
The Role of the Large T Antigen in the Molecular Pathogenesis of Merkel Cell Carcinoma

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction to Merkel Cell Carcinoma
2. The Merkel Cell Polyomavirus
2.1. General Properties of the Virus
2.2. The Large T Antigen
3. MCPyV-Mediated Carcinogenesis
3.1. Truncating Mutations in the Large T Antigen
3.2. Somatic Integration of MCPyV Viral DNA
3.3. Carcinogenic Effects of truncLT
3.3.1. Disruption of the Binding of pRb and E2F Transcription Factors
3.3.2. Interaction with p53
3.3.3. Deregulation of mi-375 by Protein Atonal Homolog 1 (ATOH1)
4. Immune Escape Mechanisms of MCPyV
4.1. Downregulation of TLR9 Maintains Chronic Infection of the Cell
4.2. MHC-Dependent Immune Escape
5. LT-Based Treatment Strategies in MCC
5.1. T-Cell-Based Immunotherapy
5.2. Large T Antigen-Based Vaccine Approaches
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Houben, R.; Celikdemir, B.; Kervarrec, T.; Schrama, D. Merkel Cell Polyomavirus: Infection, Genome, Transcripts and Its Role in Development of Merkel Cell Carcinoma. Cancers 2023, 15, 444. [Google Scholar] [CrossRef]
- Thibault, K. Evidence of an epithelial origin of Merkel cell carcinoma. Mod. Pathol. 2022, 35, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Rosado, P.; Junquera, L.; Vivanco, B.; Garcia-Consuegra, L.; Gallego, L. Merkel cell carcinoma: Our experience in this rare pathology. Med. Oral Patol. Oral Cir. Bucal 2011, 16, e736–e739. [Google Scholar] [CrossRef] [PubMed]
- Kervarrec, T.; Samimi, M.; Guyetant, S.; Sarma, B.; Cheret, J.; Blanchard, E.; Berthon, P.; Schrama, D.; Houben, R.; Touze, A. Histogenesis of Merkel Cell Carcinoma: A Comprehensive Review. Front. Oncol. 2019, 9, 451. [Google Scholar] [CrossRef]
- Harms, P.W.; Harms, K.L.; Moore, P.S.; DeCaprio, J.A.; Nghiem, P.; Wong, M.K.K.; Brownell, I. The biology and treatment of Merkel cell carcinoma: Current understanding and research priorities. Nat. Rev. Clin. Oncol. 2018, 15, 763–776. [Google Scholar] [CrossRef]
- Toker, C. Trabecular carcinoma of the skin. Arch. Dermatol. Res. 1972, 105, 107–110. [Google Scholar] [CrossRef]
- Munde, P.B.; Khandekar, S.P.; Dive, A.M.; Sharma, A. Pathophysiology of merkel cell. J. Oral Maxillofac. Pathol. 2013, 17, 408–412. [Google Scholar] [CrossRef]
- Becker, J.C.; Stang, A.; DeCaprio, J.A.; Cerroni, L.; Lebbe, C.; Veness, M.; Nghiem, P. Merkel cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17077. [Google Scholar] [CrossRef]
- Tang, C.K.; Toker, C. Trabecular carcinoma of the skin: An ultrastructural study. Cancer 1978, 42, 2311–2321. [Google Scholar] [CrossRef]
- Pulitzer, M.P.; Amin, B.D.; Busam, K.J. Merkel cell carcinoma: Review. Adv. Anat. Pathol. 2009, 16, 135–144. [Google Scholar] [CrossRef]
- Sunshine, J.C.; Jahchan, N.S.; Sage, J.; Choi, J. Are there multiple cells of origin of Merkel cell carcinoma? Oncogene 2018, 37, 1409–1416. [Google Scholar] [CrossRef] [PubMed]
- Sample, A.; He, Y.Y. Mechanisms and prevention of UV-induced melanoma. Photodermatol. Photoimmunol. Photomed. 2018, 34, 13–24. [Google Scholar] [CrossRef]
- Hedberg, M.L.; Berry, C.T.; Moshiri, A.S.; Xiang, Y.; Yeh, C.J.; Attilasoy, C.; Capell, B.C.; Seykora, J.T. Molecular Mechanisms of Cutaneous Squamous Cell Carcinoma. Int. J. Mol. Sci. 2022, 23, 3478. [Google Scholar] [CrossRef]
- Watson, M.; Holman, D.M.; Maguire-Eisen, M. Ultraviolet Radiation Exposure and Its Impact on Skin Cancer Risk. Semin. Oncol. Nurs. 2016, 32, 241–254. [Google Scholar] [CrossRef]
- Hoashi, T.; Kanda, N.; Saeki, H. Molecular Mechanisms and Targeted Therapies of Advanced Basal Cell Carcinoma. Int. J. Mol. Sci. 2022, 23, 1968. [Google Scholar] [CrossRef]
- Feng, H.; Taylor, J.L.; Benos, P.V.; Newton, R.; Waddell, K.; Lucas, S.B.; Chang, Y.; Moore, P.S. Human transcriptome subtraction by using short sequence tags to search for tumor viruses in conjunctival carcinoma. J. Virol. 2007, 81, 11332–11340. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef]
- Chang, Y.; Moore, P.S. Merkel cell carcinoma: A virus-induced human cancer. Annu. Rev. Pathol. 2012, 7, 123–144. [Google Scholar] [CrossRef] [PubMed]
- Czech-Sioli, M.; Günther, T.; Therre, M.; Spohn, M.; Indenbirken, D.; Theiss, J.; Riethdorf, S.; Qi, M.; Alawi, M.; Wülbeck, C.; et al. High-resolution analysis of Merkel Cell Polyomavirus in Merkel Cell Carcinoma reveals distinct integration patterns and suggests NHEJ and MMBIR as underlying mechanisms. PLoS Pathog. 2020, 16, e1008562. [Google Scholar] [CrossRef]
- Stang, A.; Becker, J.C.; Nghiem, P.; Ferlay, J. The association between geographic location and incidence of Merkel cell carcinoma in comparison to melanoma: An international assessment. Eur. J. Cancer 2018, 94, 47–60. [Google Scholar] [CrossRef]
- Youlden, D.R.; Soyer, H.P.; Youl, P.H.; Fritschi, L.; Baade, P.D. Incidence and survival for Merkel cell carcinoma in Queensland, Australia, 1993–2010. JAMA Dermatol. 2014, 150, 864–872. [Google Scholar] [CrossRef] [PubMed]
- BfS—UV Index Worldwide. Available online: https://www.bfs.de/en/topics/opt/uv/index/worldwide/worldwide.html (accessed on 30 March 2024).
