
Patterns in Genome-Wide Codon Usage Bias in Representative Species of Lycophytes and Ferns

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Coding Sequence Data
2.2. Analysis of Codon Composition
2.3. ENC-Plot Analysis
2.4. PR2-Plot Analysis
2.5. Neutrality Plot Analysis
2.6. Correspondence Analysis (CoA)
2.7. RSCU and Optimal Codon Analysis
2.8. Comparison of Codon Usage Preferences between Four Representative Species and Other Plants
3. Results
3.1. Codon Composition Analysis
3.2. ENC Analysis
3.3. Genomic Codon Usage Bias Analysis
3.4. Analysis of Factors Influencing Codon Usage Bias
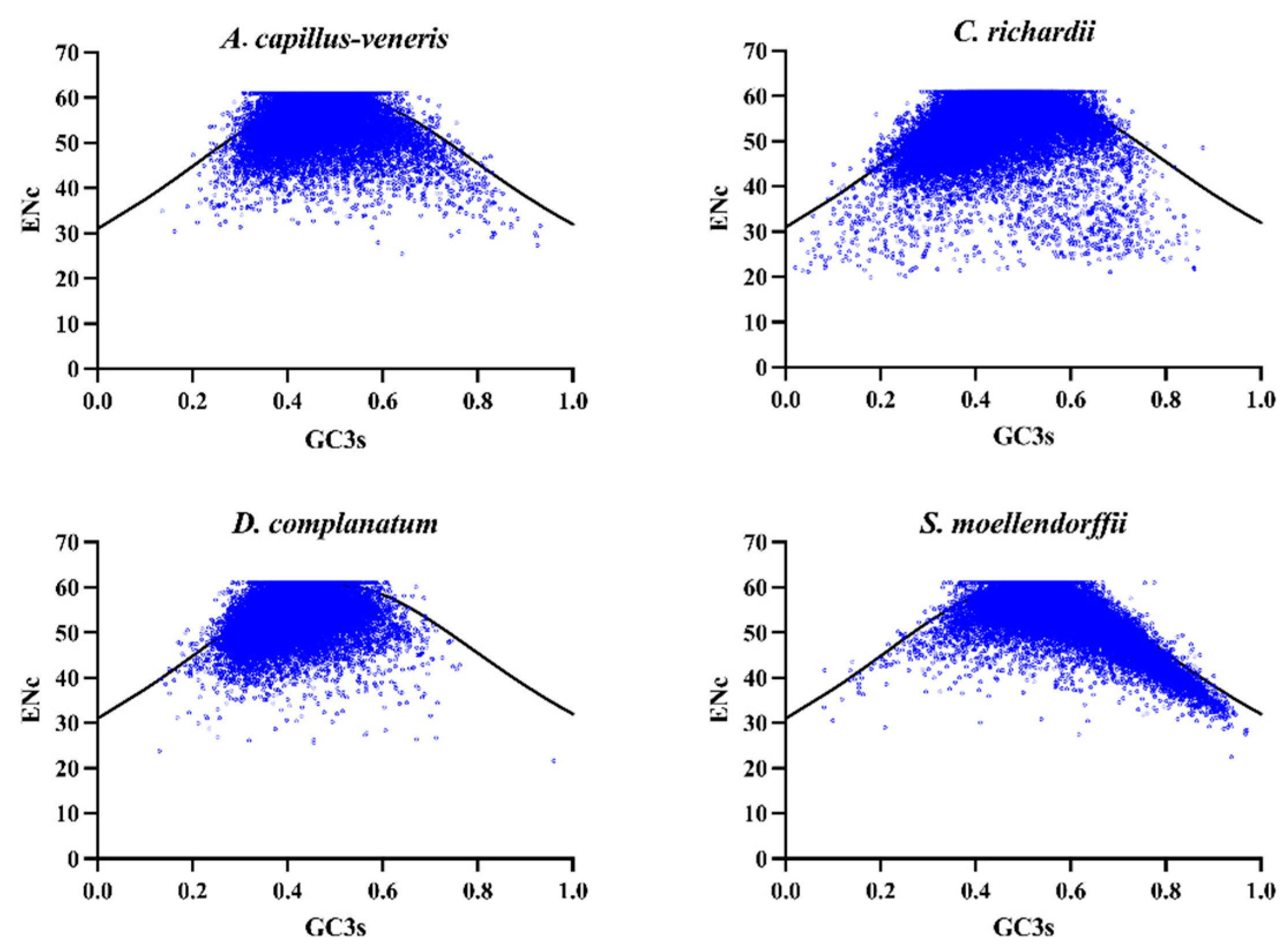
3.4.1. ENC-Plot
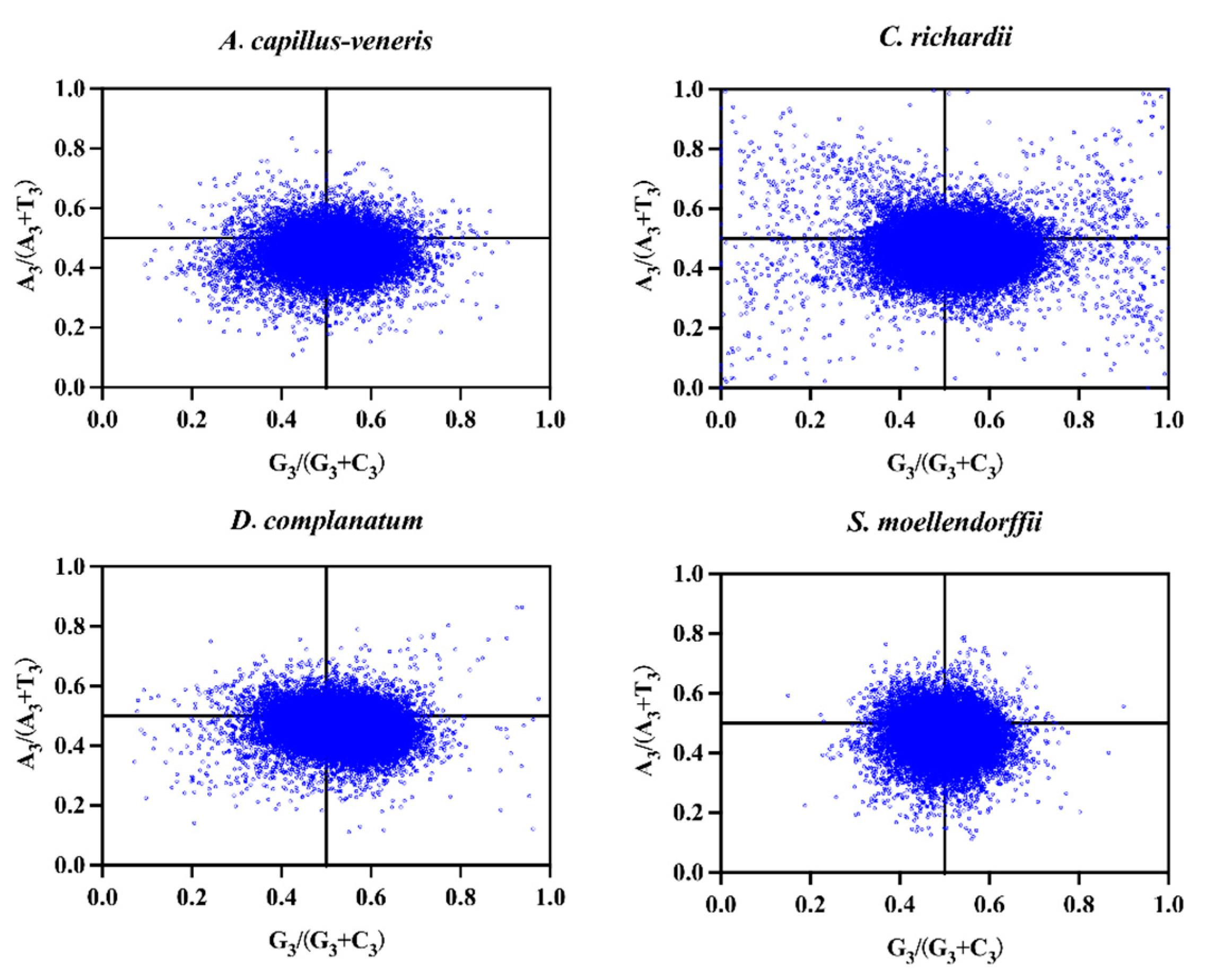
3.4.2. PR2-Plot
3.4.3. Neutrality Plot
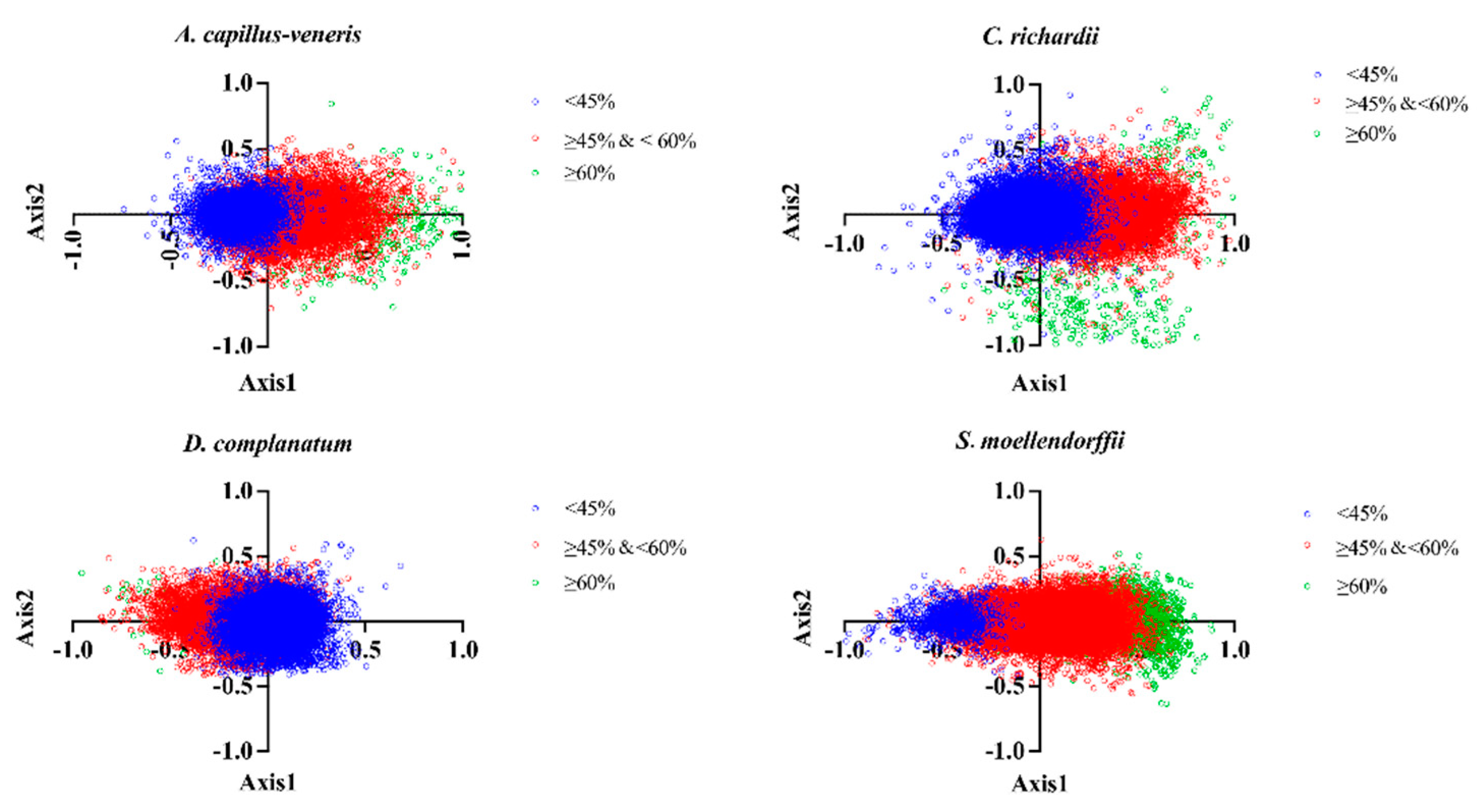
3.4.4. Correspondence Analysis (CoA)
3.5. RSCU and Optimal Codon Analysis
3.6. Comparison of Codon Usage Patterns between Four Lycophytes and Ferns and Other Species
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Pteridophyte Phylogeny Group I. A community-derived classification for extant lycophytes and ferns. J. Syst. Evol. 2016, 54, 563–603. [Google Scholar]
- Smith, A.R.; Pryer, K.M.; Schuettpelz, E.; Korall, P.; Schneider, H.; Wolf, P.G. A classification for extant ferns. Taxon 2006, 55, 705–731. [Google Scholar] [CrossRef]
- Marchant, D.B.; Sessa, E.B.; Wolf, P.G.; Heo, K.; Barbazuk, W.B.; Soltis, P.S.; Soltis, D.E. The C-Fern (Ceratopteris richardii) genome: Insights into plant genome evolution with the first partial homosporous fern genome assembly. Sci. Rep. 2019, 9, 18181. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Qin, X.; Liao, Q.; Du, R.; Luo, X.; Zhou, Q.; Li, Z.; Chen, H.; Jin, W.; Yuan, Y.; et al. The genome of homosporous maidenhair fern sheds light on the euphyllophyte evolution and defences. Nat. Plants 2022, 8, 1024–1037. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.M.; Zhang, L.B. Phylogeny, character evolution, and classification of Selaginellaceae (lycophytes). Plant Divers. 2023, 45, 630–684. [Google Scholar] [CrossRef] [PubMed]
- Ikemura, T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 1985, 2, 13–34. [Google Scholar] [PubMed]
- Zhao, Y.; Zheng, H.; Xu, A.; Yan, D.H.; Jiang, Z.J.; Sun, Q.Q. Analysis of codon usage bias of envelope glycoprotein genes in nuclear polyhedrosis virus (NPV) and its relation to evolution. BMC Genomics 2016, 17, 677. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Cai, X.N.; Chen, Q.Z.; Zhou, H.X.; Cai, Y.; Ben, A.L. Factors affecting synonymous codon usage bias in chloroplast genome of Oncidium Gower Ramsey. Evol. Bioinform. 2011, 6, 271–278. [Google Scholar]
