

A Public Mid-Density Genotyping Platform for Hexaploid Sweetpotato (Ipomoea batatas [L.] Lam)
 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Germplasm Selection and Whole-Genome Sequencing of a Sweetpotato Diversity Panel
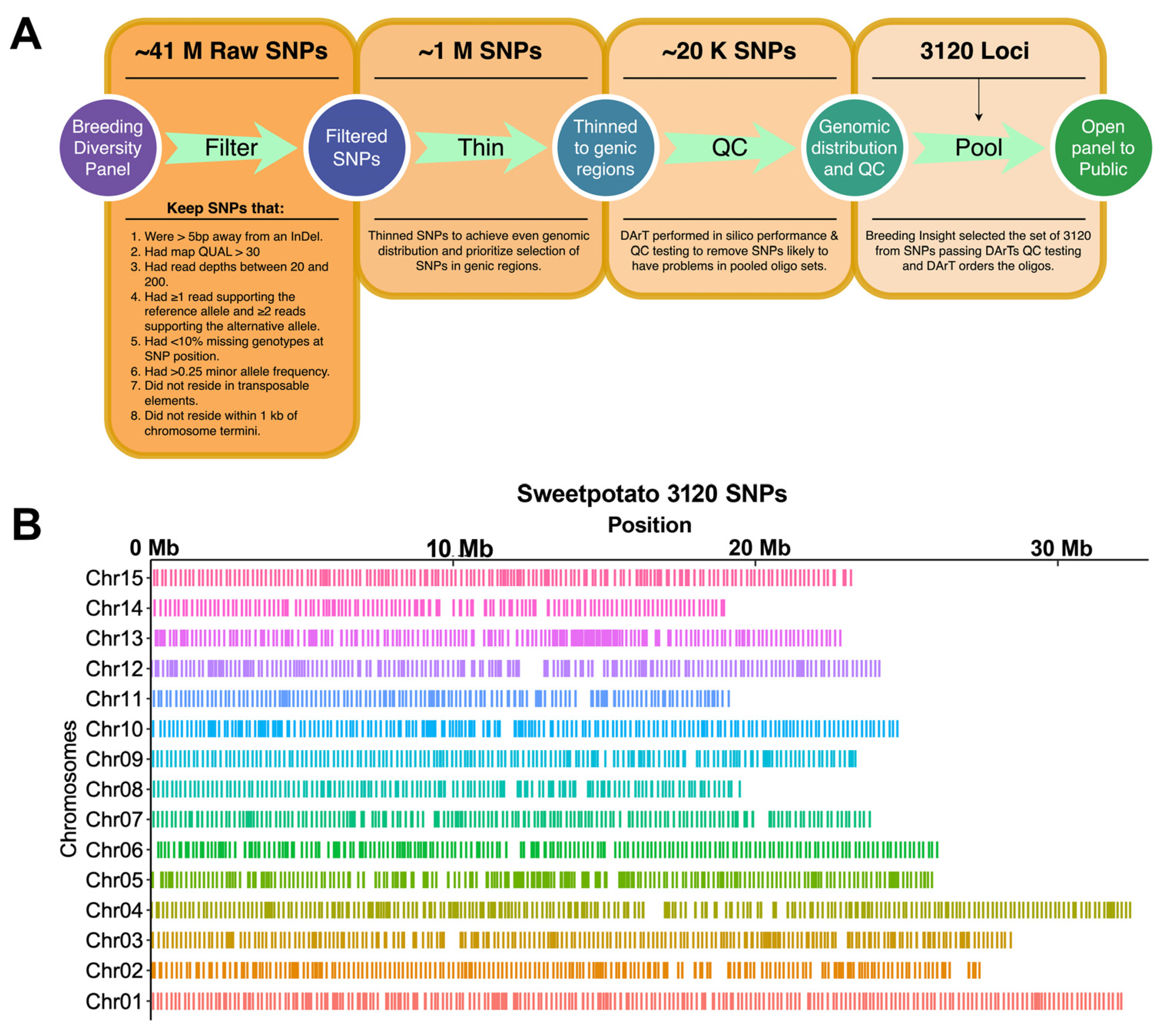
2.2. SNP Discovery and Selection of 3K Marker Loci for Building a DArTag Genotyping Panel
2.3. Principles and Implementation of the DArTag Genotyping Assay
2.4. Validation of the Sweetpotato DArTag Panel
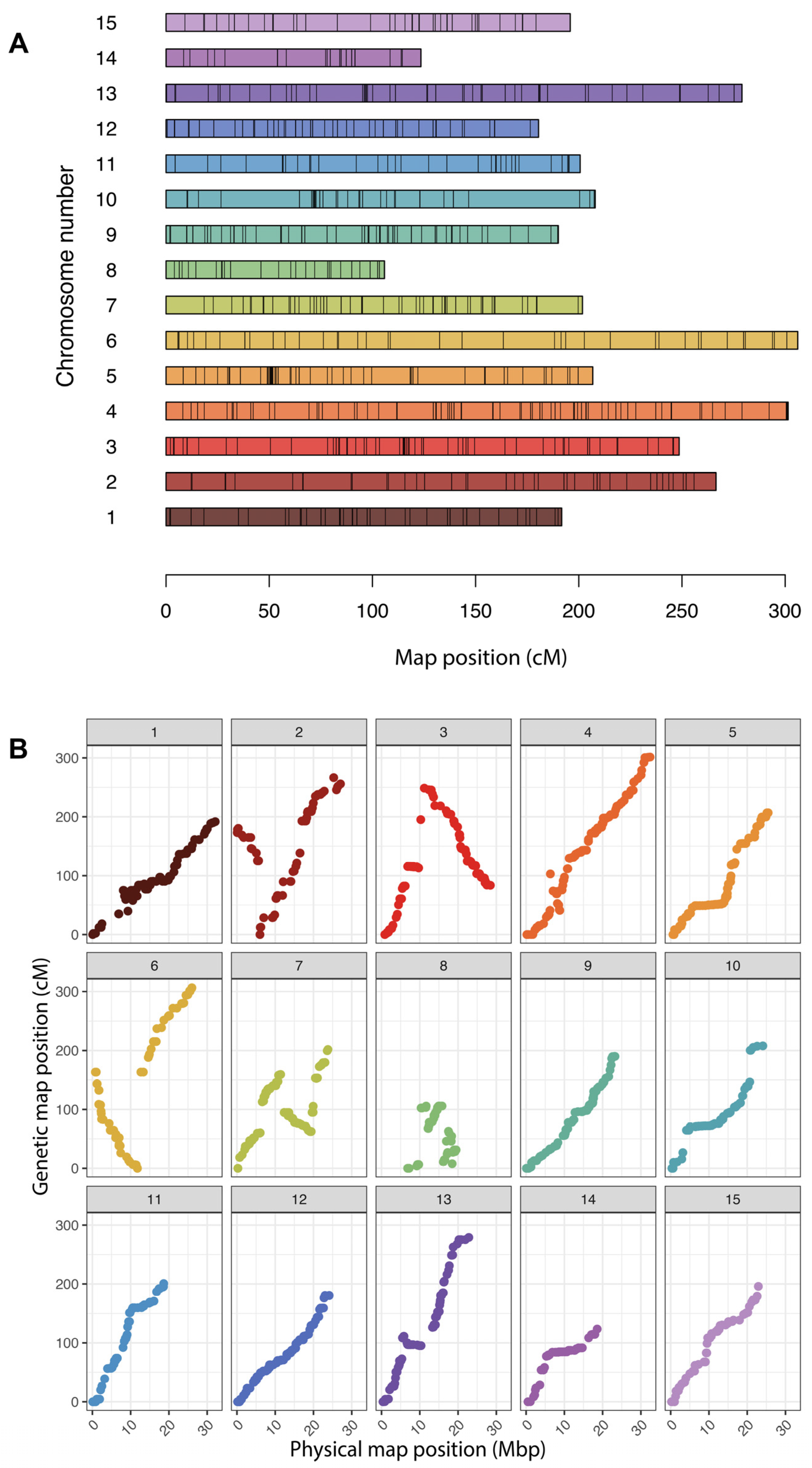
2.5. Genetic Map Construction
3. Results
3.1. The Sweetpotato 3K DArTag Genotyping Panel
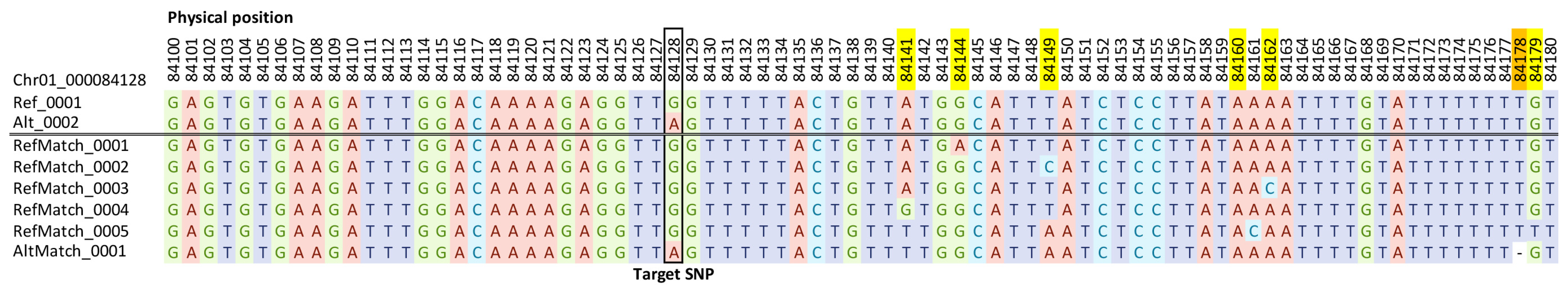
3.2. Microhaplotypes from the Sweetpotato 3K DArTag Genotyping Panel
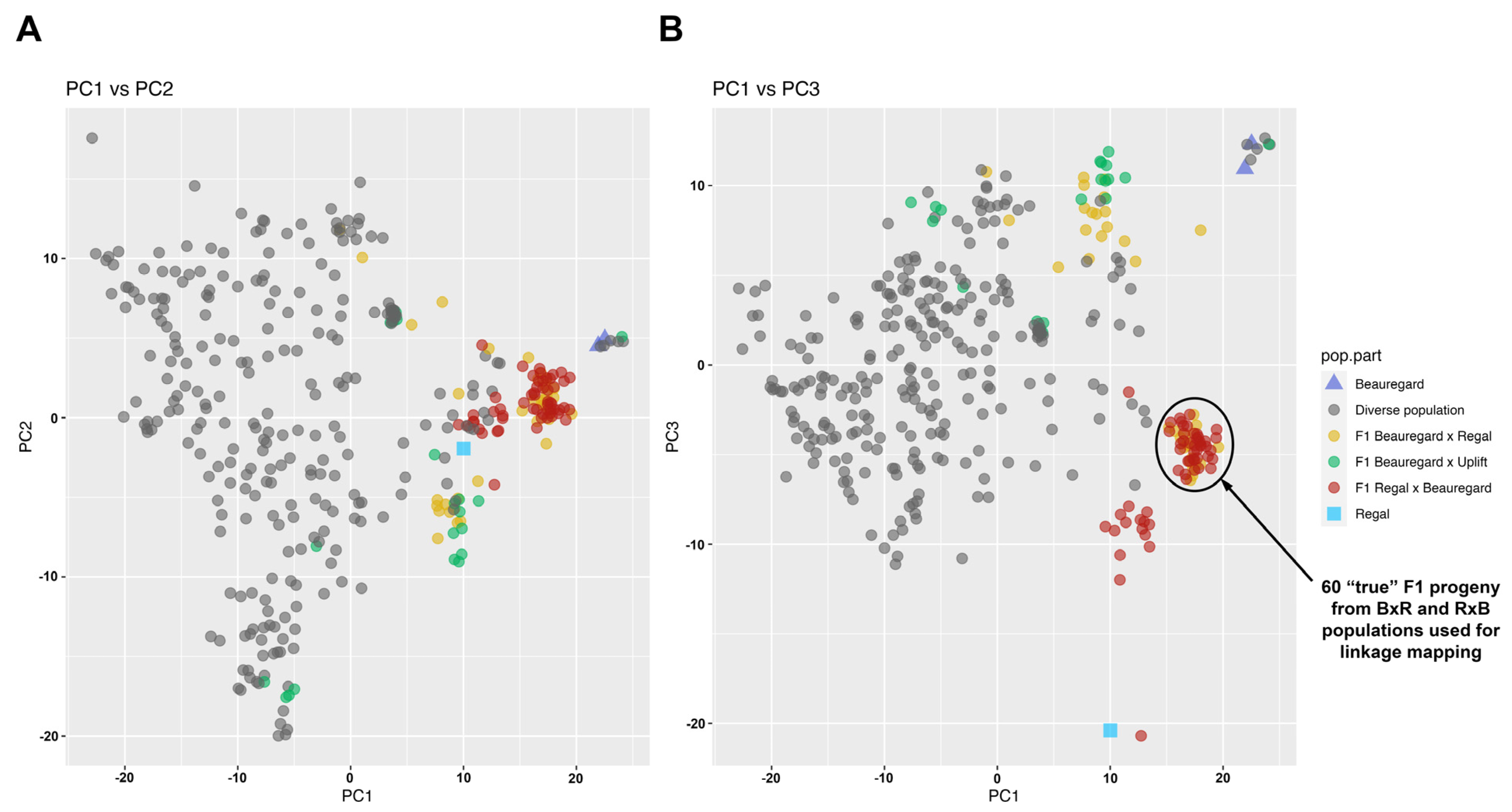
3.3. Panel Effectiveness in Extant Sweetpotato Accessions
3.4. Creation of a Linkage Map
4. Discussion
4.1. Assessment of DArTag Performance and Utility in a Complex Polyploid Genome
4.2. Improving the DArTag Panel as a Global Community
5. Conclusions
Access to and Limitations of the Sweetpotato 3K DArTag Panel
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tanksley, S.D. Molecular Markers in Plant Breeding. Plant Mol. Biol. Rep. 1983, 1, 3–8. [Google Scholar] [CrossRef]
- Helentjaris, T.; King, G.; Slocum, M.; Siedenstrang, C.; Wegman, S. Restriction Fragment Polymorphisms as Probes for Plant Diversity and Their Development as Tools for Applied Plant Breeding. Plant Mol. Biol. 1985, 5, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Feuerstein, U.; Brown, A.H.D.; Burdon, J.J. Linkage of Rust Resistance Genes from Wild Barley (Hordeum Spontaneum) with Isozyme Markers. Plant Breed. 1990, 104, 318–324. [Google Scholar] [CrossRef]