- Harms, P.W.; Vats, P.; Verhaegen, M.E.; Robinson, D.R.; Wu, Y.M.; Dhanasekaran, S.M.; Palanisamy, N.; Siddiqui, J.; Cao, X.; Su, F.; et al. The Distinctive Mutational Spectra of Polyomavirus-Negative Merkel Cell Carcinoma. Cancer Res. 2015, 75, 3720–3727. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.M.; Cushman, C.H.; DeCaprio, J.A. Merkel Cell Polyomavirus: Oncogenesis in a Stable Genome. Viruses 2021, 14, 58. [Google Scholar] [CrossRef] [PubMed]
- Viscidi, R.P.; Rollison, D.E.; Sondak, V.K.; Silver, B.; Messina, J.L.; Giuliano, A.R.; Fulp, W.; Ajidahun, A.; Rivanera, D. Age-specific seroprevalence of Merkel cell polyomavirus, BK virus, and JC virus. Clin. Vaccine Immunol. 2011, 18, 1737–1743. [Google Scholar] [CrossRef]
- Chen, T.; Hedman, L.; Mattila, P.S.; Jartti, T.; Ruuskanen, O.; Soderlund-Venermo, M.; Hedman, K. Serological evidence of Merkel cell polyomavirus primary infections in childhood. J. Clin. Virol. 2011, 50, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Bopp, L.; Wieland, U.; Hellmich, M.; Kreuter, A.; Pfister, H.; Silling, S. Natural History of Cutaneous Human Polyomavirus Infection in Healthy Individuals. Front. Microbiol. 2021, 12, 740947. [Google Scholar] [CrossRef]
- Tolstov, Y.L.; Knauer, A.; Chen, J.G.; Kensler, T.W.; Kingsley, L.A.; Moore, P.S.; Chang, Y. Asymptomatic primary Merkel cell polyomavirus infection among adults. Emerg. Infect. Dis. 2011, 17, 1371–1380. [Google Scholar] [CrossRef]
- Spurgeon, M.E.; Lambert, P.F. Merkel cell polyomavirus: A newly discovered human virus with oncogenic potential. Virology 2013, 435, 118–130. [Google Scholar] [CrossRef]
- Krump, N.A.; You, J. From Merkel Cell Polyomavirus Infection to Merkel Cell Carcinoma Oncogenesis. Front. Microbiol. 2021, 12, 739695. [Google Scholar] [CrossRef]
- Liu, W.; You, J. Molecular Mechanisms of Merkel Cell Polyomavirus Transformation and Replication. Annu. Rev. Virol. 2020, 7, 289–307. [Google Scholar] [CrossRef]
- Spurgeon, M.E.; Cheng, J.; Ward-Shaw, E.; Dick, F.A.; DeCaprio, J.A.; Lambert, P.F. Merkel cell polyomavirus large T antigen binding to pRb promotes skin hyperplasia and tumor development. PLoS Pathog. 2022, 18, e1010551. [Google Scholar] [CrossRef]
- Czech-Sioli, M.; Siebels, S.; Radau, S.; Zahedi, R.P.; Schmidt, C.; Dobner, T.; Grundhoff, A.; Fischer, N. The Ubiquitin-Specific Protease Usp7, a Novel Merkel Cell Polyomavirus Large T-Antigen Interaction Partner, Modulates Viral DNA Replication. J. Virol. 2020, 94, 101128. [Google Scholar] [CrossRef] [PubMed]
- Wendzicki, J.A.; Moore, P.S.; Chang, Y. Large T and small T antigens of Merkel cell polyomavirus. Curr. Opin. Virol. 2015, 11, 38–43. [Google Scholar] [CrossRef]
- Neu, U.; Hengel, H.; Blaum, B.S.; Schowalter, R.M.; Macejak, D.; Gilbert, M.; Wakarchuk, W.W.; Imamura, A.; Ando, H.; Kiso, M.; et al. Structures of Merkel cell polyomavirus VP1 complexes define a sialic acid binding site required for infection. PLoS Pathog. 2012, 8, e1002738. [Google Scholar] [CrossRef]
- Topalis, D.; Andrei, G.; Snoeck, R. The large tumor antigen: A “Swiss Army knife” protein possessing the functions required for the polyomavirus life cycle. Antivir. Res. 2013, 97, 122–136. [Google Scholar] [CrossRef]
- DeCaprio, J.A.; Garcea, R.L. A cornucopia of human polyomaviruses. Nat. Rev. Microbiol. 2013, 11, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Cho, U.S.; Morrone, S.; Sablina, A.A.; Arroyo, J.D.; Hahn, W.C.; Xu, W. Structural basis of PP2A inhibition by small t antigen. PLoS Biol. 2007, 5, e202. [Google Scholar] [CrossRef] [PubMed]
- Kwun, H.J.; Guastafierro, A.; Shuda, M.; Meinke, G.; Bohm, A.; Moore, P.S.; Chang, Y. The minimum replication origin of merkel cell polyomavirus has a unique large T-antigen loading architecture and requires small T-antigen expression for optimal replication. J. Virol. 2009, 83, 12118–12128. [Google Scholar] [CrossRef]
- Liu, X.; Hein, J.; Richardson, S.C.; Basse, P.H.; Toptan, T.; Moore, P.S.; Gjoerup, O.V.; Chang, Y. Merkel cell polyomavirus large T antigen disrupts lysosome clustering by translocating human Vam6p from the cytoplasm to the nucleus. J. Biol. Chem. 2011, 286, 17079–17090. [Google Scholar] [CrossRef]
- Cotsiki, M.; Lock, R.L.; Cheng, Y.; Williams, G.L.; Zhao, J.; Perera, D.; Freire, R.; Entwistle, A.; Golemis, E.A.; Roberts, T.M.; et al. Simian virus 40 large T antigen targets the spindle assembly checkpoint protein Bub1. Proc. Natl. Acad. Sci. USA 2004, 101, 947–952. [Google Scholar] [CrossRef]
- Lane, D.P.; Crawford, L.V. T antigen is bound to a host protein in SV40-transformed cells. Nature 1979, 278, 261–263. [Google Scholar] [CrossRef]
- DeAngelis, T.; Chen, J.; Wu, A.; Prisco, M.; Baserga, R. Transformation by the simian virus 40 T antigen is regulated by IGF-I receptor and IRS-1 signaling. Oncogene 2006, 25, 32–42. [Google Scholar] [CrossRef]
- Dias, D.C.; Dolios, G.; Wang, R.; Pan, Z.Q. CUL7: A DOC domain-containing cullin selectively binds Skp1.Fbx29 to form an SCF-like complex. Proc. Natl. Acad. Sci. USA 2002, 99, 16601–16606. [Google Scholar] [CrossRef]
