
In Silico Exploration of AHR-HIF Pathway Interplay: Implications for Therapeutic Targeting in ccRCC



Abstract
:1. Introduction
2. Materials and Methods
2.1. Network Generation
- STRING: interactions with an “experimental score” > 0 and no text mining;
- BIOGRID: interactions derived from experiments with a “Biogrid_score” > 0;
- KEGG: manually curated interactions from pathways including the ARNT;
- HIPPIE: this database uses a specific scoring system; we selected interactions with a score > 0.5;
- IntAct: interactors defined by the terms “association” and “physical association”.
2.2. NCBI-GEO Expression Profiles
2.3. DEG Definition
2.4. Survival Analysis of DEGs in Renal Cell Carcinoma
3. Results
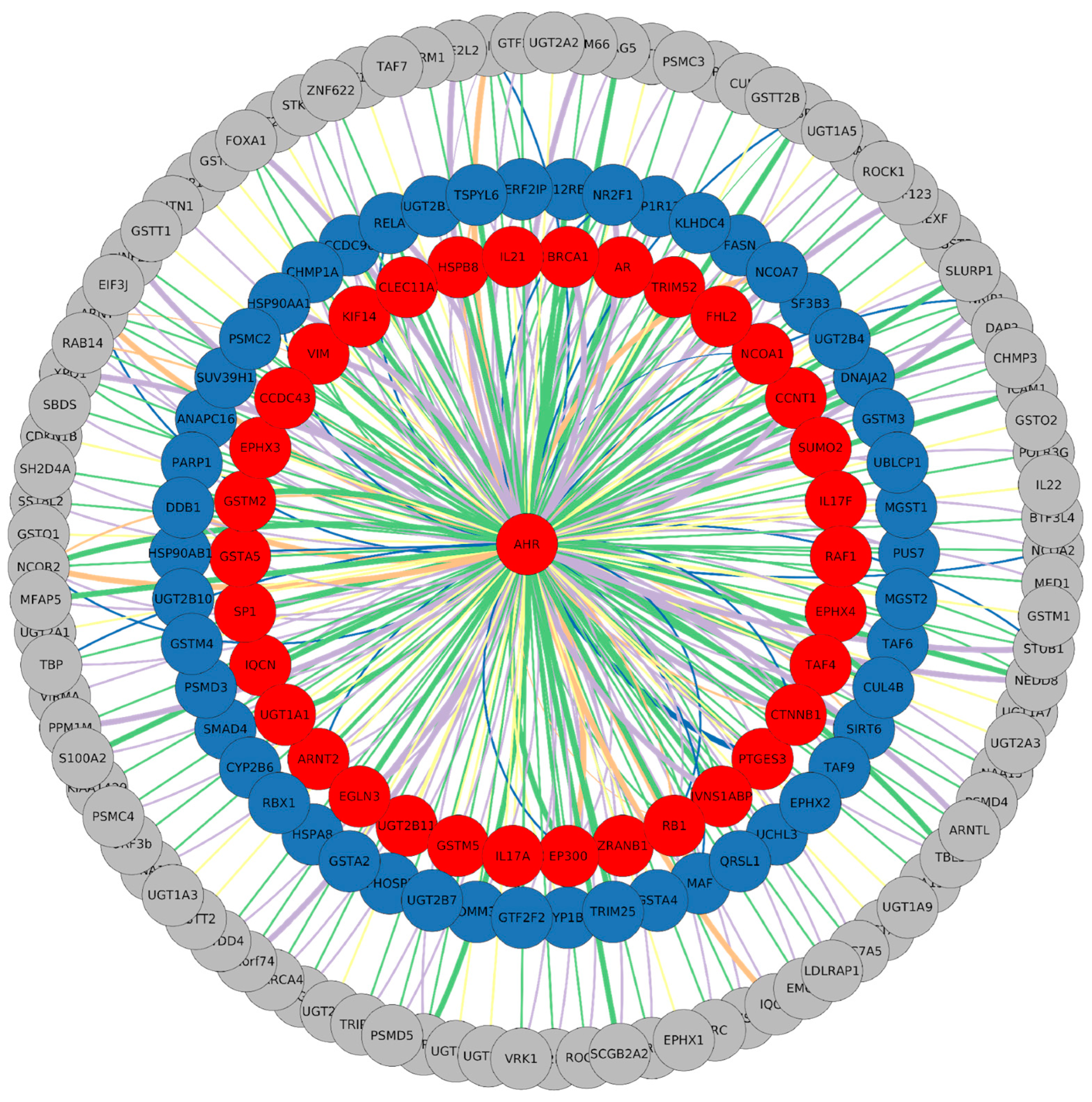
3.1. Description of PPIN Features
3.2. DEG Network Representation
3.3. Analysis of the DEGs by a Kaplan–Meier Plotter
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| VHL, pVHL | von Hippel–Lindau tumour suppressor |
| HIFs | Hypoxia-inducible factors |
| ccRCC | Clear cell renal cell carcinoma |
| AHR | Aryl hydrocarbon receptor |
| HIF-1β, ARNT | Aryl hydrocarbon receptor nuclear translocator |
| TCDD | Dioxin, 2,3,7,8-tetrachlorodibenzo-p-dioxin |
| AR | Androgen receptor |
| RB1 | Retinoblastoma-associated protein |
| SAhRMs | Selective AHR modulators |
| bHLH-PAS | Basic helix–loop–helix-PER-ARNT-SIM transcription factors |
| pNETs | Non-functioning pancreatic neuroendocrine tumours |
| RCC | Renal cell carcinoma |
| VCB complex | Complex formed by pVHL, elongin-B, elongin-C, and Cullin-2 |
| ELOB | Elongin-B |
| ELOC | Elongin-C |
| CUL2 | Cullin-2 |
| HIF-1α | Hypoxia-inducible factor 1-α |
| HSP90 | Heat shock protein 90 |
| p23 | Prostaglandin E synthase 3 |
| XAP2 | AH receptor-interacting protein |
| XREs | Xenobiotic-responsive elements |
| DREs | Dioxin-responsive elements |
| Cyp1a | Cytochrome P450, family 1, subfamily A, polypeptide 1 |
| Cyp1b | Cytochrome P450, family 1, subfamily B, polypeptide 1 |
| GSTA1 | Glutathione S-transferase A1 |
| EPHX1 | Epoxide hydrolase 1 |
| PAI2 | Plasminogen activator inhibitor 2 |
| p300 | Histone acetyltransferase p300 |
| CREBBP | CREB-binding protein |
| NCOA1/2/3 | Nuclear receptor coactivator 1/2/3 |
| NRIP1 | Nuclear receptor-interacting protein 1 |
| ER | Estrogen receptor |
| HepaRG | Human hepatic in vitro line |
| MCF7 | Human breast cancer cell line (Michigan Cancer Foundation-7) |
| HepG2 | Human liver cancer cell line |
| 786-O | Human renal cancer cell line |
| EGA | European Genome–Phenome Archive |
| TCGA | The Cancer Genome Atlas |
| GEO | Gene Expression Omnibus |