- Hasan, N.; Choudhary, S.; Naaz, N.; Sharma, N.; Laskar, R.A. Recent Advancements in Molecular Marker-Assisted Selection and Applications in Plant Breeding Programmes. J. Genet. Eng. Biotechnol. 2021, 19, 128. [Google Scholar] [CrossRef] [PubMed]
- Eathington, S.R.; Crosbie, T.M.; Edwards, M.D.; Reiter, R.S.; Bull, J.K. Molecular Markers in a Commercial Breeding Program. Crop Sci. 2007, 47, S-154–S-163. [Google Scholar] [CrossRef]
- Lorenzana, R.E.; Bernardo, R. Accuracy of Genotypic Value Predictions for Marker-Based Selection in Biparental Plant Populations. Theor. Appl. Genet. 2009, 120, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Heffner, E.L.; Sorrells, M.E.; Jannink, J. Genomic Selection for Crop Improvement. Crop Sci. 2009, 49, 1–12. [Google Scholar] [CrossRef]
- Johnson, A.C.; Gurr, G.M. Invertebrate Pests and Diseases of Sweetpotato (Ipomoea Batatas): A Review and Identification of Research Priorities for Smallholder Production: Invertebrate Pests and Diseases of Sweetpotato Smallholder Production. Ann. Appl. Biol. 2016, 168, 291–320. [Google Scholar] [CrossRef]
- Sorensen, K.A. Sweetpotato Insects: Identification, Biology and Management. In The Sweetpotato; Springer: Dordrecht, The Netherlands, 2009; pp. 161–188. ISBN 9781402094750. [Google Scholar]
- Cuthbert, F.P.; Reid, W.J. Four Little-Known Pests of Sweetpotato Roots. J. Econ. Entomol. 1965, 58, 581–583. [Google Scholar] [CrossRef]
