

Genome-Wide Identification and Characterization of Maize Long-Chain Acyl-CoA Synthetases and Their Expression Profiles in Different Tissues and in Response to Multiple Abiotic Stresses
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Identification of LACS Genes in Maize
2.2. Bioinformatics Analysis of ZmLACS Genes
2.3. Characterization of Chromosomal Locations and Gene Duplication
2.4. Multiple Sequence Alignment and Construction of the Phylogenetic Tree
2.5. Conserved Motifs and Gene Structure Analysis
2.6. Cis-Acting Element Analysis of ZmLACSs
2.7. Protein–Protein Interaction Network
2.8. Y2H Assay
2.9. Multiple Stress Treatments
2.10. RNA Isolation and Quantitative RT-PCR Analysis
2.11. Statistical Analysis
3. Results
3.1. Identification and Sequence Analysis of ZmLACS Genes in Maize
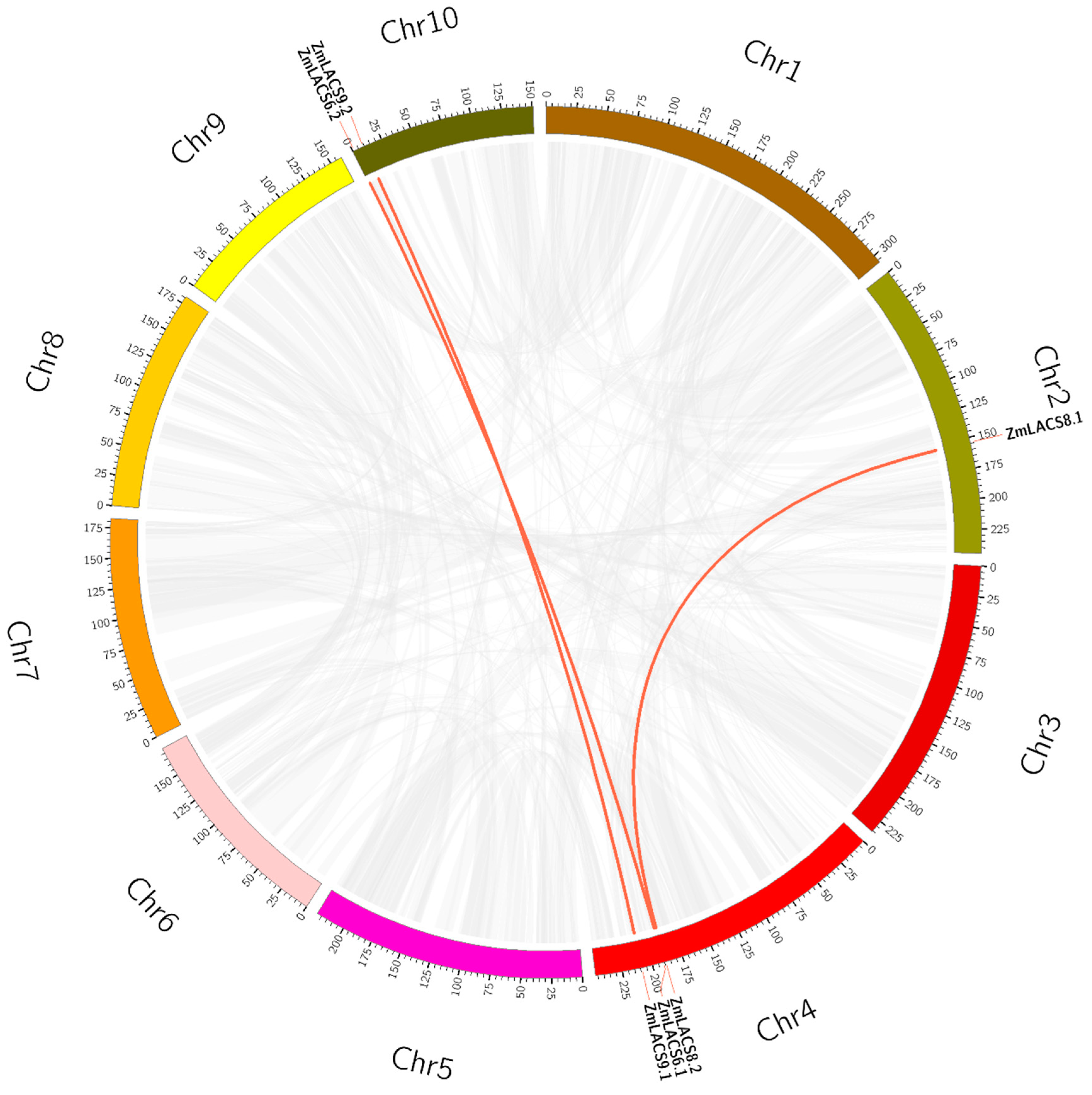
3.2. Chromosomal Localization and Syntenic Analysis of ZmLACS Genes
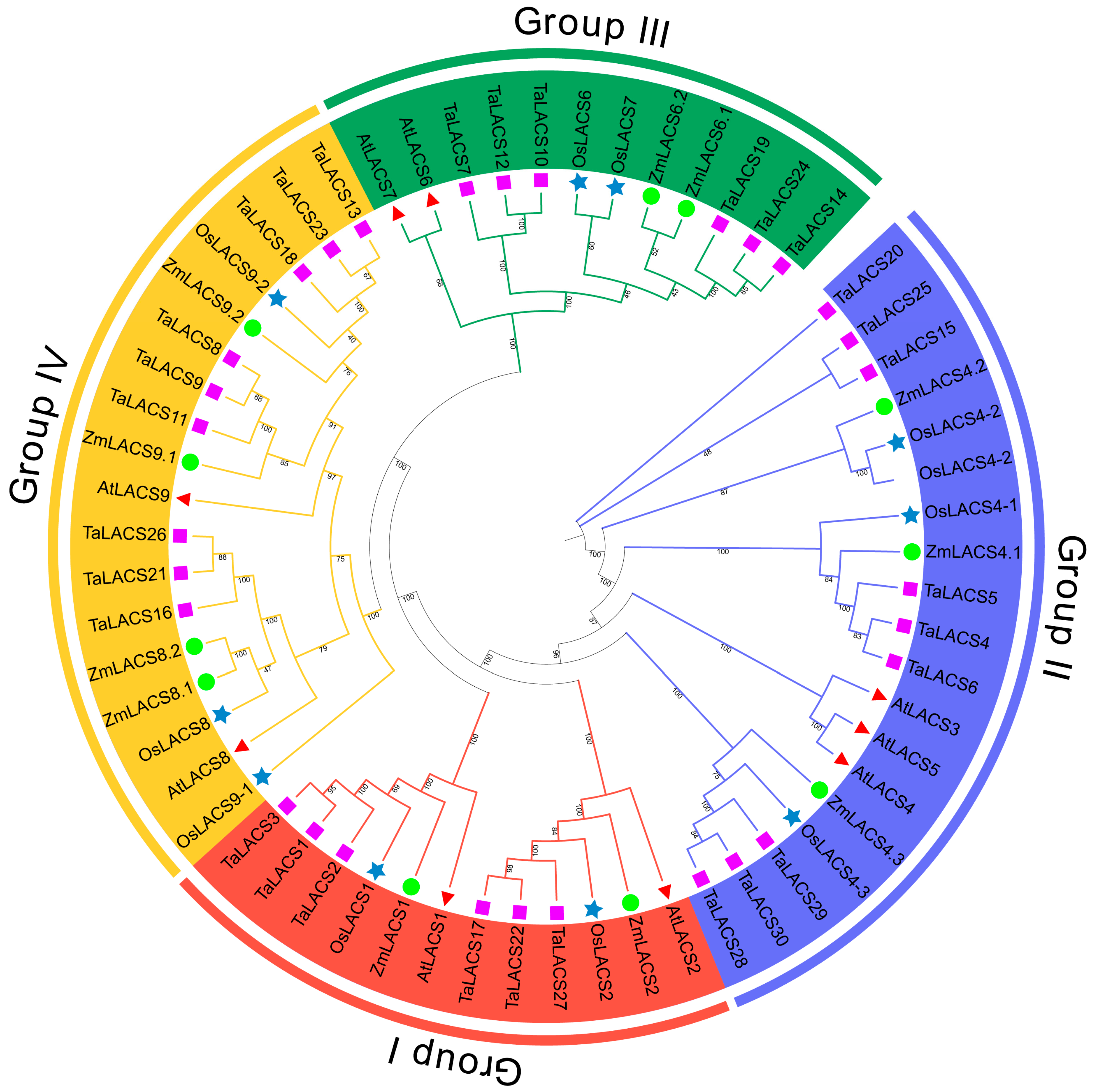
3.3. Phylogenetic Analysis of LACS Genes in Maize
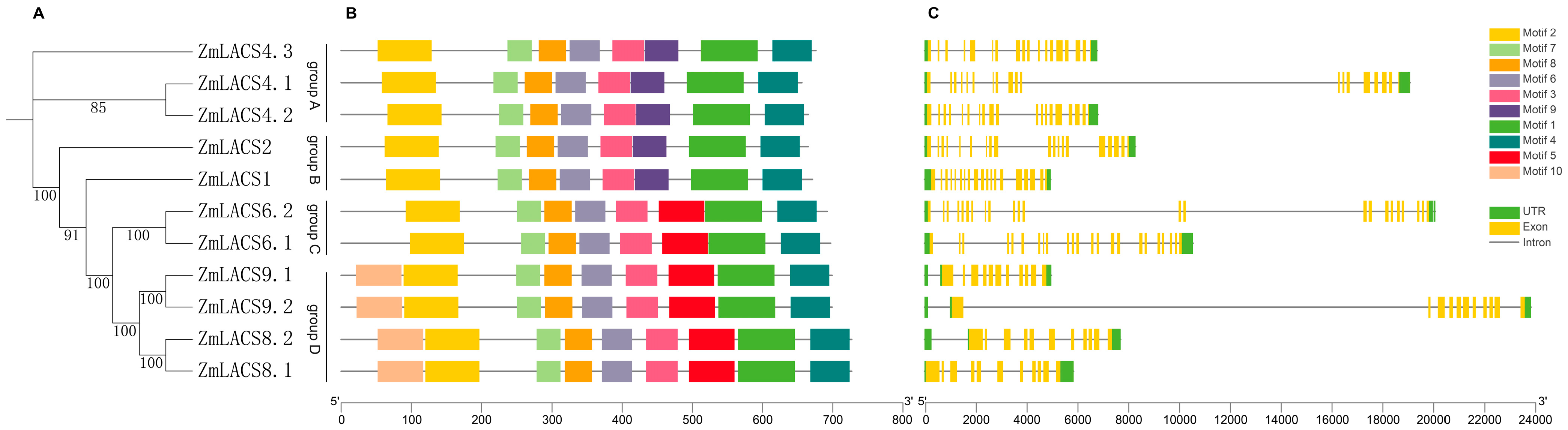
3.4. Conserved Motifs and Gene Structures of the ZmLACS Genes
3.5. Cis-Element Analysis in the Promoter Regions of ZmLACS Genes in Maize
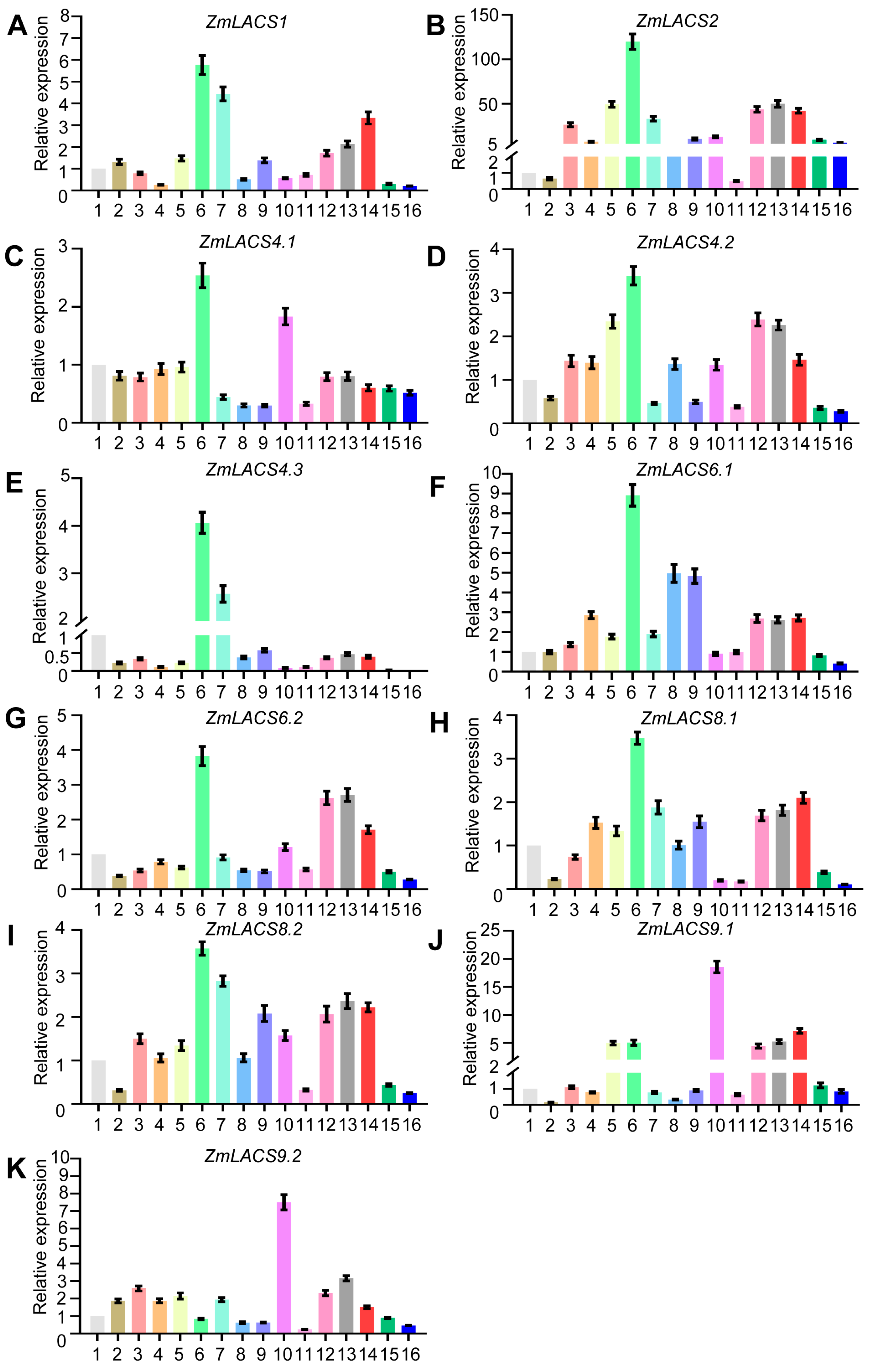
3.6. Expression Analysis of Maize LACSs in Multiple Tissues and Developmental Stages
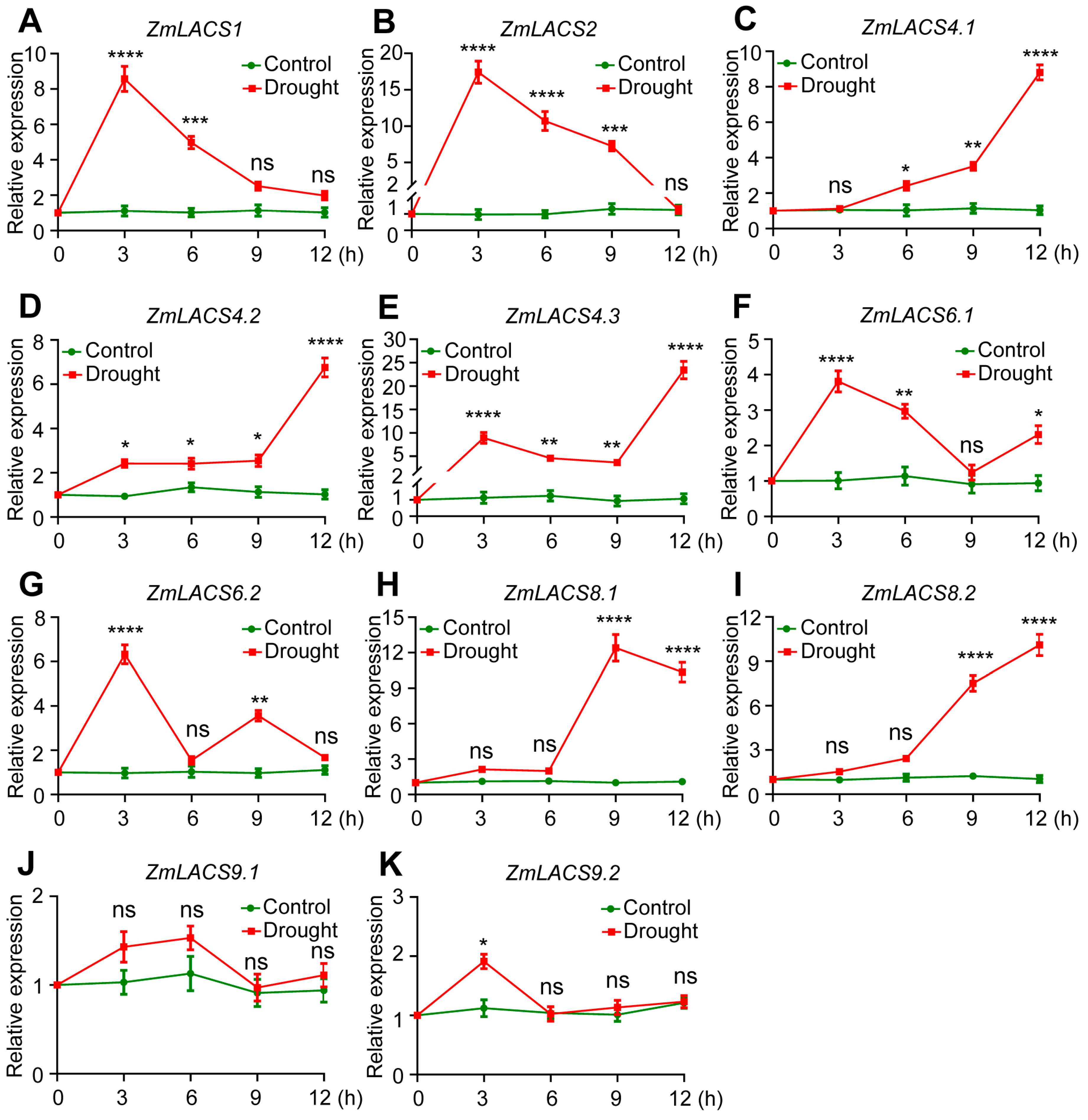
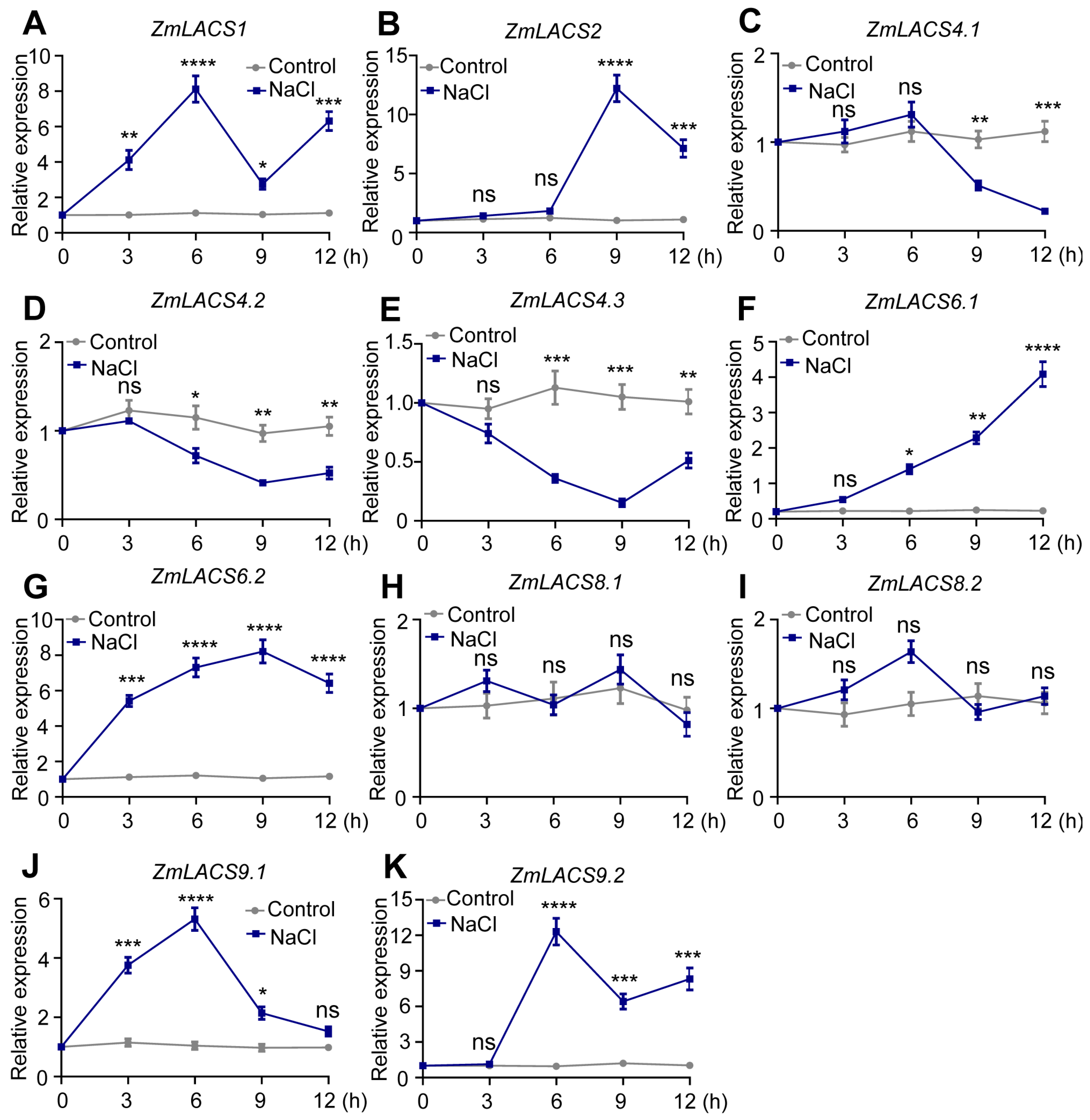
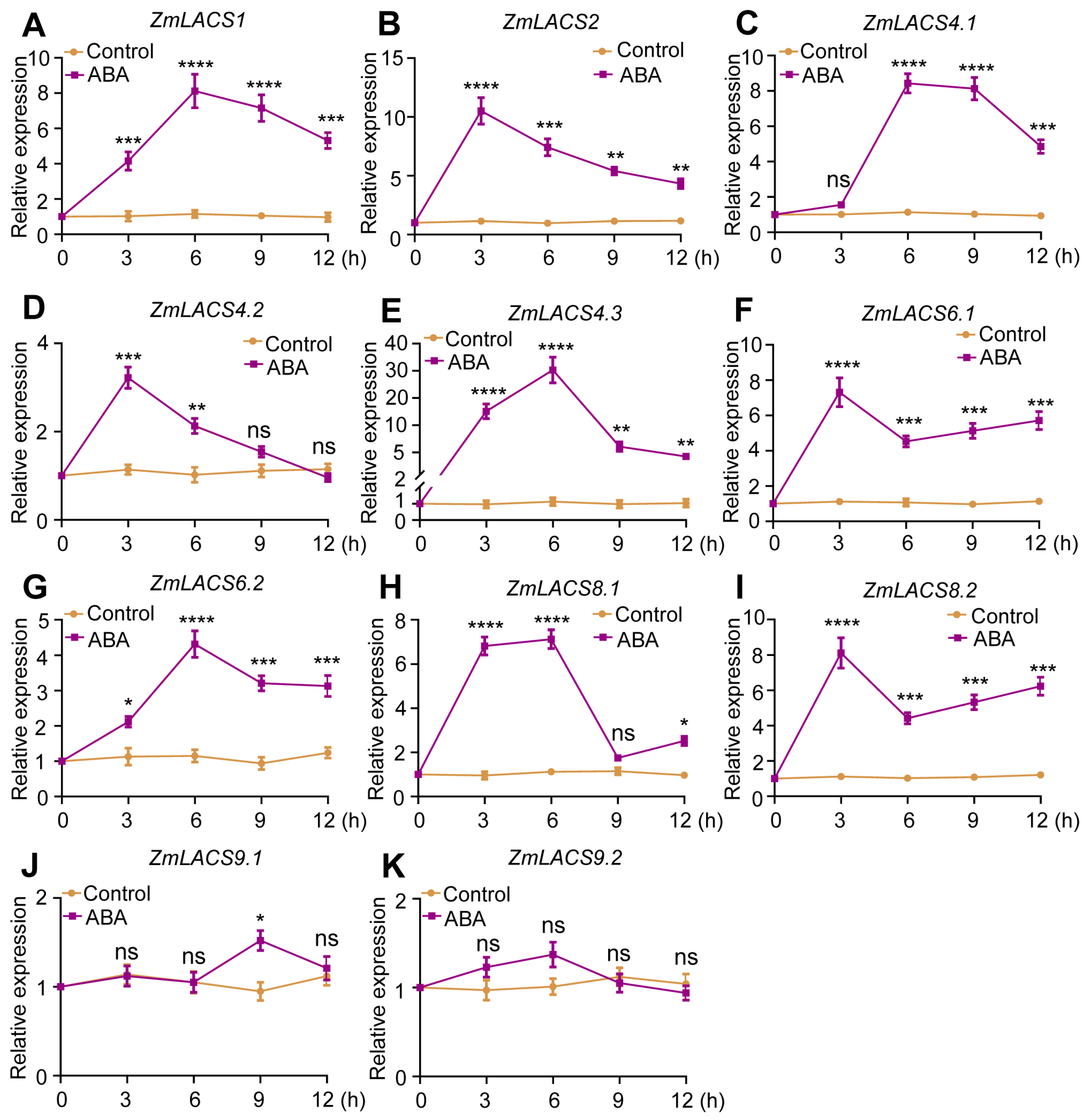
3.7. Expression Profiles of ZmLACSs under Various Stress Conditions
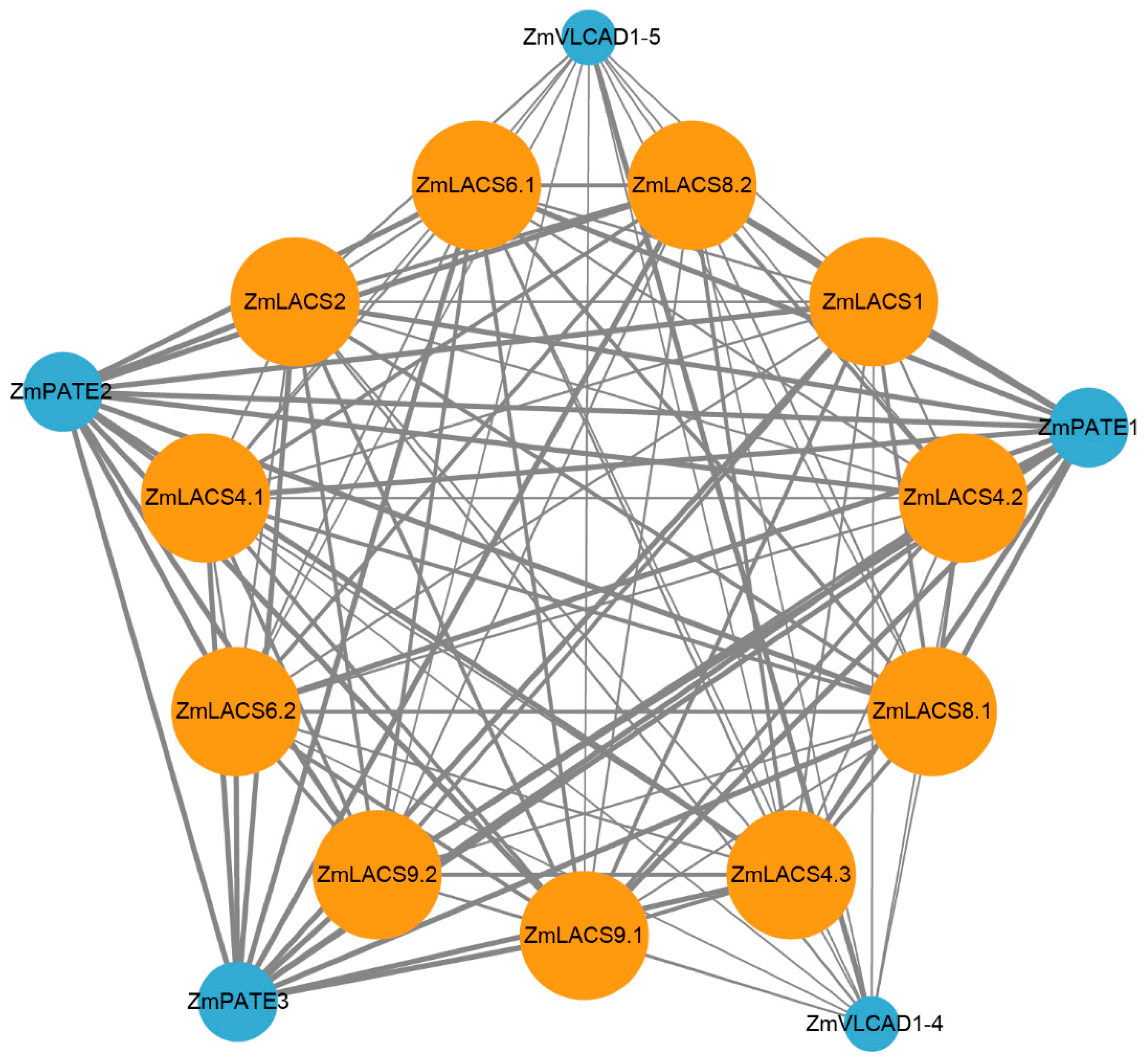
3.8. Protein Interaction Network Analysis of ZmLACS Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lü, S.; Song, T.; Kosma, D.K.; Parsons, E.P.; Rowland, O.; Jenks, M.A. Arabidopsis CER8 encodes LONG-CHAIN ACYL-COA SYNTHETASE 1 (LACS1) that has overlapping functions with LACS2 in plant wax and cutin synthesis. Plant J. 2009, 59, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Xu, C.; Li-Beisson, Y.; Philippar, K. Fatty Acid and Lipid Transport in Plant Cells. Trends Plant Sci. 2016, 21, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Grevengoed, T.J.; Klett, E.L.; Coleman, R.A. Acyl-CoA Metabolism and Partitioning. Annu. Rev. Nutr. 2014, 34, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Shockey, J.; Browse, J. Genome-level and biochemical diversity of the acyl-activating enzyme superfamily in plants. Plant J. 2011, 66, 143–160. [Google Scholar] [CrossRef] [PubMed]