| HR | Hazard ratio |
| PPI | Protein–protein interaction |
| DEGs | Differentially expressed genes |
| GSTA5 | Glutathione S-transferase A5 |
| GSTM2 | Glutathione S-transferase Mu 2 |
| UGT2B11 | UDP-glucuronosyltransferase 2B11 |
| PPIN | Protein–protein interaction network |
| VEGF-A/C | Vascular endothelial growth factor A/C |
| CDK2/4 | Cyclin-dependent kinase 2/4 |
References
- Nebert, D.W. Aryl Hydrocarbon Receptor (AHR): “Pioneer Member” of the Basic-Helix/Loop/Helix per-Arnt-Sim (bHLH/PAS) Family of “Sensors” of Foreign and Endogenous Signals. Prog. Lipid Res. 2017, 67, 38–57. [Google Scholar] [CrossRef] [PubMed]
- Kolonko-Adamska, M.; Uversky, V.N.; Greb-Markiewicz, B. The Participation of the Intrinsically Disordered Regions of the bHLH-PAS Transcription Factors in Disease Development. Int. J. Mol. Sci. 2021, 22, 2868. [Google Scholar] [CrossRef] [PubMed]
- Ziello, J.E.; Jovin, I.S.; Huang, Y. Hypoxia-Inducible Factor (HIF)-1 Regulatory Pathway and Its Potential for Therapeutic Intervention in Malignancy and Ischemia. Yale J. Biol. Med. 2007, 80, 51–60. [Google Scholar]
- Tamukong, P.K.; Kuhlmann, P.; You, S.; Su, S.; Wang, Y.; Yoon, S.; Gong, J.; Figlin, R.A.; Janes, J.L.; Freedland, S.J.; et al. Hypoxia-Inducible Factor Pathway Genes Predict Survival in Metastatic Clear Cell Renal Cell Carcinoma. Urol. Oncol. 2022, 40, e1–e495. [Google Scholar] [CrossRef]
- Shenoy, N.; Pagliaro, L. Sequential Pathogenesis of Metastatic VHL Mutant Clear Cell Renal Cell Carcinoma: Putting It Together with a Translational Perspective. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27, 1685–1695. [Google Scholar] [CrossRef]
- Chittiboina, P.; Lonser, R.R. Von Hippel-Lindau Disease. Handb. Clin. Neurol. 2015, 132, 139–156. [Google Scholar] [CrossRef]
- Minervini, G.; Pennuto, M.; Tosatto, S.C.E. The pVHL Neglected Functions, a Tale of Hypoxia-Dependent and -Independent Regulations in Cancer. Open Biol. 2020, 10, 200109. [Google Scholar] [CrossRef]
- Buckley, D.L.; Van Molle, I.; Gareiss, P.C.; Tae, H.S.; Michel, J.; Noblin, D.J.; Jorgensen, W.L.; Ciulli, A.; Crews, C.M. Targeting the von Hippel-Lindau E3 Ubiquitin Ligase Using Small Molecules to Disrupt the VHL/HIF-1α Interaction. J. Am. Chem. Soc. 2012, 134, 4465–4468. [Google Scholar] [CrossRef]
- Cai, W.; Yang, H. The Structure and Regulation of Cullin 2 Based E3 Ubiquitin Ligases and Their Biological Functions. Cell Div. 2016, 11, 7. [Google Scholar] [CrossRef]
- Chitrakar, A.; Budda, S.A.; Henderson, J.G.; Axtell, R.C.; Zenewicz, L.A. E3 Ubiquitin Ligase Von Hippel-Lindau Protein Promotes Th17 Differentiation. J. Immunol. 2020, 205, 1009–1023. [Google Scholar] [CrossRef]