- Ling, K.-S.; Jackson, D.M.; Harrison, H.; Simmons, A.M.; Pesic-VanEsbroeck, Z. Field Evaluation of Yield Effects on the U.S.A. Heirloom Sweetpotato Cultivars Infected by Sweet Potato Leaf Curl Virus. Crop Prot. 2010, 29, 757–765. [Google Scholar] [CrossRef]
- Meyers, S.L.; Jennings, K.M.; Schultheis, J.R.; Monks, D.W. Interference of Palmer Amaranth (Amaranthus palmeri) in Sweetpotato. Weed Sci. 2010, 58, 199–203. [Google Scholar] [CrossRef]
- Abate, T.; van Huis, A.; Ampofo, J.K.O. Pest Management Strategies in Traditional Agriculture: An African Perspective. Annu. Rev. Entomol. 2000, 45, 631–659. [Google Scholar] [CrossRef] [PubMed]
- Roullier, C.; Duputié, A.; Wennekes, P.; Benoit, L.; Fernández Bringas, V.M.; Rossel, G.; Tay, D.; McKey, D.; Lebot, V. Disentangling the Origins of Cultivated Sweet Potato (Ipomoea batatas (L.) Lam.). PLoS ONE 2013, 8, e62707. [Google Scholar] [CrossRef]
- Wu, S.; Lau, K.H.; Cao, Q.; Hamilton, J.P.; Sun, H.; Zhou, C.; Eserman, L.; Gemenet, D.C.; Olukolu, B.A.; Wang, H.; et al. Genome Sequences of Two Diploid Wild Relatives of Cultivated Sweetpotato Reveal Targets for Genetic Improvement. Nat. Commun. 2018, 9, 4580. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Nakanishi, T. Flower Induction by Top-Grafting in Sweet Potato. In Proceedings of the 5th International Symposium of the International Society of Tropical Roots and Tubers, International Society of Tropical Root and Tuber Crops, Manila, Philippines, 17–21 September 1979; pp. 1–58. [Google Scholar]