- Feldman, R.M.; Correll, C.C.; Kaplan, K.B.; Deshaies, R.J. A complex of Cdc4p, Skp1p, and Cdc53p/cullin catalyzes ubiquitination of the phosphorylated CDK inhibitor Sic1p. Cell 1997, 91, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Jung, P.; Verdoodt, B.; Bailey, A.; Yates, J.R., 3rd; Menssen, A.; Hermeking, H. Induction of cullin 7 by DNA damage attenuates p53 function. Proc. Natl. Acad. Sci. USA 2007, 104, 11388–11393. [Google Scholar] [CrossRef]
- Kasper, J.S.; Kuwabara, H.; Arai, T.; Ali, S.H.; DeCaprio, J.A. Simian virus 40 large T antigen’s association with the CUL7 SCF complex contributes to cellular transformation. J. Virol. 2005, 79, 11685–11692. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.H.; Kasper, J.S.; Arai, T.; DeCaprio, J.A. Cul7/p185/p193 binding to simian virus 40 large T antigen has a role in cellular transformation. J. Virol. 2004, 78, 2749–2757. [Google Scholar] [CrossRef] [PubMed]
- Hesbacher, S.; Pfitzer, L.; Wiedorfer, K.; Angermeyer, S.; Borst, A.; Haferkamp, S.; Scholz, C.-J.; Wobser, M.; Schrama, D.; Houben, R. RB1 is the crucial target of the Merkel cell polyomavirus Large T antigen in Merkel cell carcinoma cells. Oncotarget 2016, 7, 32956–32968. [Google Scholar] [CrossRef]
- Houben, R.; Adam, C.; Baeurle, A.; Hesbacher, S.; Grimm, J.; Angermeyer, S.; Henzel, K.; Hauser, S.; Elling, R.; Brocker, E.B.; et al. An intact retinoblastoma protein-binding site in Merkel cell polyomavirus large T antigen is required for promoting growth of Merkel cell carcinoma cells. Int. J. Cancer 2012, 130, 847–856. [Google Scholar] [CrossRef]
- Liu, T.; Daniels, C.K.; Cao, S. Comprehensive review on the HSC70 functions, interactions with related molecules and involvement in clinical diseases and therapeutic potential. Pharmacol. Ther. 2012, 136, 354–374. [Google Scholar] [CrossRef]
- Borowiec, J.A.; Dean, F.B.; Bullock, P.A.; Hurwitz, J. Binding and unwinding—How T antigen engages the SV40 origin of DNA replication. Cell 1990, 60, 181–184. [Google Scholar] [CrossRef]
- Fanning, E.; Zhao, K. SV40 DNA replication: From the A gene to a nanomachine. Virology 2009, 384, 352–359. [Google Scholar] [CrossRef]
- Stakaityte, G.; Wood, J.J.; Knight, L.M.; Abdul-Sada, H.; Adzahar, N.S.; Nwogu, N.; Macdonald, A.; Whitehouse, A. Merkel cell polyomavirus: Molecular insights into the most recently discovered human tumour virus. Cancers 2014, 6, 1267–1297. [Google Scholar] [CrossRef] [PubMed]
- Gai, D.; Li, D.; Finkielstein, C.V.; Ott, R.D.; Taneja, P.; Fanning, E.; Chen, X.S. Insights into the oligomeric states, conformational changes, and helicase activities of SV40 large tumor antigen. J. Biol. Chem. 2004, 279, 38952–38959. [Google Scholar] [CrossRef]
- Hickman, A.B.; Dyda, F. Binding and unwinding: SF3 viral helicases. Curr. Opin. Struct. Biol. 2005, 15, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Marei, H.E.; Althani, A.; Afifi, N.; Hasan, A.; Caceci, T.; Pozzoli, G.; Morrione, A.; Giordano, A.; Cenciarelli, C. p53 signaling in cancer progression and therapy. Cancer Cell Int. 2021, 21, 703. [Google Scholar] [CrossRef] [PubMed]
- Pietropaolo, V.; Prezioso, C.; Moens, U. Merkel Cell Polyomavirus and Merkel Cell Carcinoma. Cancers 2020, 12, 1774. [Google Scholar] [CrossRef]
- Cheng, J.; Rozenblatt-Rosen, O.; Paulson, K.G.; Nghiem, P.; DeCaprio, J.A. Merkel cell polyomavirus large T antigen has growth-promoting and inhibitory activities. J. Virol. 2013, 87, 6118–6126. [Google Scholar] [CrossRef]
- Kwun, H.J.; Chang, Y.; Moore, P.S. Protein-mediated viral latency is a novel mechanism for Merkel cell polyomavirus persistence. Proc. Natl. Acad. Sci. USA 2017, 114, E4040–E4047. [Google Scholar] [CrossRef]
- Giannecchini, S. Evidence of the Mechanism by Which Polyomaviruses Exploit the Extracellular Vesicle Delivery System during Infection. Viruses 2020, 12, 585. [Google Scholar] [CrossRef]
- Chen, Y.; Williams, V.; Filippova, M.; Filippov, V.; Duerksen-Hughes, P. Viral carcinogenesis: Factors inducing DNA damage and virus integration. Cancers 2014, 6, 2155–2186. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.J.; Paulson, K.G.; Wipf, G.C.; Miranda, D.; Madeleine, M.M.; Johnson, L.G.; Lemos, B.D.; Lee, S.; Warcola, A.H.; Iyer, J.G.; et al. Association of Merkel cell polyomavirus-specific antibodies with Merkel cell carcinoma. J. Natl. Cancer Inst. 2009, 101, 1510–1522. [Google Scholar] [CrossRef]
- Shuda, M.; Feng, H.; Kwun, H.J.; Rosen, S.T.; Gjoerup, O.; Moore, P.S.; Chang, Y. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc. Natl. Acad. Sci. USA 2008, 105, 16272–16277. [Google Scholar] [CrossRef] [PubMed]
- Shuda, M.; Arora, R.; Kwun, H.J.; Feng, H.; Sarid, R.; Fernandez-Figueras, M.T.; Tolstov, Y.; Gjoerup, O.; Mansukhani, M.M.; Swerdlow, S.H.; et al. Human Merkel cell polyomavirus infection I. MCV T antigen expression in Merkel cell carcinoma, lymphoid tissues and lymphoid tumors. Int. J. Cancer 2009, 125, 1243–1249. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, S.K.; Lang, P.A.; Friebus-Kardash, J.; Duhan, V.; Bezgovsek, J.; Lang, K.S. Mechanisms of lymphatic system-specific viral replication and its potential role in autoimmune disease. Clin. Exp. Immunol. 2019, 195, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Soikkeli, A.I.; Kylaniemi, M.K.; Sihto, H.; Alinikula, J. Oncogenic Merkel Cell Polyomavirus T Antigen Truncating Mutations are Mediated by APOBEC3 Activity in Merkel Cell Carcinoma. Cancer Res. Commun. 2022, 2, 1344–1354. [Google Scholar] [CrossRef]