- Bennett, P.; Ramsden, D.B.; Williams, A.C. Complete Structural Characterisation of the Human Aryl Hydrocarbon Receptor Gene. Mol. Pathol. 1996, 49, M12–M16. [Google Scholar] [CrossRef]
- Gargaro, M.; Scalisi, G.; Manni, G.; Mondanelli, G.; Grohmann, U.; Fallarino, F. The Landscape of AhR Regulators and Coregulators to Fine-Tune AhR Functions. Int. J. Mol. Sci. 2021, 22, 757. [Google Scholar] [CrossRef] [PubMed]
- Hao, N.; Whitelaw, M.L.; Shearwin, K.E.; Dodd, I.B.; Chapman-Smith, A. Identification of Residues in the N-terminal PAS Domains Important for Dimerization of Arnt and AhR. Nucleic Acids Res. 2011, 39, 3695–3709. [Google Scholar] [CrossRef] [PubMed]
- Pansoy, A.; Ahmed, S.; Valen, E.; Sandelin, A.; Matthews, J. 3-Methylcholanthrene Induces Differential Recruitment of Aryl Hydrocarbon Receptor to Human Promoters. Toxicol. Sci. 2010, 117, 90–100. [Google Scholar] [CrossRef]
- Bock, K.W. From TCDD-Mediated Toxicity to Searches of Physiologic AHR Functions. Biochem. Pharmacol. 2018, 155, 419–424. [Google Scholar] [CrossRef]
- Tuomisto, J. Dioxins and Dioxin-like Compounds: Toxicity in Humans and Animals, Sources, and Behaviour in the Environment. WikiJournal Med. 2019, 6, 8. [Google Scholar] [CrossRef]
- Bersten, D.C.; Sullivan, A.E.; Peet, D.J.; Whitelaw, M.L. bHLH–PAS Proteins in Cancer. Nat. Rev. Cancer 2013, 13, 827–841. [Google Scholar] [CrossRef]
- Wang, S.; Hankinson, O. Functional Involvement of the Brahma/SWI2-Related Gene 1 Protein in Cytochrome P4501A1 Transcription Mediated by the Aryl Hydrocarbon Receptor Complex*. J. Biol. Chem. 2002, 277, 11821–11827. [Google Scholar] [CrossRef]
- Barhoover, M.A.; Hall, J.M.; Greenlee, W.F.; Thomas, R.S. Aryl Hydrocarbon Receptor Regulates Cell Cycle Progression in Human Breast Cancer Cells via a Functional Interaction with Cyclin-Dependent Kinase 4. Mol. Pharmacol. 2010, 77, 195–201. [Google Scholar] [CrossRef]
- Elson, D.J.; Kolluri, S.K. Tumor-Suppressive Functions of the Aryl Hydrocarbon Receptor (AhR) and AhR as a Therapeutic Target in Cancer. Biology 2023, 12, 526. [Google Scholar] [CrossRef]
- Tsai, M.-J.; O’Malley, B.W. Molecular Mechanisms of Action of Steroid/Thyroid Receptor Superfamily Members. Annu. Rev. Biochem. 1994, 63, 451–486. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, F.; Takeyama, K.; Matsumoto, T.; Kitagawa, H.; Yamamoto, Y.; Nohara, K.; Tohyama, C.; Krust, A.; Mimura, J.; Chambon, P.; et al. Modulation of Oestrogen Receptor Signalling by Association with the Activated Dioxin Receptor. Nature 2003, 423, 545–550. [Google Scholar] [CrossRef]
