
Analysis of the 5′ Untranslated Region Length-Dependent Control of Gene Expression in Maize: A Case Study with the ZmLAZ1 Gene Family
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genome-Wide Identification of the UTRs from Maize, Rice, and Arabidopsis
2.2. Gene Ontology (GO) Enrichment Analysis on UTR-Containing Genes
2.3. Sample Preparation
2.4. Gene 5′ UTR Cloning
2.5. GUS Histochemical Staining
2.6. GUS Activity Measurement
2.7. Analysis of Cis-Acting Elements
2.8. Statistical Analysis
3. Results and Discussion
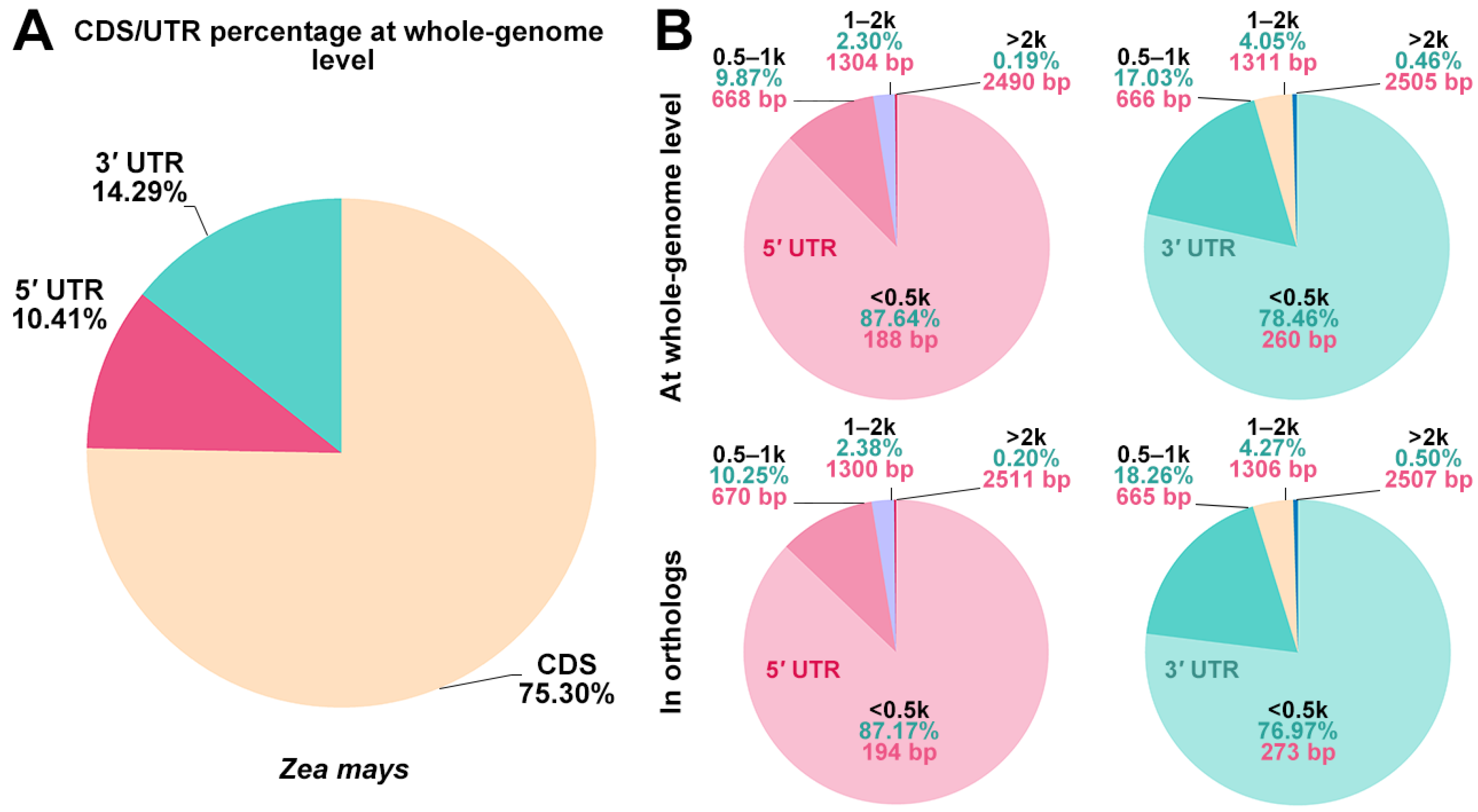
3.1. Whole-Genome Survey of UTRs in Maize
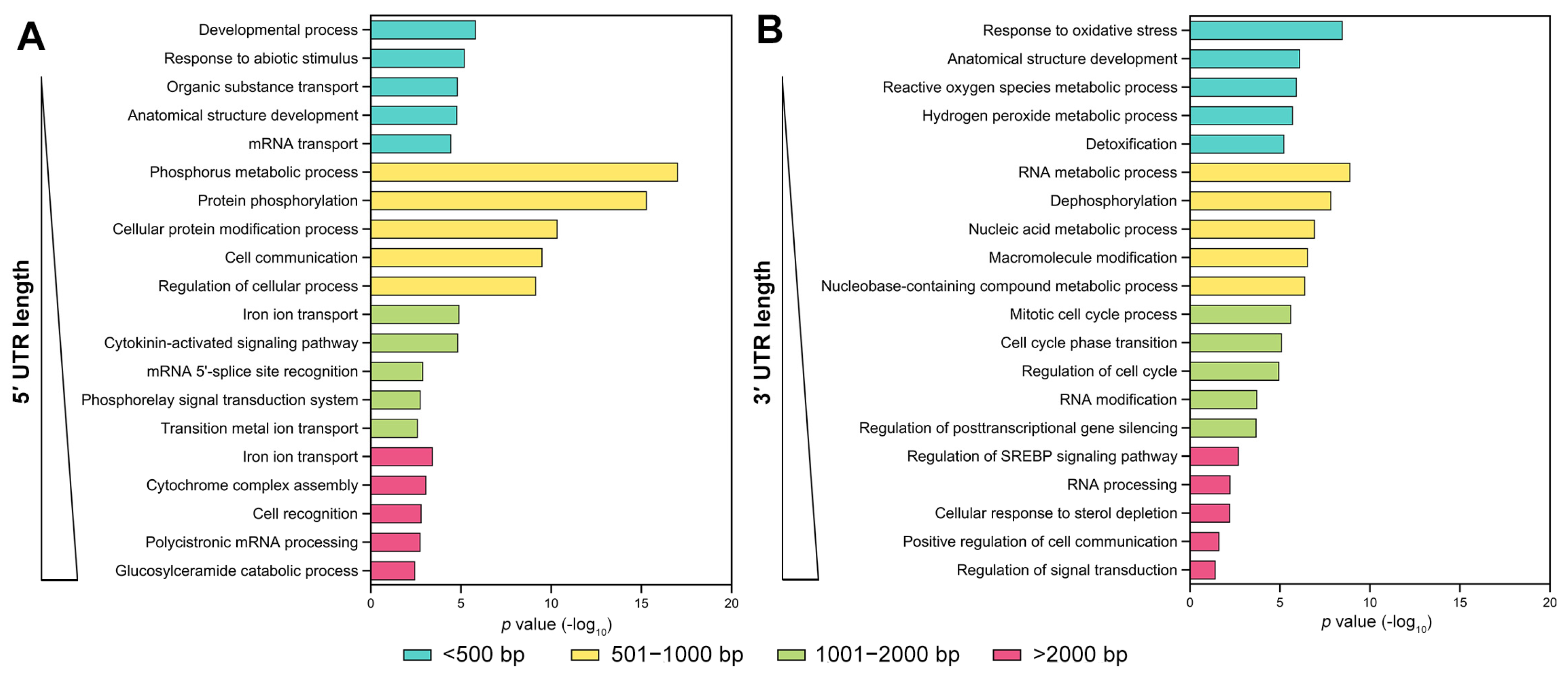
3.2. Functional Enrichment Analysis of UTR-Containing Genes in Maize
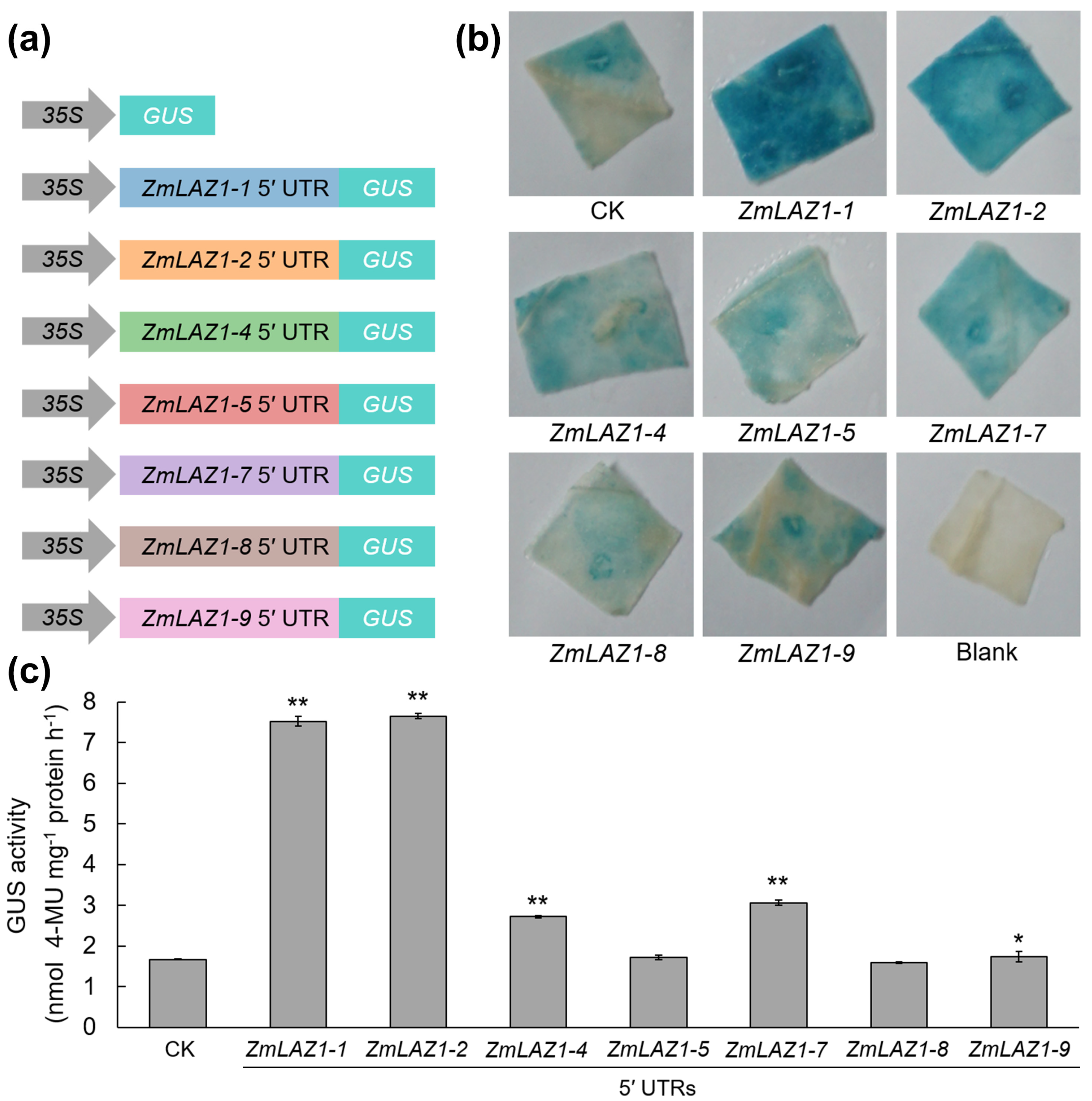
3.3. Characterization of the 5′ UTR Region of the ZmLAZ1 Gene Family
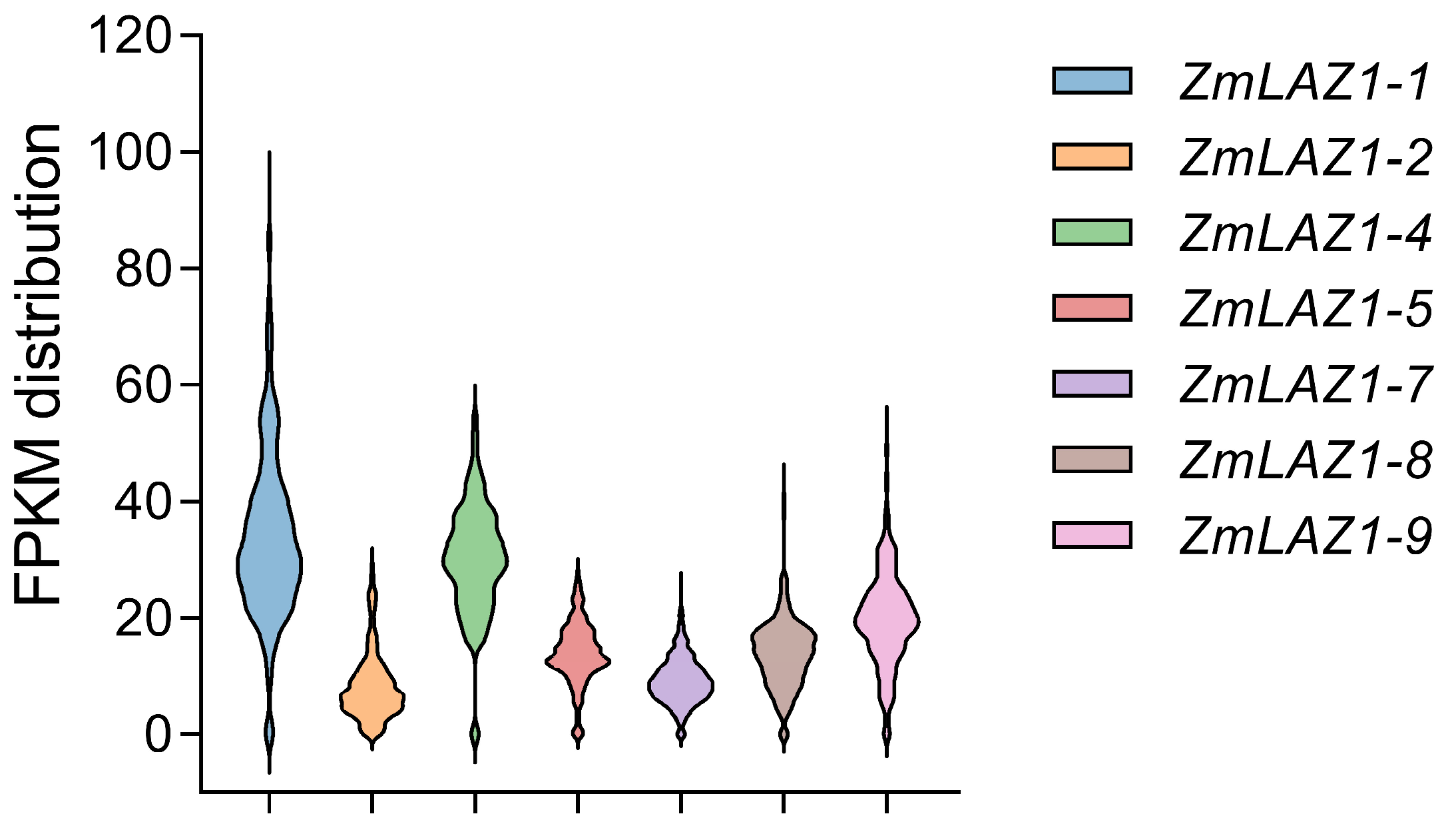
3.4. Effect of 5′ UTR Length on Gene Expression
3.5. Cis-Elements Analysis of 5′ UTR of ZmLAZ1 Gene Family
3.6. The Potential Relationship between 5′ UTR Length and Gene Function in the LAZ1 Gene Family
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mortimer, S.A.; Kidwell, M.A.; Doudna, J.A. Insights into RNA structure and function from genome-wide studies. Nat. Rev. Genet. 2014, 15, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Cerezo, M.; Robert, C.; Liu, L.; Shen, S. The Role of mRNA Translational Control in Tumor Immune Escape and Immunotherapy Resistance. Cancer Res. 2021, 81, 5596–5604. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, L.; Zhang, Y.; Wang, H.; Dong, D.; Zhu, J.; Fu, W.; Liu, T. Adipose-derived stem cells enriched with therapeutic mRNA TGF-β3 and IL-10 synergistically promote scar-less wound healing in preclinical models. Bioeng. Transl. Med. 2023, 9, e10620. [Google Scholar] [CrossRef] [PubMed]
- Wissink, E.M.; Fogarty, E.A.; Grimson, A. High-throughput discovery of post-transcriptional cis-regulatory elements. BMC Genom. 2016, 17, 177. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.Y.W.; Simons, C.; Firth, A.E.; Brown, C.M.; Hellens, R.P. Effect of 5′UTR introns on gene expression in Arabidopsis thaliana. BMC Genom. 2006, 7, 120. [Google Scholar] [CrossRef]
- Hardy, E.C.; Balcerowicz, M. Untranslated yet indispensable—UTRs act as key regulators in the environmental control of gene expression. J. Exp. Bot. 2024, 75, 4314–4331. [Google Scholar] [CrossRef]
- Holcik, M.; Sonenberg, N. Translational control in stress and apoptosis. Nat. Rev. Mol. Cell Biol. 2005, 6, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.; Tian, S.; Fujii, K.; Kladwang, W.; Das, R.; Barna, M. RNA regulons in HOX 5′ UTRs confer ribosome specificity to gene regulation. Nature 2015, 517, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Michaud, M.; Ubrig, E.; Filleur, S.; Erhardt, M.; Ephritikhine, G.; Maréchal-Drouard, L.; Duchêne, A.-M. Differential targeting of VDAC3 mRNA isoforms influences mitochondria morphology. Proc. Natl. Acad. Sci. USA 2014, 111, 8991–8996. [Google Scholar] [CrossRef]