- Carlini, D.B.; Chen, Y.; Stephan, W. The relationship between third-codon position nucleotide content, codon bias, mRNA secondary structure and gene expression in the drosophilid alcohol dehydrogenase genes Adh and Adhr. Genetics 2001, 159, 623–633. [Google Scholar] [CrossRef]
- Olejniczak, M.; Uhlenbeck, O.C. tRNA residues that have coevolved with their anticodon to ensure uniform and accurate codon recognition. Biochimie 2006, 88, 943–950. [Google Scholar] [CrossRef]
- Shah, P.; Gilchrist, M.A. Effect of correlated tRNA abundances on translation errors and evolution of codon usage bias. PLoS Genet. 2010, 6, e1001128. [Google Scholar] [CrossRef] [PubMed]
- Pek, H.B.; Klement, M.; Ang, K.S.; Chung, B.K.S.; Ow, D.S.W.; Lee, D.Y. Exploring codon context bias for synthetic gene design of a thermostable invertase in Escherichia coli. Enzyme Microb. Tech. 2015, 75–76, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.L.; Wang, Y.; Hu, J.H.; Ding, Z.T.; Li, C. Analysis of codon use features of stearoyl-acyl carrier protein desaturase gene in Camellia sinensis. J. Theor. Biol. 2013, 334, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Kessler, M.; Karger, N.D.; Kluge, J. Elevational diversity patterns as an example for evolutionary and ecological dynamics in ferns and lycophytes. J. Syst. Evol. 2016, 54, 617–625. [Google Scholar] [CrossRef]
- Xu, Z.C.; Xin, T.Y.; Bartels, D.; Li, Y.; Gu, W.; Yao, H. Genome analysis of the ancient tracheophyte Selaginella tamariscina reveals evolutionary features relevant to the acquisition of desiccation tolerance. Mol. Plant 2018, 11, 983–994. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.P.; Kuo, L.Y.; Guo, C.C.; Li, H.; Li, Z.Y.; Qi, J. A well-resolved fern nuclear phylogeny reveals the evolution history of numerous transcription factor families. Mol. Phylogenet Evol. 2018, 127, 961–977. [Google Scholar] [CrossRef]
- Shen, H.; Jin, D.; Shu, J.P.; Zhou, X.L.; Lei, M.; Wei, R. Large-scale phylogenomic analysis resolves a backbone phylogeny in ferns. GigaScience 2018, 7, gix116. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Yan, Y.H.; Harris, A.; Kang, J.S.; Shen, H.; Xiang, Q.P. Plastid phylogenomics resolve deep relationships among eupolypod II ferns with rapid radiation and rate heterogeneity. Genome Biol. Evol. 2017, 9, 1646–1657. [Google Scholar] [CrossRef]