- Beischlag, T.V.; Perdew, G.H. ERα-AHR-ARNT Protein-Protein Interactions Mediate Estradiol-Dependent Transrepression of Dioxin-Inducible Gene Transcription. J. Biol. Chem. 2005, 280, 21607–21611. [Google Scholar] [CrossRef]
- Barnes-Ellerbe, S.; Knudsen, K.E.; Puga, A. 2,3,7,8-Tetrachlorodibenzo- p -Dioxin Blocks Androgen-Dependent Cell Proliferation of LNCaP Cells through Modulation of pRB Phosphorylation. Mol. Pharmacol. 2004, 66, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, C.; Kaina, B. The Aryl Hydrocarbon Receptor (AhR) in the Regulation of Cell-Cell Contact and Tumor Growth. Carcinogenesis 2010, 31, 1319–1328. [Google Scholar] [CrossRef]
- Hanieh, H.; Bani Ismail, M.; Alfwuaires, M.A.; Ibrahim, H.-I.M.; Farhan, M. Aryl Hydrocarbon Receptor as an Anticancer Target: An Overview of Ten Years Odyssey. Molecules 2023, 28, 3978. [Google Scholar] [CrossRef]
- O’Donnell, E.F.; Kopparapu, P.R.; Koch, D.C.; Jang, H.S.; Phillips, J.L.; Tanguay, R.L.; Kerkvliet, N.I.; Kolluri, S.K. The Aryl Hydrocarbon Receptor Mediates Leflunomide-Induced Growth Inhibition of Melanoma Cells. PLoS ONE 2012, 7, e40926. [Google Scholar] [CrossRef]
- O’Donnell, E.F.; Koch, D.C.; Bisson, W.H.; Jang, H.S.; Kolluri, S.K. The Aryl Hydrocarbon Receptor Mediates Raloxifene-Induced Apoptosis in Estrogen Receptor-Negative Hepatoma and Breast Cancer Cells. Cell Death Dis. 2014, 5, e1038. [Google Scholar] [CrossRef] [PubMed]
- Koch, D.C.; Jang, H.S.; O’Donnell, E.F.; Punj, S.; Kopparapu, P.R.; Bisson, W.H.; Kerkvliet, N.I.; Kolluri, S.K. Anti-Androgen Flutamide Suppresses Hepatocellular Carcinoma Cell Proliferation via the Aryl Hydrocarbon Receptor Mediated Induction of Transforming Growth Factor-Β1. Oncogene 2015, 34, 6092–6104. [Google Scholar] [CrossRef]
- Safe, S.; Cheng, Y.; Jin, U.-H. The Aryl Hydrocarbon Receptor (AhR) as a Drug Target for Cancer Chemotherapy. Curr. Opin. Toxicol. 2017, 2, 24–29. [Google Scholar] [CrossRef]
- Baker, J.R.; Sakoff, J.A.; McCluskey, A. The Aryl Hydrocarbon Receptor (AhR) as a Breast Cancer Drug Target. Med. Res. Rev. 2020, 40, 972–1001. [Google Scholar] [CrossRef]
- Vogel, C.F.A.; Lazennec, G.; Kado, S.Y.; Dahlem, C.; He, Y.; Castaneda, A.; Ishihara, Y.; Vogeley, C.; Rossi, A.; Haarmann-Stemmann, T.; et al. Targeting the Aryl Hydrocarbon Receptor Signaling Pathway in Breast Cancer Development. Front. Immunol. 2021, 12, 625346. [Google Scholar] [CrossRef] [PubMed]
- Paris, A.; Tardif, N.; Galibert, M.-D.; Corre, S. AhR and Cancer: From Gene Profiling to Targeted Therapy. Int. J. Mol. Sci. 2021, 22, 752. [Google Scholar] [CrossRef]