- Watkins, P.A.; Maiguel, D.; Jia, Z.; Pevsner, J. Evidence for 26 distinct acyl-coenzyme A synthetase genes in the human genome. J. Lipid Res. 2007, 48, 2736–2750. [Google Scholar] [CrossRef] [PubMed]
- Watkins, P.A.; Ellis, J.M. Peroxisomal acyl-CoA synthetases. BBA-Mol. Basis Dis. 2012, 1822, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Aznar-Moreno, J.A.; Venegas Calerón, M.; Martínez-Force, E.; Garcés, R.; Mullen, R.; Gidda, S.K.; Salas, J.J. Sunflower (Helianthus annuus) long-chain acyl-coenzyme A synthetases expressed at high levels in developing seeds. Physiol. Plantarum 2013, 150, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Shockey, J.M.; Fulda, M.S.; Browse, J.A. Arabidopsis contains nine long-chain acyl-coenzyme A synthetase genes that participate in fatty acid and glycerolipid metabolism. Plant Physiol. 2002, 129, 1710–1722. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Haslam, T.M.; Sonntag, A.; Molina, I.; Kunst, L. Functional Overlap of Long-Chain Acyl-CoA Synthetases in Arabidopsis. Plant Cell Physiol. 2019, 60, 1041–1054. [Google Scholar] [CrossRef]
- Fulda, M.; Shockey, J.; Werber, M.; Wolter, F.P.; Heinz, E. Two long-chain acyl-CoA synthetases from Arabidopsis thaliana involved in peroxisomal fatty acid β-oxidation. Plant J. 2002, 32, 93–103. [Google Scholar] [CrossRef]
- Jessen, D.; Olbrich, A.; Knüfer, J.; Krüger, A.; Hoppert, M.; Polle, A.; Fulda, M. Combined activity of LACS1 and LACS4 is required for proper pollen coat formation in Arabidopsis. Plant J. 2011, 68, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Katavic, V.; Li, F.; Haughn, G.W.; Kunst, L. Insertional mutant analysis reveals that long-chain acyl-CoA synthetase 1 (LACS1), but not LACS8, functionally overlaps with LACS9 in Arabidopsis seed oil biosynthesis. Plant J. 2010, 64, 1048–1058. [Google Scholar] [CrossRef] [PubMed]