- He, Y.; Doyle, M.R.; Amasino, R.M. PAF1-complex-mediated histone methylation of FLOWERING LOCUS C chromatin is required for the vernalization-responsive, winter-annual habit in Arabidopsis. Genes Dev. 2004, 18, 2774–2784. [Google Scholar] [CrossRef]
- Singla, B.; Khurana, J.P.; Khurana, P. Structural Characterization and Expression Analysis of the SERK/SERL Gene Family in Rice (Oryza sativa). Int. J. Plant Genom. 2009, 2009, 539402. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Qiao, H.; Qiu, W.; Xu, X.; Liu, T.; Jiang, Q.; Liu, R.; Jiao, Z.; Zhang, K.; Bi, L.; et al. Identification and functional characterization of intermediate-size non-coding RNAs in maize. BMC Genom. 2018, 19, 730. [Google Scholar] [CrossRef] [PubMed]
- McCue, A.D.; Nuthikattu, S.; Reeder, S.H.; Slotkin, R.K. Gene Expression and Stress Response Mediated by the Epigenetic Regulation of a Transposable Element Small RNA. PLoS Genet. 2012, 8, e1002474. [Google Scholar] [CrossRef] [PubMed]
- Cailliatte, R.; Schikora, A.; Briat, J.-F.; Mari, S.; Curie, C. High-Affinity Manganese Uptake by the Metal Transporter NRAMP1 Is Essential for Arabidopsis Growth in Low Manganese Conditions. Plant Cell 2010, 22, 904–917. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Wang, Y.; Li, B.; Chang, J.; Chen, M.; Li, K.; Yang, G.; He, G. TaNAC29, a NAC transcription factor from wheat, enhances salt and drought tolerance in transgenic Arabidopsis. BMC Plant Biol. 2015, 15, 268. [Google Scholar] [CrossRef] [PubMed]