- Lafleur, V.N.; Halim, S.; Choudhry, H.; Ratcliffe, P.J.; Mole, D.R. Multi-Level Interaction between HIF and AHR Transcriptional Pathways in Kidney Carcinoma. Life Sci. Alliance 2023, 6, e202201756. [Google Scholar] [CrossRef] [PubMed]
- Athanasios, A.; Charalampos, V.; Vasileios, T.; Ashraf, G.M. Protein-Protein Interaction (PPI) Network: Recent Advances in Drug Discovery. Curr. Drug Metab. 2017, 18, 5–10. [Google Scholar] [CrossRef]
- Dhasmana, A.; Uniyal, S.; Anukriti; Kashyap, V.K.; Somvanshi, P.; Gupta, M.; Bhardwaj, U.; Jaggi, M.; Yallapu, M.M.; Haque, S.; et al. Topological and System-Level Protein Interaction Network (PIN) Analyses to Deduce Molecular Mechanism of Curcumin. Sci. Rep. 2020, 10, 12045. [Google Scholar] [CrossRef]
- Ba, Q.; Li, J.; Huang, C.; Li, J.; Chu, R.; Wu, Y.; Wang, H. Topological, Functional, and Dynamic Properties of the Protein Interaction Networks Rewired by Benzo(a)Pyrene. Toxicol. Appl. Pharmacol. 2015, 283, 83–91. [Google Scholar] [CrossRef]
- Foersch, S.; Schindeldecker, M.; Keith, M.; Tagscherer, K.E.; Fernandez, A.; Stenzel, P.J.; Pahernik, S.; Hohenfellner, M.; Schirmacher, P.; Roth, W.; et al. Prognostic Relevance of Androgen Receptor Expression in Renal Cell Carcinomas. Oncotarget 2017, 8, 78545–78555. [Google Scholar] [CrossRef]
- Chang, C.; Lee, S.O.; Yeh, S.; Chang, T.M. Androgen Receptor (AR) Differential Roles in Hormone-Related Tumors Including Prostate, Bladder, Kidney, Lung, Breast and Liver. Oncogene 2014, 33, 3225–3234. [Google Scholar] [CrossRef]
- Zhao, H.; Leppert, J.T.; Peehl, D.M. A Protective Role for Androgen Receptor in Clear Cell Renal Cell Carcinoma Based on Mining TCGA Data. PLoS ONE 2016, 11, e0146505. [Google Scholar] [CrossRef]
- Macleod, K.F. The RB Tumor Suppressor: A Gatekeeper to Hormone Independence in Prostate Cancer? J. Clin. Investig. 2010, 120, 4179–4182. [Google Scholar] [CrossRef]
- Harlander, S.; Schönenberger, D.; Toussaint, N.C.; Prummer, M.; Catalano, A.; Brandt, L.; Moch, H.; Wild, P.J.; Frew, I.J. Combined Vhl, Trp53 and Rb1 Mutation Causes Clear Cell Renal Cell Carcinoma in Mice. Nat. Med. 2017, 23, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.-J.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The BioGRID Database: A Comprehensive Biomedical Resource of Curated Protein, Genetic, and Chemical Interactions. Protein Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef]
- Martin, H.; Schaefer, G.A.-L. HIPPIE v2.0: Enhancing Meaningfulness and Reliability of Protein–Protein Interaction Networks|Nucleic Acids Research|Oxford Academic. Available online: https://academic.oup.com/nar/article/45/D1/D408/2290937 (accessed on 18 January 2024).