- Suh, M.C.; Samuels, A.L.; Jetter, R.; Kunst, L.; Pollard, M.; Ohlrogge, J.; Beisson, F. Cuticular Lipid Composition, Surface Structure, and Gene Expression in Arabidopsis Stem Epidermis. Plant Physiol. 2005, 139, 1649–1665. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Li, X.B. The GhACS1 gene encodes an acyl-CoA synthetase which is essential for normal microsporogenesis in early anther development of cotton. Plant J. 2009, 57, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Tochtrop, G.P.; Yu, L.; Tan, X.; Jiang, B.; Sun, X.; Gu, S.; Han, T.; Hou, W. A Peroxisomal Long-Chain Acyl-CoA Synthetase from Glycine max Involved in Lipid Degradation. PLoS ONE 2014, 9, e100144. [Google Scholar]
- He, X.; Chen, G.Q.; Kang, S.T.; McKeon, T.A. Ricinus communis Contains an Acyl-CoA Synthetase that Preferentially Activates Ricinoleate to Its CoA Thioester. Lipids 2007, 42, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Holic, R.; Li, D.; Pan, X.; Mietkiewska, E.; Chen, G.; Ozga, J.; Weselake, R.J. Substrate preferences of long-chain acyl-CoA synthetase and diacylglycerol acyltransferase contribute to enrichment of flax seed oil with α-linolenic acid. Biochem. J. 2018, 475, 1473–1489. [Google Scholar] [CrossRef]
- Kitajima-Koga, A.; Baslam, M.; Hamada, Y.; Ito, N.; Taniuchi, T.; Takamatsu, T.; Oikawa, K.; Kaneko, K.; Mitsui, T. Functional Analysis of Rice Long-Chain Acyl-CoA Synthetase 9 (OsLACS9) in the Chloroplast Envelope Membrane. Int. J. Mol. Sci. 2020, 21, 2223. [Google Scholar] [CrossRef]
- Ding, L.-N.; Gu, S.-L.; Zhu, F.-G.; Ma, Z.-Y.; Li, J.; Li, M.; Wang, Z.; Tan, X.-L. Long-chain acyl-CoA synthetase 2 is involved in seed oil production in Brassica napus. BMC Plant Biol. 2020, 20, 21. [Google Scholar] [CrossRef]
- Zhang, C.-L.; Hu, X.; Zhang, Y.-L.; Liu, Y.; Wang, G.-L.; You, C.-X.; Li, Y.-Y.; Hao, Y.-J.; Coleman, H. An apple long-chain acyl-CoA synthetase 2 gene enhances plant resistance to abiotic stress by regulating the accumulation of cuticular wax. Tree Physiol. 2020, 40, 1450–1465. [Google Scholar] [CrossRef]