- Wei, R.; Yang, J.; He, L.J.; Liu, H.M.; Hu, J.Y.; Liang, S.Q. Plastid phylogenomics provides novel insights into the infrafamilial relationship of polypodiaceae. Cladistics 2021, 37, 717–727. [Google Scholar] [CrossRef]
- Zhao, C.F.; Wei, R.; Zhang, X.C.; Xiang, Q.P. Backbone phylogeny of Lepisorus (polypodiaceae) and a novel infrageneric classification based on the total evidence from plastid and morphological data. Cladistics 2020, 36, 235–258. [Google Scholar] [CrossRef]
- Zhang, M.H.; Xiang, Q.P.; Zhang, X.C. Plastid phylogenomic analyses of the Selaginella sanguinolenta group (selaginellaceae) reveal conflict signatures resulting from sequence types, outlier genes, and pervasive RNA editing. Mol. Phylogenet Evol. 2022, 173, 107507. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.H.; Lu, H.T.; Yang, J.; Tran, G.; Zhang, X.C. Selaginella pseudotamariscina(selaginellaceae), an overlooked rosette-forming resurrection spikemoss from vietnam. Guihaia 2022, 42, 1632–1640. [Google Scholar]
- Zhou, X.M.; Zhao, J.; Yang, J.J.; Péchon, T.L.; Zhang, L.; He, Z.R. Plastome structure, evolution, and phylogeny of Selaginella. Mol. Phylogenet Evol. 2022, 169, 107410. [Google Scholar] [CrossRef]
- He, B.; Dong, H.; Jiang, C.; Cao, F.L.; Tao, S.T.; Xu, L.A. Analysis of codon usage patterns in Ginkgo biloba reveals codon usage tendency from A/U-ending to G/C-ending. Sci. Rep. 2016, 6, 35927. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.S.; Qiao, Z.Q.; Wang, X.M.; Zeng, H.J.; Li, Y.X.; Cai, N.; Chen, Y. Analysis of codon usage patterns in “Lonicerae Flos” (Lonicera macranthoides Hand. -Mazz.) based on transcriptome data. Gene 2019, 705, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Wright, F. The ‘effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.J.; Zhou, J.; Li, Z.F.; Wang, L.; Xun, G.; Zhong, Y. Comparative analysis of codon usage patterns among mitochondrion, chloroplast and nuclear genes in Triticum aestivum L. J. Integr. Plant Biol. 2007, 49, 246–254. [Google Scholar] [CrossRef]
- Shields, D.C.; Sharp, P.M. Synonymous codon usage in Bacillus subtilis reffects both translational selection and mutational biases. Nucleic Acids Res. 1987, 15, 8023–8040. [Google Scholar] [CrossRef] [PubMed]