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING Database in 2023: Protein-Protein Association Networks and Functional Enrichment Analyses for Any Sequenced Genome of Interest. Nucleic Acids Res 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Otasek, D.; Morris, J.H.; Bouças, J.; Pico, A.R.; Demchak, B. Cytoscape Automation: Empowering Workflow-Based Network Analysis. Genome Biol. 2019, 20, 185. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Racine, J.S. RStudio: A Platform-Independent IDE for R and Sweave. J. Appl. Econom. 2012, 27, 167–172. [Google Scholar] [CrossRef]
- Tenenbaum, D.; Volkening, J.; Maintainer, B.P. KEGGREST: Client-Side REST Access to the Kyoto Encyclopedia of Genes and Genomes (KEGG), Bioconductor Version: Release (3.14). Available online: https://bioconductor.org/packages/release/bioc/html/KEGGREST.html (accessed on 27 August 2024).
- Feng, H.; Gu, Z.-Y.; Li, Q.; Liu, Q.-H.; Yang, X.-Y.; Zhang, J.-J. Identification of Significant Genes with Poor Prognosis in Ovarian Cancer via Bioinformatical Analysis. J. Ovarian Res. 2019, 12, 35. [Google Scholar] [CrossRef]
- De Abrew, K.N.; Kainkaryam, R.M.; Shan, Y.K.; Overmann, G.J.; Settivari, R.S.; Wang, X.; Xu, J.; Adams, R.L.; Tiesman, J.P.; Carney, E.W.; et al. Grouping 34 Chemicals Based on Mode of Action Using Connectivity Mapping. Toxicol. Sci. 2016, 151, 447–461. [Google Scholar] [CrossRef]
- Peña-Llopis, S.; Brugarolas, J. Simultaneous Isolation of High-Quality DNA, RNA, miRNA and Proteins from Tissues for Genomic Applications. Nat. Protoc. 2013, 8, 2240–2255. [Google Scholar] [CrossRef]
- Peña-Llopis, S.; Vega-Rubín-de-Celis, S.; Liao, A.; Leng, N.; Pavía-Jiménez, A.; Wang, S.; Yamasaki, T.; Zhrebker, L.; Sivanand, S.; Spence, P.; et al. BAP1 Loss Defines a New Class of Renal Cell Carcinoma. Nat. Genet. 2012, 44, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Tan, J.; Lim, K.J.; Koh, J.; Ooi, W.F.; Li, Z.; Huang, D.; Xing, M.; Chan, Y.S.; Qu, J.Z.; et al. VHL Deficiency Drives Enhancer Activation of Oncogenes in Clear Cell Renal Cell Carcinoma. Cancer Discov. 2017, 7, 1284–1305. [Google Scholar] [CrossRef]
- Chen, S.-C.; Chen, F.-W.; Hsu, Y.-L.; Kuo, P.-L. Systematic Analysis of Transcriptomic Profile of Renal Cell Carcinoma under Long-Term Hypoxia Using Next-Generation Sequencing and Bioinformatics. Int. J. Mol. Sci. 2017, 18, 2657. [Google Scholar] [CrossRef]
- Wang, X.; Hu, J.; Fang, Y.; Fu, Y.; Liu, B.; Zhang, C.; Feng, S.; Lu, X. Multi-Omics Profiling to Assess Signaling Changes upon VHL Restoration and Identify Putative VHL Substrates in Clear Cell Renal Cell Carcinoma Cell Lines. Cells 2022, 11, 472. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for Functional Genomics Data Sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Nagy, Á.; Munkácsy, G.; Győrffy, B. Pancancer Survival Analysis of Cancer Hallmark Genes. Sci. Rep. 2021, 11, 6047. [Google Scholar] [CrossRef] [PubMed]
- Posta, M.; Győrffy, B. Analysis of a Large Cohort of Pancreatic Cancer Transcriptomic Profiles to Reveal the Strongest Prognostic Factors. Clin. Transl. Sci. 2023, 16, 1479–1491. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- He, D.; Li, L.; Zhu, G.; Liang, L.; Guan, Z.; Chang, L.; Chen, Y.; Yeh, S.; Chang, C. ASC-J9 Suppresses Renal Cell Carcinoma Progression by Targeting an Androgen Receptor-Dependent HIF2α/VEGF Signaling Pathway. Cancer Res. 2014, 74, 4420–4430. [Google Scholar] [CrossRef]