- Schnable, J.C. Genome Evolution in Maize: From Genomes Back to Genes. Annu. Rev. Plant Biol. 2015, 66, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Veljković, V.B.; Biberdžić, M.O.; Banković-Ilić, I.B.; Djalović, I.G.; Tasić, M.B.; Nježić, Z.B.; Stamenković, O.S. Biodiesel production from corn oil: A review. Renew. Sust. Energ. Rev. 2018, 91, 531–548. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Hidden Markov models. Curr. Opin. Struc Biol. 1996, 6, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018, 46, D493–D496. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Gonzales, N.R.; Gwadz, M.; Lu, S.; Marchler, G.H.; Song, J.S.; Thanki, N.; Yamashita, R.A.; et al. The conserved domain database in 2023. Nucleic Acids Res. 2023, 51, D384–D388. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Voorrips, R.E. MapChart: Software for the Graphical Presentation of Linkage Maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant. 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Pearce, M.; Tivey, A.R.N.; Basutkar, P.; Lee, J.; Edbali, O.; Madhusoodanan, N.; Kolesnikov, A.; Lopez, R. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 2022, 50, W276–W279. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Peterson, D.; Tamura, K. MEGA-CC: Computing core of molecular evolutionary genetics analysis program for automated and iterative data analysis. Bioinformatics 2012, 28, 2685–2686. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L. Discovering novel sequence motifs with MEME. Curr. Protoc. Bioinform. 2002, 2, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Jin, J.; Guo, A.-Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef] [PubMed]
- Rombauts, S.; Déhais, P.; Van Montagu, M.; Rouzé, P. PlantCARE, a plant cis-acting regulatory element database. Nucleic Acids Res. 1999, 27, 295–296. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Hurley, I.; Hale, M.E.; Prince, V.E. Duplication events and the evolution of segmental identity. Evol. Dev. 2010, 7, 556–567. [Google Scholar] [CrossRef]