- Majeed, A.; Kaur, H.; Bhardwaj, P. Selection constraints determine preference for A/U-ending codons in Taxus contorta. Genome 2020, 63, 215–224. [Google Scholar] [CrossRef]
- Sueoka, N. Translation-coupled violation of Parity Rule 2 in human genes is not the cause of heterogeneity of the DNA G + C content of third codon position. Gene 1999, 238, 53–58. [Google Scholar] [CrossRef]
- Sueoka, N. Directional mutation pressure and neutral molecular evolution. Proc. Natl. Acad. Sci. USA 1988, 85, 2653–2657. [Google Scholar] [CrossRef] [PubMed]
- Fennoy, S.L.; Bailey-Serres, J. Synonymous codon usage in Zea mays L. nuclear genes is varied by levels of C and G ending codons. Nucleic Acids Res. 1993, 21, 5294–5300. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, A.T.; Sharp, P.M. Condon usage in Aspergillus nidulans. Mol. Gen. Genet. 1991, 230, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Oliver, J.L.; Marin, A.; Matinez-Zapater, J.M. Chloroplast genes transferred to the nuclear plant genome have adjusted to nuclear base composition and codon usage. Nucleic Acids Res. 1990, 18, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Shields, D.C.; Sharp, P.M.; Higgins, D.G.; Wriqht, F. “Silent” sites in Drosophila genes are not neutral: Evidence of selection among synonymous codons. Mol. Biol. Evol. 1988, 5, 704–716. [Google Scholar] [PubMed]
- Sharp, P.M.; Li, W.H. The codon adaptation index—A measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef]
- Parvathy, S.T.; Udayasuriyan, V.; Bhadana, V. Codon usage bias. Mol. Biol. Rep. 2022, 49, 539–565. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Zhao, W.M.; Zheng, Y.; Wang, H.; Qi, M.; Yu, X.P. Analysis of synonymous codon usage bias in Chlamydia. Acta Biochim. Et Biophys. Sin. 2005, 37, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Peden, J.F. Analysis of Codon Usage. Ph.D. Dissertation, University of Nottingham, Nottingham, UK, 1999. [Google Scholar]
- Morton, B. Chloroplast DNA codon use: Evidence for selection at the psbA locus based on tRNA availability. J. Mol. Evol. 1993, 37, 273–280. [Google Scholar] [CrossRef]
- Lavner, Y.; Kotlar, D. Codon bias as a factor in regulating expression via translation rate in the human genome. Gene 2005, 35, 127–138. [Google Scholar] [CrossRef]
- Yang, M.; Liu, J.; Yang, W.; Li, Z.; Hai, Y.; Duan, B.; Zhang, H.; Yang, X.; Xia, C. Analysis of codon usage patterns in 48 Aconitum species. BMC Genom. 2023, 24, 703. [Google Scholar] [CrossRef] [PubMed]