- Shumbe, L.; Soares, E.; Muhovski, Y.; Smit, I.; Vanderschuren, H. Mutation of the Vinv 5′ UTR regulatory region reduces acrylamide levels in processed potato to reach EU food-safety standards. Plant Biotechnol. J. 2024. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Lin, S.; Zhang, Q.; Lei, Y.; Zhang, Z. Functional analysis of 5′ untranslated region of a TIR-NBS-encoding gene from triploid white poplar. Mol. Genet. Genom. 2009, 282, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Araujo, P.R.; Yoon, K.; Ko, D.; Smith, A.D.; Qiao, M.; Suresh, U.; Burns, S.C.; Penalva, L.O.F. Before It Gets Started: Regulating Translation at the 5′ UTR. Comp. Funct. Genom. 2012, 2012, 475731. [Google Scholar] [CrossRef] [PubMed]
- Davuluri, R.V.; Suzuki, Y.; Sugano, S.; Zhang, M.Q. CART classification of human 5′ UTR sequences. Genome Res. 2000, 10, 1807–1816. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Lu, Y.; Zinta, G.; Lang, Z.; Zhu, J.-K. UTR-Dependent Control of Gene Expression in Plants. Trends Plant Sci. 2018, 23, 248–259. [Google Scholar] [CrossRef]
- Palma, K.; Thorgrimsen, S.; Malinovsky, F.G.; Fiil, B.K.; Nielsen, H.B.; Brodersen, P.; Hofius, D.; Petersen, M.; Mundy, J. Autoimmunity in Arabidopsis acd11 Is Mediated by Epigenetic Regulation of an Immune Receptor. PLoS Pathog. 2010, 6, e1001137. [Google Scholar] [CrossRef]
- Liu, Q.; Vain, T.; Viotti, C.; Doyle, S.M.; Tarkowská, D.; Novák, O.; Zipfel, C.; Sitbon, F.; Robert, S.; Hofius, D. Vacuole Integrity Maintained by DUF300 Proteins Is Required for Brassinosteroid Signaling Regulation. Mol. Plant 2018, 11, 553–567. [Google Scholar] [CrossRef]
- Liu, B.L.; Yu, H.Q.; Wen, Q.; Fu, F.L.; Li, W.C. Genome-Wide Analysis of LAZ1 Gene Family from Maize. J. Plant Growth Regul. 2019, 39, 656–668. [Google Scholar] [CrossRef]