- Petricka, J.J.; Winter, C.M.; Benfey, P.N. Control of Arabidopsis root development. Annu. Rev. Plant Biol. 2012, 63, 563–590. [Google Scholar] [CrossRef]
- Hu, T.; Banzhaf, W. Nonsynonymous to Synonymous Substitution Ratio ka/ks: Measurement for Rate of Evolution in Evolutionary Computation. In Parallel Problem Solving from Nature—PPSN X, 10th International Conference, Dortmund, Germany, 13–17 September 2008; Springer: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Liu, Y.; Liu, Z.; Zhang, H.; Yuan, S.; Li, Y.; Zhang, T.; Bai, J.; Zhang, L. Genome-Wide Identification and Expression Profiling Analysis of the Long-Chain Acyl-CoA Synthetases Reveal Their Potential Roles in Wheat Male Fertility. Int. J. Mol. Sci. 2022, 23, 11942. [Google Scholar] [CrossRef]
- Zhou, Z.; Yang, J.; Zhang, C.; Zeng, L.; Meng, W.; Sun, L. Genome-wide identification and expression analyses of long-chain Acyl-CoA synthetases under abiotic stresses in Helianthus annuus. Acta Hortic. Sin. 2022, 49, 352–364. [Google Scholar]
- Travers, A.; Muskhelishvili, G. DNA structure and function. FEBS J. 2015, 282, 2279–2295. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Peng, D.; Bai, L.-P.; Ma, H.; Chen, L.-J.; Zhao, M.-H.; Xu, Z.-J.; Guo, Z.-F. Molecular switch for cold acclimation—Anatomy of the cold-inducible promoter in plants. Biochemistry 2013, 78, 342–354. [Google Scholar] [CrossRef]
- Kim, S.-J.; Zhang, Z.; Lee, Y.-C.; Mukherjee, A.B. Palmitoyl-protein thioesterase-1 deficiency leads to the activation of caspase-9 and contributes to rapid neurodegeneration in INCL. Hum. Mol. Genet. 2006, 15, 1580–1586. [Google Scholar] [CrossRef] [PubMed]
- Bernard, A.; Domergue, F.; Pascal, S.; Jetter, R.; Renne, C.; Faure, J.-D.; Haslam, R.P.; Napier, J.A.; Lessire, R.; Joubès, J. Reconstitution of Plant Alkane Biosynthesis in Yeast Demonstrates That Arabidopsis ECERIFERUM1 and ECERIFERUM3 Are Core Components of a Very-Long-Chain Alkane Synthesis Complex. Plant Cell 2012, 24, 3106–3118. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.-K.; Zhou, Y.J.; Buijs, N.A.; Nielsen, J. Functional screening of aldehyde decarbonylases for long-chain alkane production by Saccharomyces cerevisiae. Microb. Cell Fact. 2017, 16, 74. [Google Scholar] [CrossRef] [PubMed]