- Ji, D.J.; Wang, Z.L. Analysis of codon bias in Sarcozygium xanthoxylon Bunge. Mol. Plant Breed. 2021, 21, 6705–6713. [Google Scholar]
- Liang, H.H.; Fu, H.Y.; Li, Z.P.; Li, Y.Q. Analysis on codon usage bias of chloroplast genome from Chlorella. Mol. Plant Breed. 2020, 18, 5665–5673. [Google Scholar]
- Prabha, R.; Singh, D.P.; Sinha, S. Genome-wide comparative analysis of codon usage bias and codon context patterns among cyanobacterial genomes. Mar. Geonomics 2016, 32, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Kawabe, A.; Miyashita, N.T. Patterns of codon usage bias in three dicot and four monocot plant species. Genes Genet. Syst. 2003, 78, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Dong, W.; Li, W. Comparative analysis of six Lagerstroemia complete chloroplast genomes. Front. Plant Sci. 2017, 8, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Pryer, K.M.; Schneider, H.; Smith, A.R.; Cranfill, R.; Wolf, P.G.; Hunt, J.S.; Sipes, S.D. Horsetails and ferns are a monophyletic group and the closest living relatives to seed plants. Nature 2001, 409, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Wickell, D.; Kuo, L.Y.; Chen, X.Q.; Nie, B.; Liao, X.Z.; Peng, D.; Ji, J.J.; Jenkins, J.; Williams, M.; Shu, S.Q.; et al. Extraordinary preservation of gene collinearity over three hundred million years revealed in homosporous lycophytes. Proc. Natl. Acad. Sci. 2024, 121, e2312607121. [Google Scholar]
- Wang, J.; Yu, J.; Sun, P.; Li, C.; Song, X.; Lei, T.; Li, Y.; Yuan, J.; Sun, S.; Ding, H.; et al. Paleo-polyploidization in Lycophytes. Genom. Proteom. Bioinform. 2020, 18, 333–340. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GC1 | GC2 | GC3 | GCall | ENC | |
|---|---|---|---|---|---|
| A. capillus-veneris | 53.615 | 42.522 | 46.740 | 47.626 | 52.778 |
| C. richardii | 51.832 | 41.442 | 41.977 | 45.084 | 52.283 |
| D. complanatum | 52.706 | 41.724 | 40.989 | 45.140 | 53.410 |
| S. moellendorffii | 55.834 | 42.535 | 60.650 | 53.006 | 50.498 |
| Codon Index | AC | CR | DC | SM |
|---|---|---|---|---|
| T3s | 0.372 | 0.401 | 0.406 | 0.269 |
| C3s | 0.270 | 0.234 | 0.227 | 0.371 |
| A3s | 0.308 | 0.344 | 0.348 | 0.231 |
| G3s | 0.295 | 0.274 | 0.269 | 0.375 |
| CAI | 0.200 | 0.198 | 0.199 | 0.216 |
| CBI | −0.064 | −0.078 | −0.068 | 0.038 |
| Fop | 0.380 | 0.373 | 0.379 | 0.438 |