- Ortiz, L.E.; Pham, A.M.; Kwun, H.J. Identification of the Merkel Cell Polyomavirus Large Tumor Antigen Ubiquitin Conjugation Residue. Int. J. Mol. Sci. 2021, 22, 7169. [Google Scholar] [CrossRef]
- Angermeyer, S.; Hesbacher, S.; Becker, J.C.; Schrama, D.; Houben, R. Merkel cell polyomavirus-positive Merkel cell carcinoma cells do not require expression of the viral small T antigen. J. Investig. Dermatol. 2013, 133, 2059–2064. [Google Scholar] [CrossRef]
- Shuda, M.; Chang, Y.; Moore, P.S. Merkel cell polyomavirus-positive Merkel cell carcinoma requires viral small T-antigen for cell proliferation. J. Investig. Dermatol. 2014, 134, 1479–1481. [Google Scholar] [CrossRef]
- Landry, S.; Narvaiza, I.; Linfesty, D.C.; Weitzman, M.D. APOBEC3A can activate the DNA damage response and cause cell-cycle arrest. EMBO Rep. 2011, 12, 444–450. [Google Scholar] [CrossRef]
- Petljak, M.; Maciejowski, J. Molecular origins of APOBEC-associated mutations in cancer. DNA Repair. 2020, 94, 102905. [Google Scholar] [CrossRef] [PubMed]
- Kuraoka, M.; McWilliams, L.; Kelsoe, G. AID expression during B-cell development: Searching for answers. Immunol. Res. 2011, 49, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.S. Insights into the Structures and Multimeric Status of APOBEC Proteins Involved in Viral Restriction and Other Cellular Functions. Viruses 2021, 13, 497. [Google Scholar] [CrossRef]
- Poulain, F.; Lejeune, N.; Willemart, K.; Gillet, N.A. Footprint of the host restriction factors APOBEC3 on the genome of human viruses. PLoS Pathog. 2020, 16, e1008718. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Dudley, J.P. APOBECs and virus restriction. Virology 2015, 479–480, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Owiti, N.; Stokdyk, K.; Kim, N. The etiology of uracil residues in the Saccharomyces cerevisiae genomic DNA. Curr. Genet. 2019, 65, 393–399. [Google Scholar] [CrossRef]
- Verhalen, B.; Starrett, G.J.; Harris, R.S.; Jiang, M. Functional Upregulation of the DNA Cytosine Deaminase APOBEC3B by Polyomaviruses. J. Virol. 2016, 90, 6379–6386. [Google Scholar] [CrossRef]
- Mineeva-Sangwo, O.; Van Loon, E.; Andrei, G.; Kuypers, D.; Naesens, M.; Snoeck, R. Time-dependent variations in BK polyomavirus genome from kidney transplant recipients with persistent viremia. Sci. Rep. 2023, 13, 13534. [Google Scholar] [CrossRef]
- Tognon, M.; Corallini, A.; Manfrini, M.; Taronna, A.; Butel, J.S.; Pietrobon, S.; Trevisiol, L.; Bononi, I.; Vaccher, E.; Barbanti-Brodano, G.; et al. Specific Antibodies Reacting with SV40 Large T Antigen Mimotopes in Serum Samples of Healthy Subjects. PLoS ONE 2016, 11, e0145720. [Google Scholar] [CrossRef]
- Abend, J.R.; Joseph, A.E.; Das, D.; Campbell-Cecen, D.B.; Imperiale, M.J. A truncated T antigen expressed from an alternatively spliced BK virus early mRNA. J. Gen. Virol. 2009, 90, 1238–1245. [Google Scholar] [CrossRef]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.; Wieland, U.; Kreuter, A.; Pawlita, M. C-terminal deletions of Merkel cell polyomavirus large T-antigen, a highly specific surrogate marker for virally induced malignancy. Int. J. Cancer 2012, 131, 2863–2868. [Google Scholar] [CrossRef]
- Schrama, D.; Peitsch, W.K.; Zapatka, M.; Kneitz, H.; Houben, R.; Eib, S.; Haferkamp, S.; Moore, P.S.; Shuda, M.; Thompson, J.F.; et al. Merkel cell polyomavirus status is not associated with clinical course of Merkel cell carcinoma. J. Investig. Dermatol. 2011, 131, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Schrama, D.; Sarosi, E.M.; Adam, C.; Ritter, C.; Kaemmerer, U.; Klopocki, E.; Konig, E.M.; Utikal, J.; Becker, J.C.; Houben, R. Characterization of six Merkel cell polyomavirus-positive Merkel cell carcinoma cell lines: Integration pattern suggest that large T antigen truncating events occur before or during integration. Int. J. Cancer 2019, 145, 1020–1032. [Google Scholar] [CrossRef]
- Starrett, G.J.; Thakuria, M.; Chen, T.; Marcelus, C.; Cheng, J.; Nomburg, J.; Thorner, A.R.; Slevin, M.K.; Powers, W.; Burns, R.T.; et al. Clinical and molecular characterization of virus-positive and virus-negative Merkel cell carcinoma. Genome Med. 2020, 12, 30. [Google Scholar] [CrossRef]
- Hashida, Y.; Imajoh, M.; Nemoto, Y.; Kamioka, M.; Taniguchi, A.; Taguchi, T.; Kume, M.; Orihashi, K.; Daibata, M. Detection of Merkel cell polyomavirus with a tumour-specific signature in non-small cell lung cancer. Br. J. Cancer 2013, 108, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Laude, H.C.; Jonchere, B.; Maubec, E.; Carlotti, A.; Marinho, E.; Couturaud, B.; Peter, M.; Sastre-Garau, X.; Avril, M.F.; Dupin, N.; et al. Distinct merkel cell polyomavirus molecular features in tumour and non tumour specimens from patients with merkel cell carcinoma. PLoS Pathog. 2010, 6, e1001076. [Google Scholar] [CrossRef]
- Topacio, B.R.; Zatulovskiy, E.; Cristea, S.; Xie, S.; Tambo, C.S.; Rubin, S.M.; Sage, J.; Koivomagi, M.; Skotheim, J.M. Cyclin D-Cdk4,6 Drives Cell-Cycle Progression via the Retinoblastoma Protein’s C-terminal Helix. Mol. Cell 2019, 74, 758–770.e4. [Google Scholar] [CrossRef]
- Ahuja, D.; Saenz-Robles, M.T.; Pipas, J.M. SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene 2005, 24, 7729–7745. [Google Scholar] [CrossRef]