- Schubert, S.; Neubert, A.; Schierholt, A.; Sümer, A.; Zörb, C. Development of salt-resistant maize hybrids: The combination of physiological strategies using conventional breeding methods. Plant Sci. 2009, 177, 196–202. [Google Scholar] [CrossRef]
- Bennett, M.D. Comparisons with Caenorhabditis (100 Mb) and Drosophila (175 Mb) Using Flow Cytometry Show Genome Size in Arabidopsis to be 157 Mb and thus 25% Larger than the Arabidopsis Genome Initiative Estimate of 125 Mb. Ann. Bot. 2003, 91, 547–557. [Google Scholar] [CrossRef]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.P.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef]
- Velasco, R.; Zharkikh, A.; Affourtit, J.; Dhingra, A.; Cestaro, A.; Kalyanaraman, A.; Fontana, P.; Bhatnagar, S.K.; Troggio, M.; Pruss, D.; et al. The genome of the domesticated apple (Malus × domestica Borkh.). Nat. Genet. 2010, 42, 833–839. [Google Scholar] [CrossRef]
- Luo, M.-C.; Gu, Y.Q.; Puiu, D.; Wang, H.; Twardziok, S.O.; Deal, K.R.; Huo, N.; Zhu, T.; Wang, L.; Wang, Y.; et al. Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature 2017, 551, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Bano, N.; Fakhrah, S.; Mohanty, C.S.; Bag, S.K. Genome-Wide Identification and Evolutionary Analysis of Gossypium Tubby-Like Protein (TLP) Gene Family and Expression Analyses During Salt and Drought Stress. Front. Plant Sci. 2021, 12, 6679929. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; White, M.J.; MacRae, T.H. Transcription factors and their genes in higher plants. Eur. J. Biochem. 2001, 262, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Flagel, L.E.; Wendel, J.F. Gene duplication and evolutionary novelty in plants. New Phytol. 2009, 183, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Zhu, K.; Zhao, J.; Chen, M.; Wei, L.; Qiao, Z.; Tan, P.; Peng, F. Genome-Wide Identification, Characterization, and Expression Analysis of Long-Chain Acyl-CoA Synthetases in Carya illinoinensis under Different Treatments. Int. J. Mol. Sci. 2023, 24, 11558. [Google Scholar] [CrossRef] [PubMed]