| GC3s | 0.446 | 0.397 | 0.388 | 0.592 |
| GC | 0.477 | 0.452 | 0.452 | 0.531 |
| L_sym | 413.202 | 462.340 | 526.286 | 414.89 |
| L_aa | 429.631 | 480.550 | 545.783 | 431.26 |
| Gravy | −0.253 | −0.266 | −0.269 | −0.226 |
| Aromo | 0.079 | 0.082 | 0.080 | 0.082 |
| Class Limit | Class Value | Frequency | Frequency Rate | ||||||
|---|---|---|---|---|---|---|---|---|---|
| AC | CR | DC | SM | AC | CR | DC | SM | ||
| −0.25~−0.15 | −0.2 | 2 | 12 | 15 | 4 | 0.00008 | 0.00017 | 0.00022 | 0.00015 |
| −0.15~−0.05 | −0.1 | 254 | 847 | 780 | 180 | 0.00967 | 0.01207 | 0.01154 | 0.00665 |
| −0.05~0.05 | 0 | 9499 | 27,130 | 27,822 | 11,517 | 0.36176 | 0.38664 | 0.41163 | 0.42541 |
| 0.05~0.15 | 0.1 | 13,932 | 37,721 | 35,779 | 13,681 | 0.53058 | 0.53757 | 0.52935 | 0.50534 |
| 0.15~0.25 | 0.2 | 2043 | 3414 | 2746 | 1513 | 0.07780 | 0.04865 | 0.04063 | 0.05589 |
| 0.25~0.35 | 0.3 | 461 | 496 | 379 | 157 | 0.01756 | 0.00707 | 0.00561 | 0.00580 |
| 0.35~0.45 | 0.4 | 60 | 309 | 51 | 17 | 0.00229 | 0.00440 | 0.00075 | 0.00063 |
| 0.45~0.55 | 0.5 | 7 | 240 | 18 | 4 | 0.00027 | 0.00342 | 0.00027 | 0.00015 |
| Total | 26,258 | 70,169 | 67,590 | 27,073 | 1 | 1 | 1 | 1 | |
| Amino Acid | Codon | Number | RSCU | ||||||
|---|---|---|---|---|---|---|---|---|---|
| AC | CR | DC | SM | AC | CR | DC | SM | ||
| Ala | GCA | 328,432 | 989,237 | 1,115,071 | 214,300 | 1.466 | 1.618 | 1.578 | 0.871 |
| GCC | 173,863 | 376,265 | 438,127 | 244,188 | 0.776 | 0.615 | 0.62 | 0.993 | |
| GCG | 91,978 | 216,035 | 220,899 | 248,354 | 0.411 | 0.353 | 0.313 | 1.01 | |
| GCU | 301,610 | 864,565 | 1,051,993 | 277,044 | 1.347 | 1.414 | 1.489 | 1.126 | |
| Cys | UGC | 119,373 | 318,043 | 311,419 | 145,304 | 1.022 | 0.89 | 0.922 | 1.325 |
| UGU | 114,153 | 396,826 | 364,193 | 73,974 | 0.978 | 1.11 | 1.078 | 0.675 | |
| Asp | GAC | 200,175 | 514,792 | 569,822 | 311,951 | 0.691 | 0.573 | 0.599 | 1.012 |
| GAU | 378,798 | 1,281,555 | 1,331,887 | 304,291 | 1.309 | 1.427 | 1.401 | 0.988 | |
| Glu | GAA | 328,776 | 1,157,275 | 1,341,654 | 272,737 | 0.911 | 1.043 | 1.089 | 0.721 |
| GAG | 393,172 | 1,061,377 | 1,123,251 | 483,614 | 1.089 | 0.957 | 0.911 | 1.279 | |
| Phe | UUC | 173,387 | 536,715 | 507,514 | 268,206 | 0.794 | 0.814 | 0.716 | 1.129 |
| UUU | 263,340 | 782,464 | 910,388 | 206,909 | 1.206 | 1.186 | 1.284 | 0.871 | |
| Gly | GGA | 213,205 | 714,541 | 837,986 | 250,272 | 1.135 | 1.317 | 1.386 | 1.256 |