- Yang, J.; Moeinzadeh, M.-H.; Kuhl, H.; Helmuth, J.; Xiao, P.; Haas, S.; Liu, G.; Zheng, J.; Sun, Z.; Fan, W.; et al. Haplotype-Resolved Sweet Potato Genome Traces Back Its Hexaploidization History. Nat. Plants 2017, 3, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Telfer, E.; Graham, N.; Macdonald, L.; Li, Y.; Klápště, J.; Resende, M.; Neves, L.G.; Dungey, H.; Wilcox, P. A High-Density Exome Capture Genotype-by-Sequencing Panel for Forestry Breeding in Pinus Radiata. PLoS ONE 2019, 14, e0222640. [Google Scholar] [CrossRef] [PubMed]
- Darrier, B.; Russell, J.; Milner, S.G.; Hedley, P.E.; Shaw, P.D.; Macaulay, M.; Ramsay, L.D.; Halpin, C.; Mascher, M.; Fleury, D.L.; et al. A Comparison of Mainstream Genotyping Platforms for the Evaluation and Use of Barley Genetic Resources. Front. Plant Sci. 2019, 10, 544. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Yuan, Y.; Wang, H.; Yu, D.; Liu, Y.; Zhang, A.; Gowda, M.; Nair, S.K.; Hao, Z.; Lu, Y.; et al. Applications of Genotyping-by-Sequencing (GBS) in Maize Genetics and Breeding. Sci. Rep. 2020, 10, 16308. [Google Scholar] [CrossRef] [PubMed]
- Milner, S.G.; Jost, M.; Taketa, S.; Mazón, E.R.; Himmelbach, A.; Oppermann, M.; Weise, S.; Knüpffer, H.; Basterrechea, M.; König, P.; et al. Genebank Genomics Highlights the Diversity of a Global Barley Collection. Nat. Genet. 2018, 51, 319–326. [Google Scholar] [CrossRef]
- Blyton, M.D.J.; Brice, K.L.; Heller-Uszynska, K.; Pascoe, J.; Jaccoud, D.; Leigh, K.A.; Moore, B.D. A New Genetic Method for Diet Determination from Faeces That Provides Species Level Resolution in the Koala. bioRxiv 2023. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar] [CrossRef]