- Marand, A.P.; Eveland, A.L.; Kaufmann, K.; Springer, N.M. Cis-Regulatory Elements in Plant Development, Adaptation, and Evolution. Annu. Rev. Plant Biol. 2023, 74, 111–137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-L.; Mao, K.; Zhou, L.-J.; Wang, G.-L.; Zhang, Y.-L.; Li, Y.-Y.; Hao, Y.-J. Genome-wide identification and characterization of apple long-chain Acyl-CoA synthetases and expression analysis under different stresses. Plant Physiol. Biochem. 2018, 132, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Wang, Y.; Li, P.; Gong, W.; Wang, X.; Yan, H.; Ge, Q.; Liu, A.; Shi, Y.; Shang, H.; et al. Genome-Wide Analysis and Functional Characterization of LACS Gene Family Associated with Lipid Synthesis in Cotton (Gossypium spp.). Int. J. Mol. Sci. 2023, 24, 8530. [Google Scholar] [CrossRef] [PubMed]
- Weng, H.; Molina, I.; Shockey, J.; Browse, J. Organ fusion and defective cuticle function in a lacs1 lacs2 double mutant of Arabidopsis. Planta 2010, 231, 1089–1100. [Google Scholar] [CrossRef] [PubMed]
- Seo, P.J.; Lee, S.B.; Suh, M.C.; Park, M.-J.; Go, Y.S.; Park, C.-M. The MYB96 Transcription Factor Regulates Cuticular Wax Biosynthesis under Drought Conditions in Arabidopsis. Plant Cell 2011, 23, 1138–1152. [Google Scholar] [CrossRef]
- Bernard, A.; Joubès, J. Arabidopsis cuticular waxes: Advances in synthesis, export and regulation. Prog. Lipid Res. 2013, 52, 110–129. [Google Scholar] [CrossRef] [PubMed]
- Kosma, D.K.; Bourdenx, B.; Bernard, A.L.; Parsons, E.P.; Lü, S.; Joubès, J.; Jenks, M.A. The Impact of Water Deficiency on Leaf Cuticle Lipids of Arabidopsis. Plant Physiol. 2009, 151, 1918–1929. [Google Scholar] [CrossRef]
- Engeseth, N.J.; Pacovsky, R.S.; Newman, T.; Ohlrogge, J.B. Characterization of an acyl-CoA-binding protein from Arabidopsis thaliana. Arch. Biochem. Biophys. 1996, 331, 55–62. [Google Scholar] [CrossRef]