| GGC | 182,091 | 422,746 | 489,520 | 242,831 | 0.97 | 0.779 | 0.81 | 1.219 | |
| GGG | 155,260 | 404,290 | 427,736 | 157,826 | 0.827 | 0.745 | 0.708 | 0.792 | |
| GGU | 200,691 | 629,334 | 662,767 | 145,882 | 1.069 | 1.16 | 1.096 | 0.732 | |
| His | CAC | 108,927 | 272,492 | 306,282 | 165,967 | 0.712 | 0.594 | 0.648 | 1.189 |
| CAU | 197,221 | 645,662 | 639,124 | 113,122 | 1.288 | 1.406 | 1.352 | 0.811 | |
| Ile | AUA | 132,939 | 495,964 | 476,885 | 89,876 | 0.763 | 0.854 | 0.788 | 0.493 |
| AUC | 155,759 | 472,970 | 492,669 | 282,736 | 0.894 | 0.815 | 0.814 | 1.55 | |
| AUU | 233,749 | 772,433 | 845,577 | 174,532 | 1.342 | 1.331 | 1.398 | 0.957 | |
| Lys | AAA | 264,644 | 906,517 | 1,046,153 | 208,904 | 0.853 | 0.954 | 0.998 | 0.663 |
| AAG | 356,189 | 994,699 | 1,049,801 | 421,403 | 1.147 | 1.046 | 1.002 | 1.337 | |
| Leu | CUA | 114,756 | 342,395 | 398,912 | 92,206 | 0.616 | 0.644 | 0.688 | 0.409 |
| CUC | 168,150 | 414,007 | 386,053 | 331,224 | 0.903 | 0.778 | 0.666 | 1.47 | |
| CUG | 182,510 | 514,216 | 600,222 | 277,257 | 0.98 | 0.967 | 1.036 | 1.231 | |
| CUU | 279,707 | 857,324 | 932,495 | 200,373 | 1.502 | 1.612 | 1.609 | 0.889 | |
| UUA | 123,649 | 451,252 | 468,975 | 60,664 | 0.629 | 0.745 | 0.698 | 0.44 | |
| UUG | 269,356 | 759,794 | 874,982 | 215,282 | 1.371 | 1.255 | 1.302 | 1.56 | |
| Met | AUG | 282,681 | 866,305 | 860,493 | 276,469 | 1 | 1 | 1 | 1 |
| Asn | AAC | 163,371 | 483,812 | 520,991 | 238,044 | 0.772 | 0.696 | 0.707 | 1.195 |
| AAU | 259,882 | 906,830 | 953,288 | 160,282 | 1.228 | 1.304 | 1.293 | 0.805 | |
| Pro | CCA | 184,259 | 582,251 | 663,628 | 178,328 | 1.322 | 1.494 | 1.505 | 1.253 |
| CCC | 109,951 | 234,875 | 252,967 | 115,033 | 0.789 | 0.603 | 0.574 | 0.808 | |
| CCG | 61,060 | 153,096 | 151,889 | 144,537 | 0.438 | 0.393 | 0.344 | 1.016 | |
| CCU | 202,036 | 588,760 | 695,197 | 131,240 | 1.45 | 1.511 | 1.577 | 0.922 | |
| Gln | CAA | 247,609 | 716,536 | 854,813 | 184,836 | 1.049 | 1.027 | 1.043 | 0.822 |
| CAG | 224,626 | 678,388 | 783,813 | 264,729 | 0.951 | 0.973 | 0.957 | 1.178 | |
| Arg | AGA | 163,721 | 556,336 | 601,738 | 137,503 | 1.033 | 1.098 | 1.152 | 0.924 |
| AGG | 153,253 | 456,708 | 443,328 | 160,121 | 0.967 | 0.902 | 0.848 | 1.076 | |
| CGA | 82,637 | 238,749 | 295,095 | 107,147 | 1.102 | 1.157 | 1.236 | 1.062 | |
| CGC | 79,522 | 167,607 | 190,657 | 116,118 | 1.06 | 0.813 | 0.798 | 1.151 | |
| CGG | 65,558 | 173,829 | 182,712 | 111,949 | 0.874 | 0.843 | 0.765 | 1.11 | |
