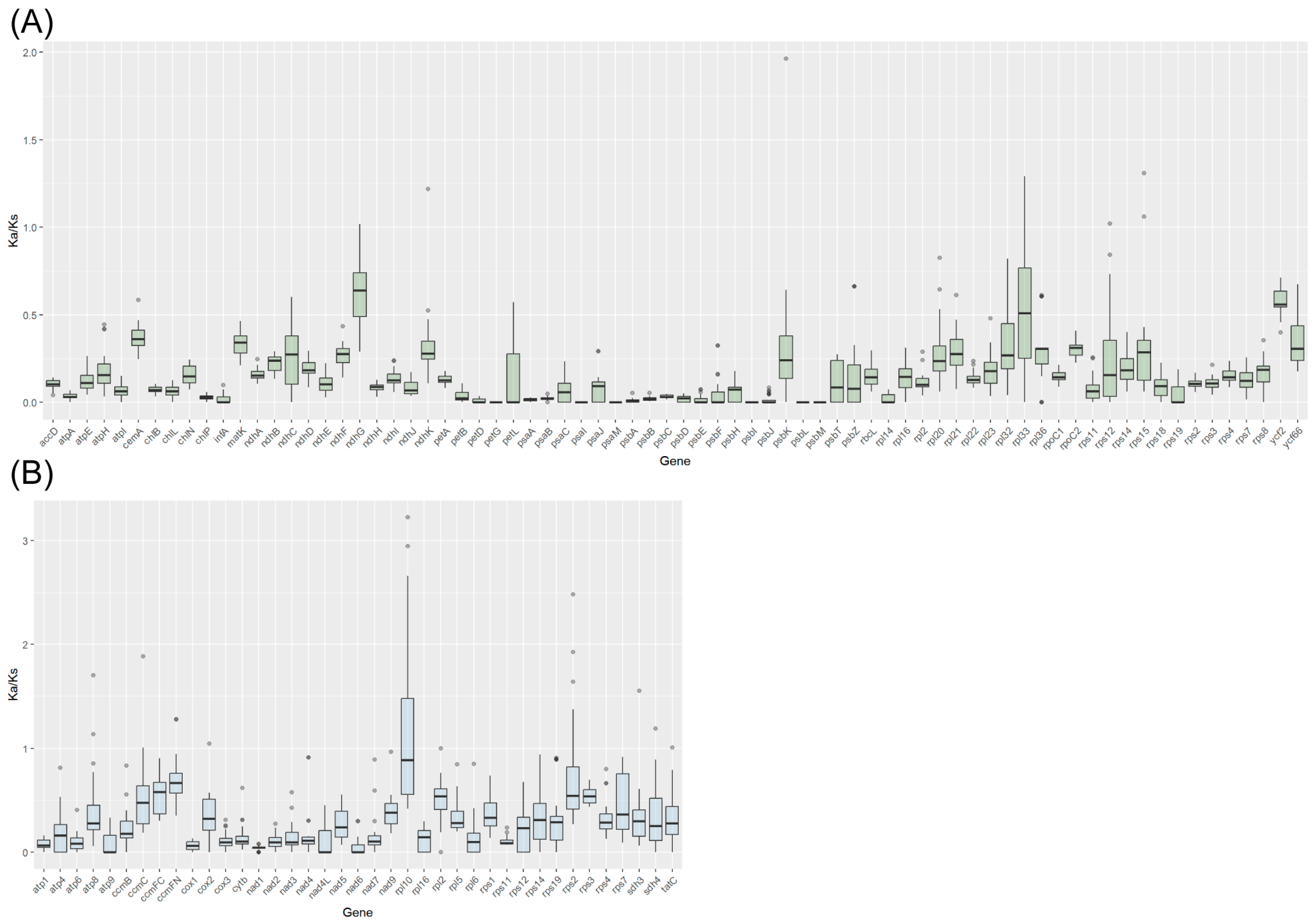
3.5. Synteny and Selective Pressure Analysis
3.6. Phylogenetic Analyses
4. Discussion
4.1. Characterization of T. atrovirens Chloroplast Genome
4.2. Characterization of T. atrovirens Mitochondrial Genome
4.3. Phylogenetic Relationships of Pottiaceae
4.4. Potential DNA Barcoding Regions for Pottiaceae
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Birky, C.W. Uniparental inheritance of mitochondrial and chloroplast genes: Mechanisms and evolution. Proc. Natl. Acad. Sci. USA 1995, 92, 11331–11338. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kan, S.; Liao, X.; Zhou, J.; Tembrock, L.R.; Daniell, H.; Jin, S.; Wu, Z. Plant organellar genomes: Much done, much more to do. Trends Plant Sci. 2024; early view. [Google Scholar] [CrossRef]
- Goffinet, B.; Buck, W.R.; Shaw, A.J. Morphology, anatomy, and classification of the Bryophyta. In Bryophyte Biology, 2nd ed.; Goffinet, B., Shaw, A.J., Eds.; Cambridge University Press: Cambridge, UK, 2009; pp. 55–138. [Google Scholar]
- Asaf, S.; Jan, R.; Asif, S.; Bilal, S.; Khan, A.L.; Kim, K.-M.; Lee, I.-J.; Ahmed, A.-H. Plastome diversity and evolution in mosses: Insights from structural characterization, comparative genomics, and phylogenetic analysis. Int. J. Biol. Macromol. 2024, 257, 128608. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Liu, Y. The mitochondrial genomes of bryophytes. Bryophyt. Divers. Evol. 2021, 43, 112–126. [Google Scholar] [CrossRef]
- Zander, R.H. Macroevolutionary versus molecular analysis: Systematics of the Didymodon segregates Aithobryum, Exobryum and Fuscobryum (Pottiaceae). Hattoria 2019, 10, 1–38. [Google Scholar] [CrossRef]
- Zander, R.H. Genera of the Pottiaceae: Mosses of harsh environments. Bull. Buffalo Soc. Nat. Sci. 1993, 32, 1–378. [Google Scholar] [CrossRef] [PubMed]
- Werner, O.; Ros, R.; Cano, M.J.; Guerra, J. Molecular phylogeny of Pottiaceae (Musci) based on chloroplast rps 4 sequence data. Plant Syst. Evol. 2004, 243, 147–164. [Google Scholar] [CrossRef]
- Alonso, M.; Jiménez, J.A.; Nylinder, S.; Hedenäs, L.; Cano, M.J. Disentangling generic limits in Chionoloma, Oxystegus, Pachyneuropsis and Pseudosymblepharis (Bryophyta: Pottiaceae): An inquiry into their phylogenetic relationships. Taxon 2016, 65, 3–18. [Google Scholar] [CrossRef]
- Cano, M.J.; Jiménez, J.A.; Gallego, M.T.; Guerra, J. A molecular approach to the phylogeny of the moss genus Pseudocrossidium (Pottiaceae, Bryopsida) and its taxonomic implications. J. Syst. Evol. 2022, 60, 914–931. [Google Scholar] [CrossRef]