- Sihto, H.; Kukko, H.; Koljonen, V.; Sankila, R.; Bohling, T.; Joensuu, H. Merkel cell polyomavirus infection, large T antigen, retinoblastoma protein and outcome in Merkel cell carcinoma. Clin. Cancer Res. 2011, 17, 4806–4813. [Google Scholar] [CrossRef]
- Mandigo, A.C.; Tomlins, S.A.; Kelly, W.K.; Knudsen, K.E. Relevance of pRB Loss in Human Malignancies. Clin. Cancer Res. 2022, 28, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, X.; Diaz, J.; Tsang, S.H.; Buck, C.B.; You, J. Merkel cell polyomavirus large T antigen disrupts host genomic integrity and inhibits cellular proliferation. J. Virol. 2013, 87, 9173–9188. [Google Scholar] [CrossRef] [PubMed]
- Park, D.E.; Cheng, J.; Berrios, C.; Montero, J.; Cortes-Cros, M.; Ferretti, S.; Arora, R.; Tillgren, M.L.; Gokhale, P.C.; DeCaprio, J.A. Dual inhibition of MDM2 and MDM4 in virus-positive Merkel cell carcinoma enhances the p53 response. Proc. Natl. Acad. Sci. USA 2019, 116, 1027–1032. [Google Scholar] [CrossRef]
- Lilyestrom, W.; Klein, M.G.; Zhang, R.; Joachimiak, A.; Chen, X.S. Crystal structure of SV40 large T-antigen bound to p53: Interplay between a viral oncoprotein and a cellular tumor suppressor. Genes Dev. 2006, 20, 2373–2382. [Google Scholar] [CrossRef] [PubMed]
- Loke, A.S.W.; Lambert, P.F.; Spurgeon, M.E. Current In Vitro and In Vivo Models to Study MCPyV-Associated MCC. Viruses 2022, 14, 2204. [Google Scholar] [CrossRef] [PubMed]
- Houben, R.; Dreher, C.; Angermeyer, S.; Borst, A.; Utikal, J.; Haferkamp, S.; Peitsch, W.K.; Schrama, D.; Hesbacher, S. Mechanisms of p53 restriction in Merkel cell carcinoma cells are independent of the Merkel cell polyoma virus T antigens. J. Investig. Dermatol. 2013, 133, 2453–2460. [Google Scholar] [CrossRef]
- Kumar, S.; Xie, H.; Scicluna, P.; Lee, L.; Bjornhagen, V.; Hoog, A.; Larsson, C.; Lui, W.O. MiR-375 Regulation of LDHB Plays Distinct Roles in Polyomavirus-Positive and -Negative Merkel Cell Carcinoma. Cancers 2018, 10, 443. [Google Scholar] [CrossRef]
- Fan, K.; Gravemeyer, J.; Ritter, C.; Rasheed, K.; Gambichler, T.; Moens, U.; Shuda, M.; Schrama, D.; Becker, J.C. MCPyV Large T Antigen-Induced Atonal Homolog 1 Is a Lineage-Dependency Oncogene in Merkel Cell Carcinoma. J. Investig. Dermatol. 2020, 140, 56–65.e53. [Google Scholar] [CrossRef]
- Binder, A.K.; Bremm, F.; Dorrie, J.; Schaft, N. Non-Coding RNA in Tumor Cells and Tumor-Associated Myeloid Cells-Function and Therapeutic Potential. Int. J. Mol. Sci. 2024, 25, 7275. [Google Scholar] [CrossRef]
- Abraham, K.J.; Zhang, X.; Vidal, R.; Pare, G.C.; Feilotter, H.E.; Tron, V.A. Roles for miR-375 in Neuroendocrine Differentiation and Tumor Suppression via Notch Pathway Suppression in Merkel Cell Carcinoma. Am. J. Pathol. 2016, 186, 1025–1035. [Google Scholar] [CrossRef]
- Fan, K.; Ritter, C.; Nghiem, P.; Blom, A.; Verhaegen, M.E.; Dlugosz, A.; Odum, N.; Woetmann, A.; Tothill, R.W.; Hicks, R.J.; et al. Circulating Cell-Free miR-375 as Surrogate Marker of Tumor Burden in Merkel Cell Carcinoma. Clin. Cancer Res. 2018, 24, 5873–5882. [Google Scholar] [CrossRef]
- Renwick, N.; Cekan, P.; Masry, P.A.; McGeary, S.E.; Miller, J.B.; Hafner, M.; Li, Z.; Mihailovic, A.; Morozov, P.; Brown, M.; et al. Multicolor microRNA FISH effectively differentiates tumor types. J. Clin. Investig. 2013, 123, 2694–2702. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, S.M.; Wright, M.C.; Bolock, A.M.; Geng, X.; Maricich, S.M. Ectopic Atoh1 expression drives Merkel cell production in embryonic, postnatal and adult mouse epidermis. Development 2015, 142, 2533–2544. [Google Scholar] [CrossRef]
- Verhaegen, M.E.; Mangelberger, D.; Harms, P.W.; Eberl, M.; Wilbert, D.M.; Meireles, J.; Bichakjian, C.K.; Saunders, T.L.; Wong, S.Y.; Dlugosz, A.A. Merkel Cell Polyomavirus Small T Antigen Initiates Merkel Cell Carcinoma-like Tumor Development in Mice. Cancer Res. 2017, 77, 3151–3157. [Google Scholar] [CrossRef] [PubMed]
- Harold, A.; Amako, Y.; Hachisuka, J.; Bai, Y.; Li, M.Y.; Kubat, L.; Gravemeyer, J.; Franks, J.; Gibbs, J.R.; Park, H.J.; et al. Conversion of Sox2-dependent Merkel cell carcinoma to a differentiated neuron-like phenotype by T antigen inhibition. Proc. Natl. Acad. Sci. USA 2019, 116, 20104–20114. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Yuan, S.S.; Zhang, L.J.; Ji, Z.L.; Quan, X.J. Atonal bHLH transcription factor 1 is an important factor for maintaining the balance of cell proliferation and differentiation in tumorigenesis. Oncol. Lett. 2020, 20, 2595–2605. [Google Scholar] [CrossRef]
- Jani, S.; Church, C.D.; Nghiem, P. Insights into anti-tumor immunity via the polyomavirus shared across human Merkel cell carcinomas. Front. Immunol. 2023, 14, 1172913. [Google Scholar] [CrossRef]
- Engels, E.A.; Frisch, M.; Goedert, J.J.; Biggar, R.J.; Miller, R.W. Merkel cell carcinoma and HIV infection. Lancet 2002, 359, 497–498. [Google Scholar] [CrossRef]
- Paulson, K.G.; Iyer, J.G.; Blom, A.; Warton, E.M.; Sokil, M.; Yelistratova, L.; Schuman, L.; Nagase, K.; Bhatia, S.; Asgari, M.M.; et al. Systemic immune suppression predicts diminished Merkel cell carcinoma-specific survival independent of stage. J. Investig. Dermatol. 2013, 133, 642–646. [Google Scholar] [CrossRef]