- Huang, Q.; Sun, Y.; Zhai, W.; Ma, X.; Shen, D.; Du, S.; You, B.; Niu, Y.; Huang, C.-P.; Zhang, X.; et al. Androgen Receptor Modulates Metastatic Routes of VHL Wild-Type Clear Cell Renal Cell Carcinoma in an Oxygen-Dependent Manner. Oncogene 2020, 39, 6677–6691. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, F.; Baba, A.; Fujii-Kuriyama, Y.; Kato, S. Intrinsic AhR Function Underlies Cross-Talk of Dioxins with Sex Hormone Signalings. Biochem. Biophys. Res. Commun. 2008, 370, 541–546. [Google Scholar] [CrossRef]
- Ohtake, F.; Fujii-Kuriyama, Y.; Kato, S. AhR Acts as an E3 Ubiquitin Ligase to Modulate Steroid Receptor Functions. Biochem. Pharmacol. 2009, 77, 474–484. [Google Scholar] [CrossRef]
- Luecke-Johansson, S.; Gralla, M.; Rundqvist, H.; Ho, J.C.; Johnson, R.S.; Gradin, K.; Poellinger, L. A Molecular Mechanism To Switch the Aryl Hydrocarbon Receptor from a Transcription Factor to an E3 Ubiquitin Ligase. Mol. Cell Biol. 2017, 37, e00630-16. [Google Scholar] [CrossRef]
- Harbour, J.W.; Luo, R.X.; Dei Santi, A.; Postigo, A.A.; Dean, D.C. Cdk Phosphorylation Triggers Sequential Intramolecular Interactions That Progressively Block Rb Functions as Cells Move through G1. Cell 1999, 98, 859–869. [Google Scholar] [CrossRef]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H.; et al. Integrated Molecular Analysis of Clear-Cell Renal Cell Carcinoma. Nat. Genet. 2013, 45, 860–867. [Google Scholar] [CrossRef]
- Formosa, R.; Borg, J.; Vassallo, J. Aryl Hydrocarbon Receptor (AHR) Is a Potential Tumour Suppressor in Pituitary Adenomas. Endocr. Relat. Cancer 2017, 24, 445–457. [Google Scholar] [CrossRef]
- Safe, S.; Jin, U.; Park, H.; Chapkin, R.S.; Jayaraman, A. Aryl Hydrocarbon Receptor (AHR) Ligands as Selective AHR Modulators (SAhRMs). Int. J. Mol. Sci. 2020, 21, 6654. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.; Qin, C.; McDougal, A. Development of Selective Aryl Hydrocarbon Receptor Modulators for Treatment of Breast Cancer. Expert. Opin. Investig. Drugs 1999, 8, 1385–1396. [Google Scholar] [CrossRef]
- Dolciami, D.; Ballarotto, M.; Gargaro, M.; López-Cara, L.C.; Fallarino, F.; Macchiarulo, A. Targeting Aryl Hydrocarbon Receptor for Next-Generation Immunotherapies: Selective Modulators (SAhRMs) versus Rapidly Metabolized Ligands (RMAhRLs). Eur. J. Med. Chem. 2020, 185, 111842. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BIOGRID | HIPPIE | STRING | IntAct | KEGG | Merged | |
|---|---|---|---|---|---|---|
| Nodes | 126 | 122 | 20 | 14 | 50 | 182 |
| Edges | 150 | 122 | 52 | 17 | 49 | 327 |
| Node Name | Expression Levels in at Least Three out of the Five Datasets | Kaplan–Meier Analysis | Expression Level Validation with the ccRCC GEO Datasets | ||
|---|---|---|---|---|---|
| Protein ID | TCDD | ccRCC | Overall Survival (p-Value FDR) | LOW Surv. Expression Level | |
| AHR | HIGH | LOW | LOW | X | |
| ANAPC16 | LOW | HIGH | LOW | ||
| AR | HIGH | LOW | LOW | X | |