- Yu, H.; Liu, B.; Yang, Q.; Yang, Q.; Li, W.; Fu, F. Maize ZmLAZ1-3 gene negatively regulates drought tolerance in transgenic Arabidopsis. BMC Plant Biol. 2024, 24, 246. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Yu, H.; Yang, Q.; Ding, L.; Sun, F.; Qu, J.; Feng, W.; Yang, Q.; Li, W.; Fu, F. Zinc Transporter ZmLAZ1-4 Modulates Zinc Homeostasis on Plasma and Vacuolar Membrane in Maize. Front. Plant Sci. 2022, 13, 881055. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Peluso, P.; Shi, J.; Liang, T.; Stitzer, M.C.; Wang, B.; Campbell, M.S.; Stein, J.C.; Wei, X.; Chin, C.-S.; et al. Improved maize reference genome with single-molecule technologies. Nature 2017, 546, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Jefferson, R.A.; Kavanagh, T.A.; Bevan, M.W. GUS fusions: β-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J. 1987, 6, 3901–3907. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, E.; Godoy Herz, M.A.; Barta, A.; Kalyna, M.; Kornblihtt, A.R. Let there be light: Regulation of gene expression in plants. RNA Biol. 2014, 11, 1215–1220. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, H.; Long, Y.; Shu, Y.; Zhai, J. Plant Public RNA-seq Database: A comprehensive online database for expression analysis of ~45,000 plant public RNA-Seq libraries. Plant Biotechnol. J. 2022, 20, 806–808. [Google Scholar] [CrossRef]