- Jimenez, J.A.; Cano, M.J.; Guerra, J. A multilocus phylogeny of the moss genus Didymodon and allied genera (Pottiaceae): Generic delimitations and their implications for systematics. J. Syst. Evol. 2022, 60, 281–304. [Google Scholar] [CrossRef]
- Mishler, B.D. Ontogeny and phylogeny in Tortula (Musci: Pottiaceae). Syst. Bot. 1986, 11, 189–208. [Google Scholar] [CrossRef]
- Xu, J.; Bai, X.-L.; Yang, C.; Zhang, P. Study on diversity and binding-sand effect of moss on biotic crusts of fixed dunes. Chin. J. Plant Ecol. 2003, 27, 545–551. [Google Scholar] [CrossRef]
- Oliver, M.J.; Velten, J.; Mishler, B.D. Desiccation tolerance in bryophytes: A reflection of the primitive strategy for plant survival in dehydrating habitats? Integr. Comp. Biol. 2005, 45, 788–799. [Google Scholar] [CrossRef]
- Proctor, M.C.; Ligrone, R.; Duckett, J.G. Desiccation tolerance in the moss Polytrichum formosum: Physiological and fine-structural changes during desiccation and recovery. Ann. Bot. 2007, 99, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Zhang, D.; Li, X.; Yang, H.; Zhang, Y.; Wood, A.J. De novo transcriptome characterization and gene expression profiling of the desiccation tolerant moss Bryum argenteum following rehydration. BMC Genom. 2015, 16, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y. Establishment of Gametophyte Regeneration System of Two Mosses in Wulanhada Volcano. Master’s Thesis, Inner Mongolia University, Hohhot, China, 2021. [Google Scholar]
- Allen, G.C.; Flores-Vergara, M.; Krasynanski, S.; Kumar, S.; Thompson, W. A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat. Protoc. 2006, 1, 2320–2325. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; dePamphilis, C.; Yi, T.; Li, D.-Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Ruan, J.; Li, H. Fast and accurate long-read assembly with wtdbg2. Nat. Methods 2020, 17, 155–158. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Lewis, S.E.; Searle, S.; Harris, N.; Gibson, M.; Iyer, V.; Richter, J.; Wiel, C.; Bayraktaroglu, L.; Birney, E.; Crosby, M. Apollo: A sequence annotation editor. Genome Biol. 2002, 3, research0082.1. [Google Scholar] [CrossRef] [PubMed]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3. 1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.; Wang, G. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.; Pachter, L.; Poliakov, A.; Rubin, E.; Dubchak, I. VISTA: Computational tools for Comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Librado, P.J.R.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Benson, G. Tandem Repeats Finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ye, W.; Zhang, Y.; Xu, Y. High speed BLASTN: An accelerated MegaBLAST search tool. Nucleic Acids Res. 2015, 43, 7762–7768. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.-h.; Jin, H.; Marler, B.; Guo, H. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- RStudio Team. RStudio Team: Integrated Development for R. Available online: http://www.rstudio.com (accessed on 20 February 2024).