| ARNT2 | HIGH | HIGH | LOW | ||
| BRCA1 | HIGH | LOW | LOW | ||
| CCDC43 | HIGH | LOW | LOW | X | |
| CHMP1A | LOW | LOW | HIGH | ||
| CLEC11A | HIGH | HIGH | HIGH | X | |
| CYP1B1 | LOW | LOW | HIGH | ||
| DDB1 | LOW | HIGH | LOW | ||
| DNAJA2 | LOW | LOW | LOW | X | |
| EGLN3 | HIGH | LOW | LOW | X | |
| EP300 | HIGH | LOW | LOW | X | |
| EPHX2 | LOW | HIGH | LOW | ||
| EPHX4 | HIGH | LOW | HIGH | ||
| FASN | LOW | LOW | HIGH | ||
| GSTA4 | LOW | HIGH | LOW | ||
| GSTM2 | HIGH | HIGH | HIGH | X | |
| GSTM3 | LOW | HIGH | LOW | ||
| GSTM4 | LOW | HIGH | LOW | ||
| GSTM5 | HIGH | HIGH | LOW | ||
| HSP90AB1 | LOW | LOW | LOW | X | |
| HSPB8 | HIGH | LOW | LOW | ||
| IVNS1ABP | HIGH | HIGH | LOW | ||
| KIF14 | HIGH | LOW | HIGH | ||
| MAF | LOW | LOW | LOW | X | |
| MGST1 | LOW | HIGH | HIGH | X | |
| MGST2 | LOW | HIGH | LOW | ||
| MPHOSPH8 | LOW | LOW | LOW | X | |
| NCOA1 | HIGH | HIGH | LOW | ||
| NCOA7 | LOW | HIGH | LOW | ||
| NR2F1 | LOW | HIGH | LOW | ||
| PARP1 | LOW | LOW | HIGH | ||
| PPP1R12A | LOW | LOW | LOW | X | |
| PSMC2 | LOW | LOW | HIGH | ||
| PSMD3 | LOW | LOW | HIGH | ||
| PTGES3 | HIGH | LOW | LOW | X | |
| PUS7 | LOW | LOW | HIGH | ||
| RAF1 | HIGH | LOW | HIGH | ||
| RB1 | HIGH | LOW | LOW | X | |
| RBX1 | LOW | LOW | HIGH | ||
| RELA | LOW | LOW | HIGH | ||
| SF3B3 | LOW | LOW | LOW | X | |
| SMAD4 | LOW | HIGH | LOW | ||
| SUMO2 | HIGH | LOW | HIGH | ||
| SUV39H1 | LOW | LOW | LOW | X | |
| TERF2IP | LOW | HIGH | HIGH | X | |
| TOMM34 | LOW | HIGH | HIGH | X | |
| TRIM52 | HIGH | LOW | HIGH | ||
| UBLCP1 | LOW | LOW | LOW | X | |
| UGT1A1 | HIGH | HIGH | HIGH | ||
| VIM | HIGH | LOW | HIGH | ||
| Upregulated Nodes (TCDD 100 nM for 6 h) | Downregulated Nodes (TCDD 100 nM for 6 h) | ||||||
|---|---|---|---|---|---|---|---|
| Gene ID | Expression Levels in ccRCC | Overall Survival (p-Value FDR) | Expression Levels Correlated to Worse Survival Rate | Gene ID | Expression Levels in ccRCC | Overall Survival (p-Value FDR) | Expression Levels Correlated to Worse Survival Rate |
| CCDC43 | LOW | LOW | DNAJA2 | LOW | LOW | ||
| AR | LOW | LOW | HSP90AB1 | LOW | LOW | ||
| EP300 | LOW | LOW | MAF | LOW | LOW | ||
| RB1 | LOW | LOW | MGST1 | HIGH | HIGH | ||
| EGLN3 | LOW | LOW | MPHOSPH8 | LOW | LOW | ||
| GSTM2 | HIGH | HIGH | PPP1R12A | LOW | LOW | ||
| AHR | LOW | LOW | SF3B3 | LOW | LOW | ||
| CLEC11A | HIGH | HIGH | SUV39H1 | LOW | LOW | ||
| PTGES3 | LOW | LOW | TOMM34 | HIGH | HIGH | ||
| UBLCP1 | LOW | LOW | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gregoris, F.; Minervini, G.; Tosatto, S.C.E. In Silico Exploration of AHR-HIF Pathway Interplay: Implications for Therapeutic Targeting in ccRCC. Genes 2024, 15, 1167. https://doi.org/10.3390/genes15091167
Gregoris F, Minervini G, Tosatto SCE. In Silico Exploration of AHR-HIF Pathway Interplay: Implications for Therapeutic Targeting in ccRCC. Genes. 2024; 15(9):1167. https://doi.org/10.3390/genes15091167
Chicago/Turabian StyleGregoris, Francesco, Giovanni Minervini, and Silvio C. E. Tosatto. 2024. "In Silico Exploration of AHR-HIF Pathway Interplay: Implications for Therapeutic Targeting in ccRCC" Genes 15, no. 9: 1167. https://doi.org/10.3390/genes15091167