- Malinovsky, F.G.; Brodersen, P.; Fiil, B.K.; McKinney, L.V.; Thorgrimsen, S.; Beck, M.; Nielsen, H.B.; Pietra, S.; Zipfel, C.; Robatzek, S.; et al. Lazarus1, a DUF300 Protein, Contributes to Programmed Cell Death Associated with Arabidopsis acd11 and the Hypersensitive Response. PLoS ONE 2010, 5, e12586. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Seward, D.J.; Li, L.; Boyer, J.L.; Ballatori, N. Expression cloning of two genes that together mediate organic solute and steroid transport in the liver of a marine vertebrate. Proc. Natl. Acad. Sci. USA 2001, 98, 9431–9436. [Google Scholar] [CrossRef] [PubMed]
- Gage, J.L.; Mali, S.; McLoughlin, F.; Khaipho-Burch, M.; Monier, B.; Bailey-Serres, J.; Vierstra, R.D.; Buckler, E.S. Variation in upstream open reading frames contributes to allelic diversity in maize protein abundance. Proc. Natl. Acad. Sci. USA 2022, 119, e2112516119. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, B.; Liu, X.; Sun, M.; Sun, Y.; Liu, D.; Hao, L.; Tao, Y. Analysis of the 5′ Untranslated Region Length-Dependent Control of Gene Expression in Maize: A Case Study with the ZmLAZ1 Gene Family. Genes 2024, 15, 994. https://doi.org/10.3390/genes15080994
Liu B, Liu X, Sun M, Sun Y, Liu D, Hao L, Tao Y. Analysis of the 5′ Untranslated Region Length-Dependent Control of Gene Expression in Maize: A Case Study with the ZmLAZ1 Gene Family. Genes. 2024; 15(8):994. https://doi.org/10.3390/genes15080994
Chicago/Turabian StyleLiu, Bingliang, Xiaowei Liu, Min Sun, Yanxia Sun, Dayu Liu, Li Hao, and Yang Tao. 2024. "Analysis of the 5′ Untranslated Region Length-Dependent Control of Gene Expression in Maize: A Case Study with the ZmLAZ1 Gene Family" Genes 15, no. 8: 994. https://doi.org/10.3390/genes15080994