- Katoh, K.; Standley, D. MAFFT Multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Fu, C.-N.; Li, H.-T.; Milne, R.; Zhang, T.; Ma, P.-F.; Yang, J.; Li, D.-Z.; Gao, L.-M. Comparative analyses of plastid genomes from fourteen Cornales species: Inferences for phylogenetic relationships and genome evolution. BMC Genom. 2017, 18, 956. [Google Scholar] [CrossRef] [PubMed]
- Bell, N.E.; Boore, J.L.; Mishler, B.D.; Hyvönen, J. Organellar genomes of the four-toothed moss, Tetraphis pellucida. BMC Genom. 2014, 15, 383. [Google Scholar] [CrossRef] [PubMed]
- Sadamitsu, A.; Inoue, Y.; Sakakibara, K.; Tsubota, H.; Yamaguchi, T.; Deguchi, H.; Nishiyama, T.; Shimamura, M. The complete plastid genome sequence of the enigmatic moss, Takakia lepidozioides (Takakiopsida, Bryophyta): Evolutionary perspectives on the largest collection of genes in mosses and the intensive RNA editing. Plant Mol. Biol. 2021, 107, 431–449. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, C.; Kobayashi, Y.; Aoki, S.; Sugita, C.; Sugita, M. Complete chloroplast DNA sequence of the moss Physcomitrella patens: Evidence for the loss and relocation of rpoA from the chloroplast to the nucleus. Nucleic Acids Res. 2003, 31, 5324–5331. [Google Scholar] [CrossRef]
- Oliver, M.J.; Murdock, A.G.; Mishler, B.D.; Kuehl, J.V.; Boore, J.L.; Mandoli, D.F.; Everett, K.D.; Wolf, P.G.; Duffy, A.M.; Karol, K.G. Chloroplast genome sequence of the moss Tortula ruralis: Gene content, polymorphism, and structural arrangement relative to other green plant chloroplast genomes. BMC Genom. 2010, 11, 143. [Google Scholar] [CrossRef] [PubMed]
- Skippington, E.; Barkman, T.J.; Rice, D.W.; Palmer, J.D. Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc. Natl. Acad. Sci. USA 2015, 112, E3515–E3524. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B.; Alverson, A.J.; Chuckalovcak, J.P.; Wu, M.; McCauley, D.E.; Palmer, J.D.; Taylor, D.R. Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 2012, 10, e1001241. [Google Scholar] [CrossRef] [PubMed]
- Kwon, W.; Kim, Y.; Park, J. The complete mitochondrial genome of Dumortiera hirsuta (Sw.) Nees (Dumortieraceae, Marchantiophyta). Mitochondrial DNA Part B 2019, 4, 1586–1587. [Google Scholar] [CrossRef]