- Matzinger, P. Essay 1: The Danger model in its historical context. Scand. J. Immunol. 2001, 54, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Pradeu, T.; Cooper, E.L. The danger theory: 20 years later. Front. Immunol. 2012, 3, 287. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, N.; Shuda, M.; Gheit, T.; Kwun, H.J.; Cornet, I.; Saidj, D.; Zannetti, C.; Hasan, U.; Chang, Y.; Moore, P.S.; et al. The T antigen locus of Merkel cell polyomavirus downregulates human Toll-like receptor 9 expression. J. Virol. 2013, 87, 13009–13019. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, I.; Caux, C.; Hasan, U.; Bendriss-Vermare, N.; Olive, D. Impaired Toll-like receptor 7 and 9 signaling: From chronic viral infections to cancer. Trends Immunol. 2010, 31, 391–397. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Antiviral signaling through pattern recognition receptors. J. Biochem. 2007, 141, 137–145. [Google Scholar] [CrossRef]
- Notley, C.A.; Jordan, C.K.; McGovern, J.L.; Brown, M.A.; Ehrenstein, M.R. DNA methylation governs the dynamic regulation of inflammation by apoptotic cells during efferocytosis. Sci. Rep. 2017, 7, 42204. [Google Scholar] [CrossRef]
- Wu, Y.; Yi, M.; Niu, M.; Mei, Q.; Wu, K. Myeloid-derived suppressor cells: An emerging target for anticancer immunotherapy. Mol. Cancer 2022, 21, 184. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, F.; Suzuki, K.; Sasaki, S.; Ishii, N.; Klinman, D.M.; Ishii, K.J. Transcriptional regulation of the human TLR9 gene. J. Immunol. 2004, 173, 2552–2561. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Gade, P.; Xiao, W.; Kalvakolanu, D.V. The interferon signaling network and transcription factor C/EBP-β. Cell. Mol. Immunol. 2007, 4, 407–418. [Google Scholar]
- Lyngaa, R.; Pedersen, N.W.; Schrama, D.; Thrue, C.A.; Ibrani, D.; Met, O.; Thor, S.P.; Nghiem, P.; Becker, J.C.; Hadrup, S.R. T-cell responses to oncogenic merkel cell polyomavirus proteins distinguish patients with merkel cell carcinoma from healthy donors. Clin. Cancer Res. 2014, 20, 1768–1778. [Google Scholar] [CrossRef]
- Gavvovidis, I.; Leisegang, M.; Willimsky, G.; Miller, N.; Nghiem, P.; Blankenstein, T. Targeting Merkel Cell Carcinoma by Engineered T Cells Specific to T-Antigens of Merkel Cell Polyomavirus. Clin. Cancer Res. 2018, 24, 3644–3655. [Google Scholar] [CrossRef]
- Lee, P.C.; Klaeger, S.; Le, P.M.; Korthauer, K.; Cheng, J.; Ananthapadmanabhan, V.; Frost, T.C.; Stevens, J.D.; Wong, A.Y.; Iorgulescu, J.B.; et al. Reversal of viral and epigenetic HLA class I repression in Merkel cell carcinoma. J. Clin. Investig. 2022, 132, e151666. [Google Scholar] [CrossRef] [PubMed]
- Paulson, K.G.; Tegeder, A.; Willmes, C.; Iyer, J.G.; Afanasiev, O.K.; Schrama, D.; Koba, S.; Thibodeau, R.; Nagase, K.; Simonson, W.T.; et al. Downregulation of MHC-I expression is prevalent but reversible in Merkel cell carcinoma. Cancer Immunol. Res. 2014, 2, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Iyer, J.G.; Afanasiev, O.K.; McClurkan, C.; Paulson, K.; Nagase, K.; Jing, L.; Marshak, J.O.; Dong, L.; Carter, J.; Lai, I.; et al. Merkel cell polyomavirus-specific CD8+ and CD4+ T-cell responses identified in Merkel cell carcinomas and blood. Clin. Cancer Res. 2011, 17, 6671–6680. [Google Scholar] [CrossRef]
- Zeng, Q.; Gomez, B.P.; Viscidi, R.P.; Peng, S.; He, L.; Ma, B.; Wu, T.C.; Hung, C.F. Development of a DNA vaccine targeting Merkel cell polyomavirus. Vaccine 2012, 30, 1322–1329. [Google Scholar] [CrossRef] [PubMed]
- Dowlatshahi, M.; Huang, V.; Gehad, A.E.; Jiang, Y.; Calarese, A.; Teague, J.E.; Dorosario, A.A.; Cheng, J.; Nghiem, P.; Schanbacher, C.F.; et al. Tumor-specific T cells in human Merkel cell carcinomas: A possible role for Tregs and T-cell exhaustion in reducing T-cell responses. J. Investig. Dermatol. 2013, 133, 1879–1889. [Google Scholar] [CrossRef]
- Gerer, K.F.; Erdmann, M.; Hadrup, S.R.; Lyngaa, R.; Martin, L.M.; Voll, R.E.; Schuler-Thurner, B.; Schuler, G.; Schaft, N.; Hoyer, S.; et al. Preclinical evaluation of NF-kappaB-triggered dendritic cells expressing the viral oncogenic driver of Merkel cell carcinoma for therapeutic vaccination. Ther. Adv. Med. Oncol. 2017, 9, 451–464. [Google Scholar] [CrossRef]
- Gehrcken, L.; Sauerer, T.; Schaft, N.; Dorrie, J. T-Cell Responses in Merkel Cell Carcinoma: Implications for Improved Immune Checkpoint Blockade and Other Therapeutic Options. Int. J. Mol. Sci. 2021, 22, 8679. [Google Scholar] [CrossRef]
- Cheng, J.; Park, D.E.; Berrios, C.; White, E.A.; Arora, R.; Yoon, R.; Branigan, T.; Xiao, T.; Westerling, T.; Federation, A.; et al. Merkel cell polyomavirus recruits MYCL to the EP400 complex to promote oncogenesis. PLoS Pathog. 2017, 13, e1006668. [Google Scholar] [CrossRef]
- Sauerer, T.; Lischer, C.; Weich, A.; Berking, C.; Vera, J.; Dörrie, J. Single-Molecule RNA Sequencing Reveals IFNγ-Induced Differential Expression of Immune Escape Genes in Merkel Cell Polyomavirus–Positive MCC Cell Lines. Front. Microbiol. 2021, 12, 5662. [Google Scholar] [CrossRef]
- Samimi, M.; Benlalam, H.; Aumond, P.; Gaboriaud, P.; Fradin, D.; Kervarrec, T.; Florenceau, L.; Vignard, V.; Blom, A.; Touze, A.; et al. Viral and tumor antigen-specific CD8 T-cell responses in Merkel cell carcinoma. Cell Immunol. 2019, 344, 103961. [Google Scholar] [CrossRef]