- Chen, H.; Freed, D.; DNAscope. Sentieon DNAseq; Github: San Jose, CA, USA, 2022. [Google Scholar]
- Krishnakumar, S.; Zheng, J.; Wilhelmy, J.; Faham, M.; Mindrinos, M.; Davis, R. A Comprehensive Assay for Targeted Multiplex Amplification of Human DNA Sequences. Proc. Natl. Acad. Sci. USA 2008, 105, 9296–9301. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Mejia-Guerra, K.M.; Mollinari, M.; Samac, D.; Irish, B.; Heller-Uszynska, K.; Beil, C.T.; Sheehan, M.J. A Public Mid-Density Genotyping Platform for Alfalfa (Medicago sativa L.). Genet. Resour. 2023, 4, 55–63. [Google Scholar] [CrossRef]
- Zhao, D.; Sapkota, M.; Glaubitz, J.; Bassil, N.; Mengist, M.; Iorizzo, M.; Heller-Uszynska, K.; Mollinari, M.; Beil, C.T.; Sheehan, M.J. A Public Mid-Density Genotyping Platform for Cultivated Blueberry (Vaccinium spp.). Genet. Resour. 2024, 5, 36–44. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2021. [Google Scholar]
- Clark, L.V.; Mays, W.; Lipka, A.E.; Sacks, E.J. A Population-Level Statistic for Assessing Mendelian Behavior of Genotyping-by-Sequencing Data from Highly Duplicated Genomes. BMC Bioinform. 2022, 23, 101. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2; Springer: New York, NY, USA, 2016. [Google Scholar]
- Gerard, D.; Ferrão, L.F.V.; Garcia, A.A.F.; Stephens, M. Genotyping Polyploids from Messy Sequencing Data. Genetics 2018, 210, 789–807. [Google Scholar] [CrossRef] [PubMed]
- Mollinari, M.; Garcia, A.A.F. Linkage Analysis and Haplotype Phasing in Experimental Autopolyploid Populations with High Ploidy Level Using Hidden Markov Models. G3 Genes Genomes Genet. 2019, 9, 3297–3314. [Google Scholar] [CrossRef] [PubMed]