- Myszczyński, K.; Górski, P.; Ślipiko, M.; Sawicki, J. Sequencing of organellar genomes of Gymnomitrion concinnatum (Jungermanniales) revealed the first exception in the structure and gene order of evolutionary stable liverworts mitogenomes. BMC Plant Biol. 2018, 18, 321. [Google Scholar] [CrossRef]
- Mower, J.P.; Sloan, D.B.; Alverson, A.J. Plant mitochondrial genome diversity: The genomics revolution. In Plant Genome Divers; Wendel, J.F., Greilhuber, J., Dolezel, J., Leitch, I.J., Eds.; Springer: Vienna, Austria, 2012; Volume 1, pp. 123–144. [Google Scholar]
- Maréchal, A.; Brisson, N. Recombination and the maintenance of plant organelle genome stability. New Phytol. 2010, 186, 299–317. [Google Scholar] [CrossRef]
- Dong, S.; Zhao, C.; Zhang, S.; Zhang, L.; Wu, H.; Liu, H.; Zhu, R.; Jia, Y.; Goffinet, B.; Liu, Y. Mitochondrial genomes of the early land plant lineage liverworts (Marchantiophyta): Conserved genome structure, and ongoing low frequency recombination. BMC Genom. 2019, 20, 953. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-Z.; Ma, W.-Z.; Schneider, H.; Yu, Y.; Wu, Y.-H. Mitochondrial genome from Andreaea wangiana reveals structural conservatism and a trend of size reduction in mosses. Bryologist 2019, 122, 597–606. [Google Scholar] [CrossRef]
- Liu, Y.; Medina, R.; Goffinet, B. 350 my of mitochondrial genome stasis in mosses, an early land plant lineage. Mol. Biol. Evol. 2014, 31, 2586–2591. [Google Scholar] [CrossRef] [PubMed]
- Donoghue, P.C.; Harrison, C.J.; Paps, J.; Schneider, H. The evolutionary emergence of land plants. Current Biology 2021, 31, R1281–R1298. [Google Scholar] [CrossRef] [PubMed]
- One Thousand Plant Transcriptomes Initiative. One thousand plant transcriptomes and the phylogenomics of green plants. Nature 2019, 574, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Parenti, L.R. A phylogenetic analysis of the land plants. Biol. J. Linn. Soc. 1980, 13, 225–242. [Google Scholar] [CrossRef]
- Li, Y.-F.; Luo, L.; Liu, Y.; He, Q.; Yu, N.-N.; Gaowa, N.; Yi, Z.-Q.; Wang, J.-J.; Han, W.; Peng, T.; et al. The Bryophyte Phylogeny Group: A revised familial classification system based on plastid phylogenomic data. J. Syst. Evol. 2024; early view. [Google Scholar] [CrossRef]
- Steenwyk, J.L.; Li, Y.; Zhou, X.; Shen, X.-X.; Rokas, A. Incongruence in the phylogenomics era. Nat. Rev. Genet. 2023, 24, 834–850. [Google Scholar] [CrossRef] [PubMed]
- Bechteler, J.; Peñaloza-Bojacá, G.; Bell, D.; Burleigh, J.G.; McDaniel, S.F.; Davis, E.C.; Sessa, E.B.; Bippus, A.; Cargill, D.C.; Chantanoarrapint, S.; et al. Comprehensive phylogenomic time tree of bryophytes reveals deep relationships and uncovers gene incongruences in the last 500 million years of diversification. Am. J. Bot. 2023, 110, e16249. [Google Scholar] [CrossRef]