- Pulliam, T.; Jani, S.; Jing, L.; Ryu, H.; Jojic, A.; Shasha, C.; Zhang, J.; Kulikauskas, R.; Church, C.; Garnett-Benson, C.; et al. Circulating cancer-specific CD8 T cell frequency is associated with response to PD-1 blockade in Merkel cell carcinoma. Cell Rep. Med. 2024, 5, 101412. [Google Scholar] [CrossRef] [PubMed]
- Chi, Z.; Lu, Y.; Yang, Y.; Li, B.; Lu, P. Transcriptional and epigenetic regulation of PD-1 expression. Cell. Mol. Life Sci. 2021, 78, 3239–3246. [Google Scholar] [CrossRef]
- Heath, M.; Jaimes, N.; Lemos, B.; Mostaghimi, A.; Wang, L.C.; Penas, P.F.; Nghiem, P. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: The AEIOU features. J. Am. Acad. Dermatol. 2008, 58, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Babadzhanov, M.; Doudican, N.; Wilken, R.; Stevenson, M.; Pavlick, A.; Carucci, J. Current concepts and approaches to merkel cell carcinoma. Arch. Dermatol. Res. 2021, 313, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Fojnica, A.; Ljuca, K.; Akhtar, S.; Gatalica, Z.; Vranic, S. An Updated Review of the Biomarkers of Response to Immune Checkpoint Inhibitors in Merkel Cell Carcinoma: Merkel Cell Carcinoma and Immunotherapy. Cancers 2023, 15, 5084. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, K.; Zheng, D.X.; Agak, G.W. T-Cell Mediated Immunity in Merkel Cell Carcinoma. Cancers 2022, 14, 6058. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, M.; Nie, H.; Yuan, Y. PD-1 and PD-L1 in cancer immunotherapy: Clinical implications and future considerations. Hum. Vaccin. Immunother. 2019, 15, 1111–1122. [Google Scholar] [CrossRef]
- Iyer, J.G.; Blom, A.; Doumani, R.; Lewis, C.; Tarabadkar, E.S.; Anderson, A.; Ma, C.; Bestick, A.; Parvathaneni, U.; Bhatia, S.; et al. Response rates and durability of chemotherapy among 62 patients with metastatic Merkel cell carcinoma. Cancer Med. 2016, 5, 2294–2301. [Google Scholar] [CrossRef]
- Nghiem, P.T.; Bhatia, S.; Lipson, E.J.; Kudchadkar, R.R.; Miller, N.J.; Annamalai, L.; Berry, S.; Chartash, E.K.; Daud, A.; Fling, S.P.; et al. PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. N. Engl. J. Med. 2016, 374, 2542–2552. [Google Scholar] [CrossRef]
- Nghiem, P.; Bhatia, S.; Lipson, E.J.; Sharfman, W.H.; Kudchadkar, R.R.; Brohl, A.S.; Friedlander, P.A.; Daud, A.; Kluger, H.M.; Reddy, S.A.; et al. Three-year survival, correlates and salvage therapies in patients receiving first-line pembrolizumab for advanced Merkel cell carcinoma. J. Immunother. Cancer 2021, 9, e002478. [Google Scholar] [CrossRef]
- Chikuma, S. CTLA-4, an Essential Immune-Checkpoint for T-Cell Activation. Curr. Top. Microbiol. Immunol. 2017, 410, 99–126. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S.J.; Lopez-Martin, J.A.; Bendell, J.; Ott, P.A.; Taylor, M.; Eder, J.P.; Jager, D.; Pietanza, M.C.; Le, D.T.; de Braud, F.; et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): A multicentre, open-label, phase 1/2 trial. Lancet Oncol. 2016, 17, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Wuthrick, E.; Blakaj, D.; Eroglu, Z.; Verschraegen, C.; Thapa, R.; Mills, M.; Dibs, K.; Liveringhouse, C.; Russell, J.; et al. Combined nivolumab and ipilimumab with or without stereotactic body radiation therapy for advanced Merkel cell carcinoma: A randomised, open label, phase 2 trial. Lancet 2022, 400, 1008–1019. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Fera, D.; Schultz, D.C.; Hodawadekar, S.; Reichman, M.; Donover, P.S.; Melvin, J.; Troutman, S.; Kissil, J.L.; Huryn, D.M.; Marmorstein, R. Identification and characterization of small molecule antagonists of pRb inactivation by viral oncoproteins. Chem. Biol. 2012, 19, 518–528. [Google Scholar] [CrossRef]
- Chan, I.S.; Bhatia, S.; Kaufman, H.L.; Lipson, E.J. Immunotherapy for Merkel cell carcinoma: A turning point in patient care. J. Immunother. Cancer 2018, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Want, M.Y.; Bashir, Z.; Najar, R.A. T Cell Based Immunotherapy for Cancer: Approaches and Strategies. Vaccines 2023, 11, 835. [Google Scholar] [CrossRef]
- Wu, R.; Forget, M.A.; Chacon, J.; Bernatchez, C.; Haymaker, C.; Chen, J.Q.; Hwu, P.; Radvanyi, L.G. Adoptive T-cell therapy using autologous tumor-infiltrating lymphocytes for metastatic melanoma: Current status and future outlook. Cancer J. 2012, 18, 160–175. [Google Scholar] [CrossRef]
- Feldman, S.A.; Assadipour, Y.; Kriley, I.; Goff, S.L.; Rosenberg, S.A. Adoptive Cell Therapy--Tumor-Infiltrating Lymphocytes, T-Cell Receptors, and Chimeric Antigen Receptors. Semin. Oncol. 2015, 42, 626–639. [Google Scholar] [CrossRef]
- Chen, L.; Wang, Y.; Hu, Q.; Liu, Y.; Qi, X.; Tang, Z.; Hu, H.; Lin, N.; Zeng, S.; Yu, L. Unveiling tumor immune evasion mechanisms: Abnormal expression of transporters on immune cells in the tumor microenvironment. Front. Immunol. 2023, 14, 1225948. [Google Scholar] [CrossRef]
- Tie, Y.; Tang, F.; Wei, Y.Q.; Wei, X.W. Immunosuppressive cells in cancer: Mechanisms and potential therapeutic targets. J. Hematol. Oncol. 2022, 15, 61. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.; Bi, T.M.; Pulliam, T.H.; Sarkar, K.; Church, C.D.; Kumar, N.; Mayer-Blackwell, K.; Jani, S.; Ramchurren, N.; Hansen, U.K.; et al. Merkel cell polyomavirus-specific and CD39+CLA+ CD8 T cells as blood-based predictive biomarkers for PD-1 blockade in Merkel cell carcinoma. Cell Rep. Med. 2024, 5, 101390. [Google Scholar] [CrossRef] [PubMed]