- Mollinari, M.; Olukolu, B.A.; Pereira, G.d.S.; Khan, A.; Gemenet, D.; Yencho, G.C.; Zeng, Z.-B. Unraveling the Hexaploid Sweetpotato Inheritance Using Ultra-Dense Multilocus Mapping. G3 Genes Genomes Genet. 2020, 10, 281–292. [Google Scholar] [CrossRef]
- Amadeu, R.R.; Garcia, A.A.F.; Munoz, P.R.; Ferrão, L.F.V. AGHmatrix: Genetic Relationship Matrices in R. Bioinformatics 2023, 39, btad445. [Google Scholar] [CrossRef]
- Jighly, A. When Do Autopolyploids Need Poly-Sequencing Data? Mol. Ecol. 2022, 31, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, D.; Sandercock, A.M.; Mejia-Guerra, M.K.; Mollinari, M.; Heller-Uszynska, K.; Wadl, P.A.; Webster, S.A.; Beil, C.T.; Sheehan, M.J. A Public Mid-Density Genotyping Platform for Hexaploid Sweetpotato (Ipomoea batatas [L.] Lam). Genes 2024, 15, 1047. https://doi.org/10.3390/genes15081047
Zhao D, Sandercock AM, Mejia-Guerra MK, Mollinari M, Heller-Uszynska K, Wadl PA, Webster SA, Beil CT, Sheehan MJ. A Public Mid-Density Genotyping Platform for Hexaploid Sweetpotato (Ipomoea batatas [L.] Lam). Genes. 2024; 15(8):1047. https://doi.org/10.3390/genes15081047
Chicago/Turabian StyleZhao, Dongyan, Alexander M. Sandercock, Maria Katherine Mejia-Guerra, Marcelo Mollinari, Kasia Heller-Uszynska, Phillip A. Wadl, Seymour A. Webster, Craig T. Beil, and Moira J. Sheehan. 2024. "A Public Mid-Density Genotyping Platform for Hexaploid Sweetpotato (Ipomoea batatas [L.] Lam)" Genes 15, no. 8: 1047. https://doi.org/10.3390/genes15081047