- Zander, R.H. The Pottiaceae s. str. as an evolutionary Lazarus taxon. J. Hattori Bot. Lab. 2006, 100, 581–602. [Google Scholar]
- Inoue, Y.; Tsubota, H. On the systematic position of the genus Timmiella (Dicranidae, Bryopsida) and its allied genera, with the description of a new family Timmiellaceae. Phytotaxa 2014, 181, 151–162. [Google Scholar] [CrossRef]
- Inoue, Y.; Tsubota, H. Systematics of the family Pottiaceae (Bryophyta) with special reference to the familial and subfamilial circumscriptions. Hikobia 2016, 17, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, P.; Li, D.-Z.; Bank, M.; Twyford, A. Telling plant species apart with DNA: From barcodes to genomes. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20150338. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ge, T.C.; Ge, X.-J. A case study of DNA barcoding in Chinese Grimmiaceae and a moss recorded in China for the first time. Taxon 2011, 60, 185–193. [Google Scholar] [CrossRef]
- Lang, A.S.; Kruijer, J.; Stech, M. DNA barcoding of Arctic bryophytes: An example from the moss genus Dicranum (Dicranaceae, Bryophyta). Polar Biol. 2014, 37, 1157–1169. [Google Scholar] [CrossRef]
- Ślipiko, M.; Myszczyński, K.; Buczkowska, K.; Bączkiewicz, A.; Szczecińska, M.; Sawicki, J. Molecular delimitation of European leafy liverworts of the genus Calypogeia based on plastid super-barcodes. BMC Plant Biol. 2020, 20, 243. [Google Scholar] [CrossRef]
- Dong, W.; Xu, C.; Li, C.; Sun, J.; Zuo, Y.; Shi, S.; Cheng, T.; Guo, J.; Zhou, S. ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 2015, 5, 8348. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Y.; Zhang, L.; Yang, M.; Qi, Q.; Yang, Q.; López-Pujol, J.; Wang, L.; Zhao, D. Complete Organelle Genome of the Desiccation-Tolerant (DT) Moss Tortula atrovirens and Comparative Analysis of the Pottiaceae Family. Genes 2024, 15, 782. https://doi.org/10.3390/genes15060782
Ma Y, Zhang L, Yang M, Qi Q, Yang Q, López-Pujol J, Wang L, Zhao D. Complete Organelle Genome of the Desiccation-Tolerant (DT) Moss Tortula atrovirens and Comparative Analysis of the Pottiaceae Family. Genes. 2024; 15(6):782. https://doi.org/10.3390/genes15060782
Chicago/Turabian StyleMa, Yang, Lifang Zhang, Min Yang, Qin Qi, Qian Yang, Jordi López-Pujol, Lihong Wang, and Dongping Zhao. 2024. "Complete Organelle Genome of the Desiccation-Tolerant (DT) Moss Tortula atrovirens and Comparative Analysis of the Pottiaceae Family" Genes 15, no. 6: 782. https://doi.org/10.3390/genes15060782