- Longino, N.V.; Yang, J.; Iyer, J.G.; Ibrani, D.; Chow, I.T.; Laing, K.J.; Campbell, V.L.; Paulson, K.G.; Kulikauskas, R.M.; Church, C.D.; et al. Human CD4+ T Cells Specific for Merkel Cell Polyomavirus Localize to Merkel Cell Carcinomas and Target a Required Oncogenic Domain. Cancer Immunol. Res. 2019, 7, 1727–1739. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.J.; Barreira-Silva, P.; Boyce, S.; Powers, J.; Cavallo, K.; Behar, S.M. CD4 T cell help prevents CD8 T cell exhaustion and promotes control of Mycobacterium tuberculosis infection. Cell Rep. 2021, 36, 109696. [Google Scholar] [CrossRef]
- Kim, H.J.; Cantor, H. CD4 T-cell subsets and tumor immunity: The helpful and the not-so-helpful. Cancer Immunol. Res. 2014, 2, 91–98. [Google Scholar] [CrossRef]
- Veatch, J.R.; Lee, S.M.; Fitzgibbon, M.; Chow, I.T.; Jesernig, B.; Schmitt, T.; Kong, Y.Y.; Kargl, J.; Houghton, A.M.; Thompson, J.A.; et al. Tumor-infiltrating BRAFV600E-specific CD4+ T cells correlated with complete clinical response in melanoma. J. Clin. Investig. 2018, 128, 1563–1568. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef]
- Schrama, D.; Hesbacher, S.; Angermeyer, S.; Schlosser, A.; Haferkamp, S.; Aue, A.; Adam, C.; Weber, A.; Schmidt, M.; Houben, R. Serine 220 phosphorylation of the Merkel cell polyomavirus large T antigen crucially supports growth of Merkel cell carcinoma cells. Int. J. Cancer 2016, 138, 1153–1162. [Google Scholar] [CrossRef]
- Buchta Rosean, C.; Leyder, E.C.; Hamilton, J.; Carter, J.J.; Galloway, D.A.; Koelle, D.M.; Nghiem, P.; Heiland, T. LAMP1 targeting of the large T antigen of Merkel cell polyomavirus results in potent CD4 T cell responses and tumor inhibition. Front. Immunol. 2023, 14, 1253568. [Google Scholar] [CrossRef]
- Paulson, K.G.; Lewis, C.W.; Redman, M.W.; Simonson, W.T.; Lisberg, A.; Ritter, D.; Morishima, C.; Hutchinson, K.; Mudgistratova, L.; Blom, A.; et al. Viral oncoprotein antibodies as a marker for recurrence of Merkel cell carcinoma: A prospective validation study. Cancer 2017, 123, 1464–1474. [Google Scholar] [CrossRef]
- Paulson, K.G.; Carter, J.J.; Johnson, L.G.; Cahill, K.W.; Iyer, J.G.; Schrama, D.; Becker, J.C.; Madeleine, M.M.; Nghiem, P.; Galloway, D.A. Antibodies to merkel cell polyomavirus T antigen oncoproteins reflect tumor burden in merkel cell carcinoma patients. Cancer Res. 2010, 70, 8388–8397. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, S.; Prommersberger, S.; Pfeiffer, I.A.; Schuler-Thurner, B.; Schuler, G.; Dorrie, J.; Schaft, N. Concurrent interaction of DCs with CD4 and CD8 T cells improves secondary CTL expansion: It takes three to tango. Eur. J. Immunol. 2014, 44, 3543–3559. [Google Scholar] [CrossRef] [PubMed]
- Gambichler, T.; Schrama, D.; Kapynen, R.; Weyer-Fahlbusch, S.S.; Becker, J.C.; Susok, L.; Kreppel, F.; Abu Rached, N. Current Progress in Vaccines against Merkel Cell Carcinoma: A Narrative Review and Update. Vaccines 2024, 12, 533. [Google Scholar] [CrossRef]
- Gomez, B.P.; Wang, C.; Viscidi, R.P.; Peng, S.; He, L.; Wu, T.C.; Hung, C.F. Strategy for eliciting antigen-specific CD8+ T cell-mediated immune response against a cryptic CTL epitope of merkel cell polyomavirus large T antigen. Cell Biosci. 2012, 2, 36. [Google Scholar] [CrossRef] [PubMed]
- Gomez, B.; He, L.; Tsai, Y.C.; Wu, T.C.; Viscidi, R.P.; Hung, C.F. Creation of a Merkel cell polyomavirus small T antigen-expressing murine tumor model and a DNA vaccine targeting small T antigen. Cell Biosci. 2013, 3, 29. [Google Scholar] [CrossRef]
- Heath, W.R.; Belz, G.T.; Behrens, G.M.; Smith, C.M.; Forehan, S.P.; Parish, I.A.; Davey, G.M.; Wilson, N.S.; Carbone, F.R.; Villadangos, J.A. Cross-presentation, dendritic cell subsets, and the generation of immunity to cellular antigens. Immunol. Rev. 2004, 199, 9–26. [Google Scholar] [CrossRef]
- Steinman, R.M.; Banchereau, J. Taking dendritic cells into medicine. Nature 2007, 449, 419–426. [Google Scholar] [CrossRef]
- Dörrie, J.; Schaft, N.; Schuler, G.; Schuler-Thurner, B. Therapeutic Cancer Vaccination with Ex Vivo RNA-Transfected Dendritic Cells-An Update. Pharmaceutics 2020, 12, 92. [Google Scholar] [CrossRef]
- Bonehill, A.; Heirman, C.; Tuyaerts, S.; Michiels, A.; Breckpot, K.; Brasseur, F.; Zhang, Y.; Van Der Bruggen, P.; Thielemans, K. Messenger RNA-electroporated dendritic cells presenting MAGE-A3 simultaneously in HLA class I and class II molecules. J. Immunol. 2004, 172, 6649–6657. [Google Scholar] [CrossRef]

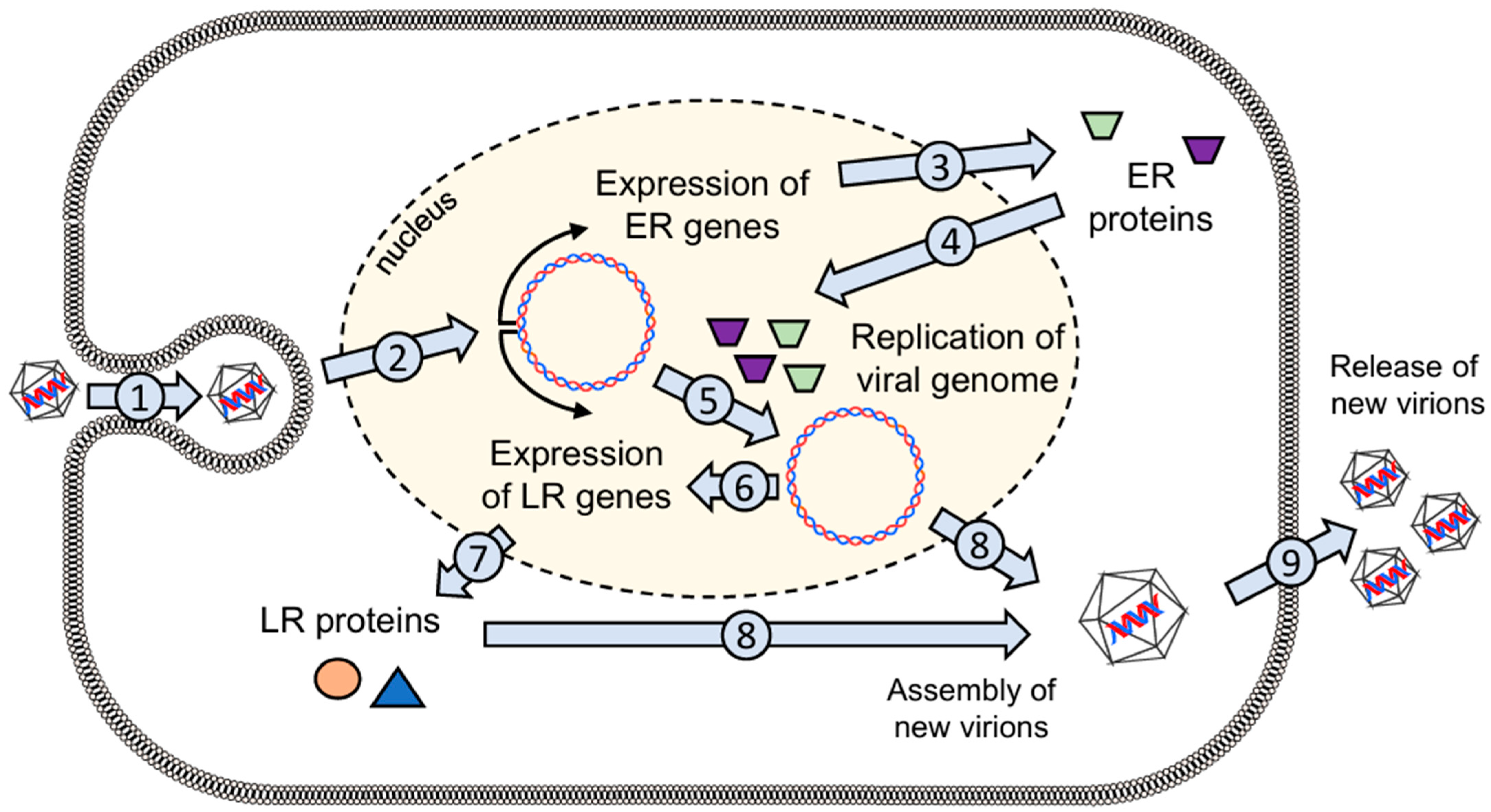
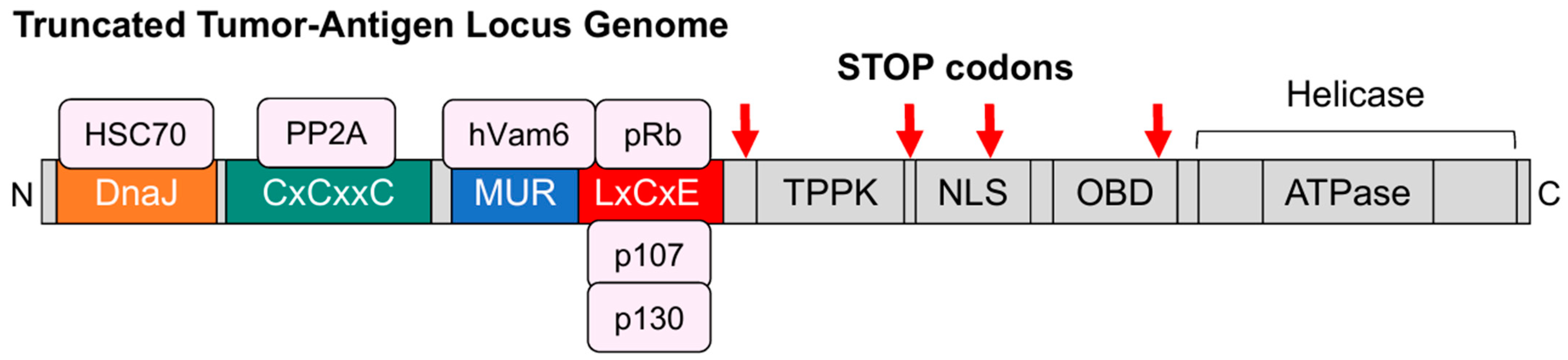
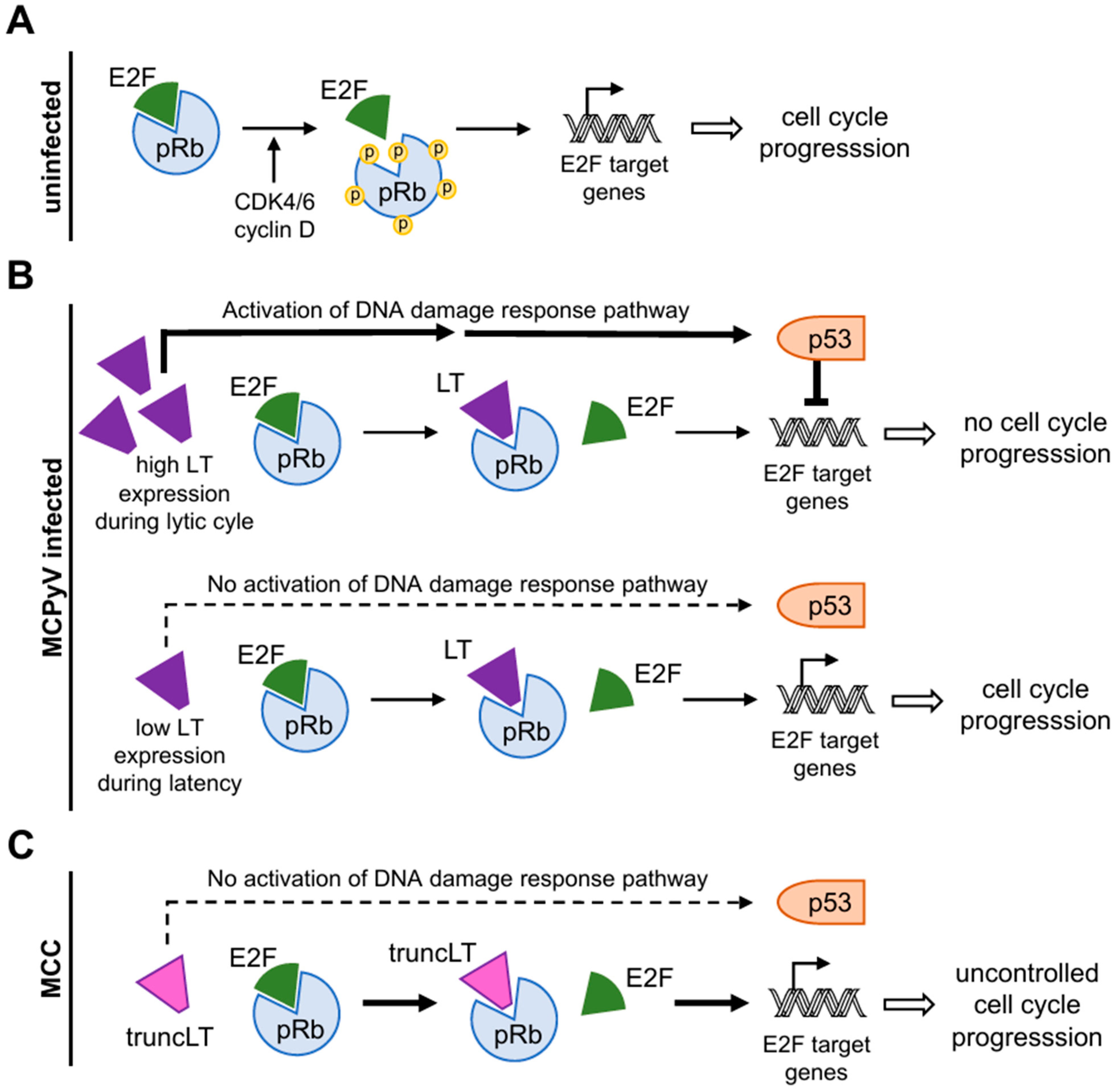
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Myrda, J.; Bremm, F.; Schaft, N.; Dörrie, J. The Role of the Large T Antigen in the Molecular Pathogenesis of Merkel Cell Carcinoma. Genes 2024, 15, 1127. https://doi.org/10.3390/genes15091127
Myrda J, Bremm F, Schaft N, Dörrie J. The Role of the Large T Antigen in the Molecular Pathogenesis of Merkel Cell Carcinoma. Genes. 2024; 15(9):1127. https://doi.org/10.3390/genes15091127
Chicago/Turabian StyleMyrda, Julia, Franziska Bremm, Niels Schaft, and Jan Dörrie. 2024. "The Role of the Large T Antigen in the Molecular Pathogenesis of Merkel Cell Carcinoma" Genes 15, no. 9: 1127. https://doi.org/10.3390/genes15091127