| CGU | 72,345 | 244,908 | 286,909 | 68,380 | 0.964 | 1.187 | 1.201 | 0.678 | |
| Ser | AGC | 179,597 | 440,360 | 505,404 | 236,996 | 1.054 | 0.901 | 0.924 | 1.39 |
| AGU | 161,155 | 537,111 | 588,387 | 104,037 | 0.946 | 1.099 | 1.076 | 0.61 | |
| UCA | 205,493 | 738,956 | 786,199 | 102,542 | 1.252 | 1.407 | 1.409 | 0.671 | |
| UCC | 136,411 | 378,592 | 385,704 | 178,370 | 0.831 | 0.721 | 0.691 | 1.168 | |
| UCG | 78,268 | 211,624 | 222,177 | 185,204 | 0.477 | 0.403 | 0.398 | 1.213 | |
| UCU | 236,121 | 772,144 | 837,411 | 144,767 | 1.439 | 1.47 | 1.501 | 0.948 | |
| Thr | ACA | 205,060 | 670,959 | 703,525 | 127,562 | 1.495 | 1.624 | 1.551 | 0.914 |
| ACC | 115,165 | 265,042 | 305,155 | 131,863 | 0.839 | 0.642 | 0.673 | 0.944 | |
| ACG | 66,861 | 184,439 | 189,383 | 154,925 | 0.487 | 0.446 | 0.418 | 1.11 | |
| ACU | 161,738 | 532,033 | 615,793 | 144,121 | 1.179 | 1.288 | 1.358 | 1.032 | |
| Val | GUA | 128,256 | 433,311 | 448,957 | 82,007 | 0.692 | 0.802 | 0.751 | 0.404 |
| GUC | 137,417 | 379,589 | 378,766 | 211,259 | 0.742 | 0.702 | 0.633 | 1.041 | |
| GUG | 250,181 | 619,010 | 715,051 | 312,706 | 1.35 | 1.145 | 1.196 | 1.54 | |
| GUU | 225,220 | 729,989 | 848,969 | 206,157 | 1.216 | 1.351 | 1.42 | 1.015 | |
| Trp | UGG | 148,751 | 416,101 | 457,356 | 168,797 | 1 | 1 | 1 | 1 |
| Tyr | UAC | 119,310 | 326,033 | 350,895 | 192,562 | 0.828 | 0.711 | 0.744 | 1.247 |
| UAU | 168,731 | 591,712 | 592,105 | 116,394 | 1.172 | 1.289 | 1.256 | 0.753 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, P.; Zhang, L.; Lu, L.; Zhu, Y.; Gao, D.; Liu, S. Patterns in Genome-Wide Codon Usage Bias in Representative Species of Lycophytes and Ferns. Genes 2024, 15, 887. https://doi.org/10.3390/genes15070887
Xu P, Zhang L, Lu L, Zhu Y, Gao D, Liu S. Patterns in Genome-Wide Codon Usage Bias in Representative Species of Lycophytes and Ferns. Genes. 2024; 15(7):887. https://doi.org/10.3390/genes15070887
Chicago/Turabian StyleXu, Piaoran, Lijuan Zhang, Liping Lu, Yanli Zhu, Dandan Gao, and Shanshan Liu. 2024. "Patterns in Genome-Wide Codon Usage Bias in Representative Species of Lycophytes and Ferns" Genes 15, no. 7: 887. https://doi.org/10.3390/genes15070887
APA StyleXu, P., Zhang, L., Lu, L., Zhu, Y., Gao, D., & Liu, S. (2024). Patterns in Genome-Wide Codon Usage Bias in Representative Species of Lycophytes and Ferns. Genes, 15(7), 887. https://doi.org/10.3390/genes15070887