- Dörmann, P.; Voelker, T.A.; Ohlrogge, J.B. Accumulation of palmitate in Arabidopsis mediated by the acyl-acyl carrier protein thioesterase FATB1. Plant Physiol. 2000, 123, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, Y.; Song, G.; Yang, D.; Xia, Z.; Sun, C.; Zhao, Y.; Hou, M.; Zhang, M.; Qi, Z.; et al. Gene expression and expression quantitative trait locianalyses uncover natural variations underlying the improvement of important agronomic traits during modern maize breeding. Plant J. 2023, 115, 772–787. [Google Scholar] [CrossRef] [PubMed]
- Malenica, N.; Dunić, J.A.; Vukadinović, L.; Cesar, V.; Šimić, D. Genetic Approaches to Enhance Multiple Stress Tolerance in Maize. Genes 2021, 12, 1760. [Google Scholar] [CrossRef]
- Masuka, B.; Araus, J.L.; Das, B.; Sonder, K.; Cairns, J.E. Phenotyping for abiotic stress tolerance in maize. J. Integr. Plant Biol. 2012, 54, 238–249. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, Z.; Hou, J.; Leng, B.; Yao, G.; Ma, C.; Sun, Y.; Liu, Q.; Zhang, F.; Mu, C.; Liu, X. Genome-Wide Identification and Characterization of Maize Long-Chain Acyl-CoA Synthetases and Their Expression Profiles in Different Tissues and in Response to Multiple Abiotic Stresses. Genes 2024, 15, 983. https://doi.org/10.3390/genes15080983
Yan Z, Hou J, Leng B, Yao G, Ma C, Sun Y, Liu Q, Zhang F, Mu C, Liu X. Genome-Wide Identification and Characterization of Maize Long-Chain Acyl-CoA Synthetases and Their Expression Profiles in Different Tissues and in Response to Multiple Abiotic Stresses. Genes. 2024; 15(8):983. https://doi.org/10.3390/genes15080983
Chicago/Turabian StyleYan, Zhenwei, Jing Hou, Bingying Leng, Guoqi Yao, Changle Ma, Yue Sun, Qiantong Liu, Fajun Zhang, Chunhua Mu, and Xia Liu. 2024. "Genome-Wide Identification and Characterization of Maize Long-Chain Acyl-CoA Synthetases and Their Expression Profiles in Different Tissues and in Response to Multiple Abiotic Stresses" Genes 15, no. 8: 983. https://doi.org/10.3390/genes15080983
APA StyleYan, Z., Hou, J., Leng, B., Yao, G., Ma, C., Sun, Y., Liu, Q., Zhang, F., Mu, C., & Liu, X. (2024). Genome-Wide Identification and Characterization of Maize Long-Chain Acyl-CoA Synthetases and Their Expression Profiles in Different Tissues and in Response to Multiple Abiotic Stresses. Genes, 15(8), 983. https://doi.org/10.3390/genes15080983