
Transcriptomic Analysis of Leaves from Two Maize Hybrids Under Heat Stress During the Early Generative Stage

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Heat Treatments
2.2. Measurement of Heat-Stress-Related Physiological Indexes
2.3. RNA Library Construction and Sequencing
2.4. Transcriptome Analysis
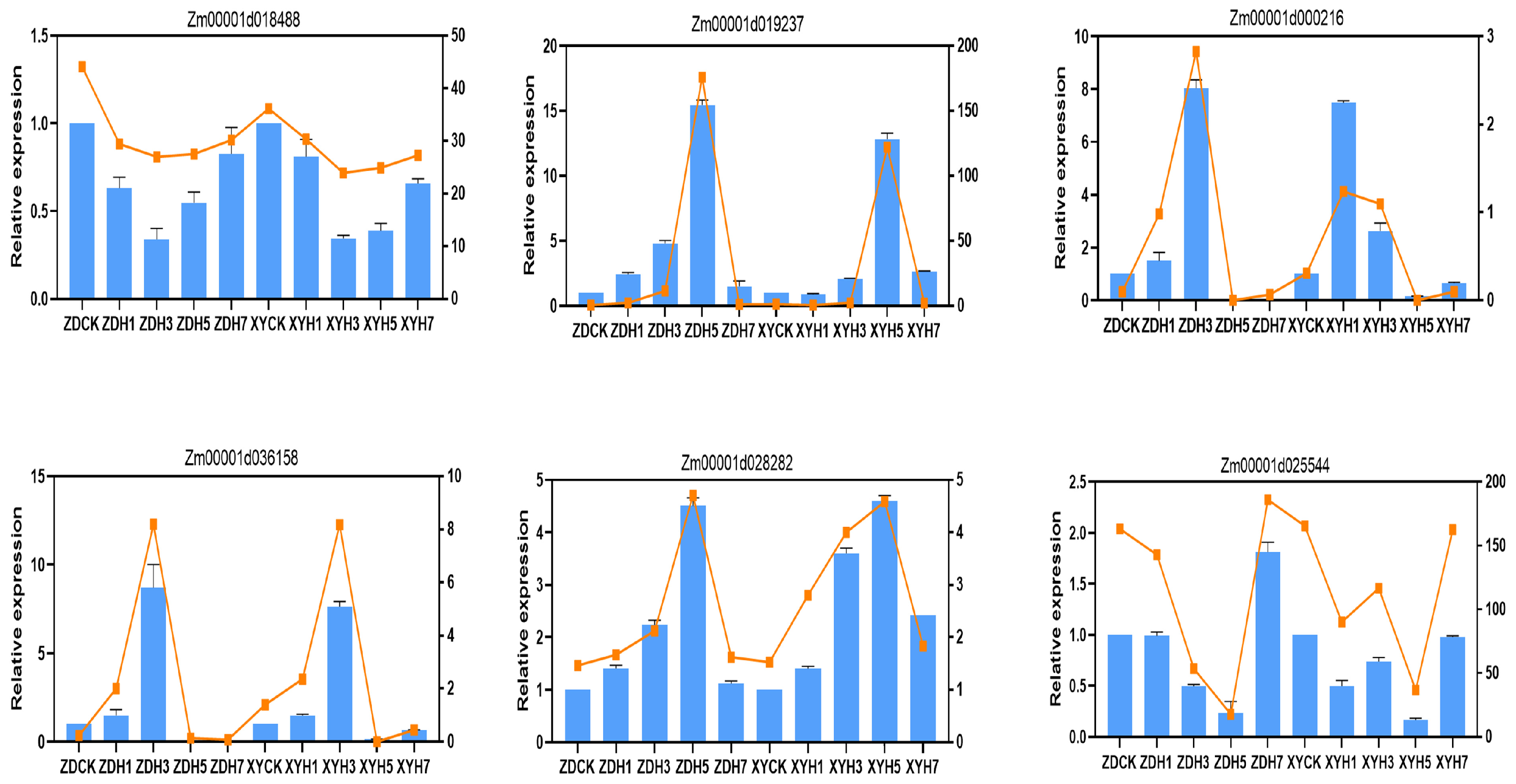
2.5. Quantitative Real-Time PCR
2.6. Statistical Analysis
3. Results
3.1. Analysis of Physiological Indicators Under High-Temperature Stress
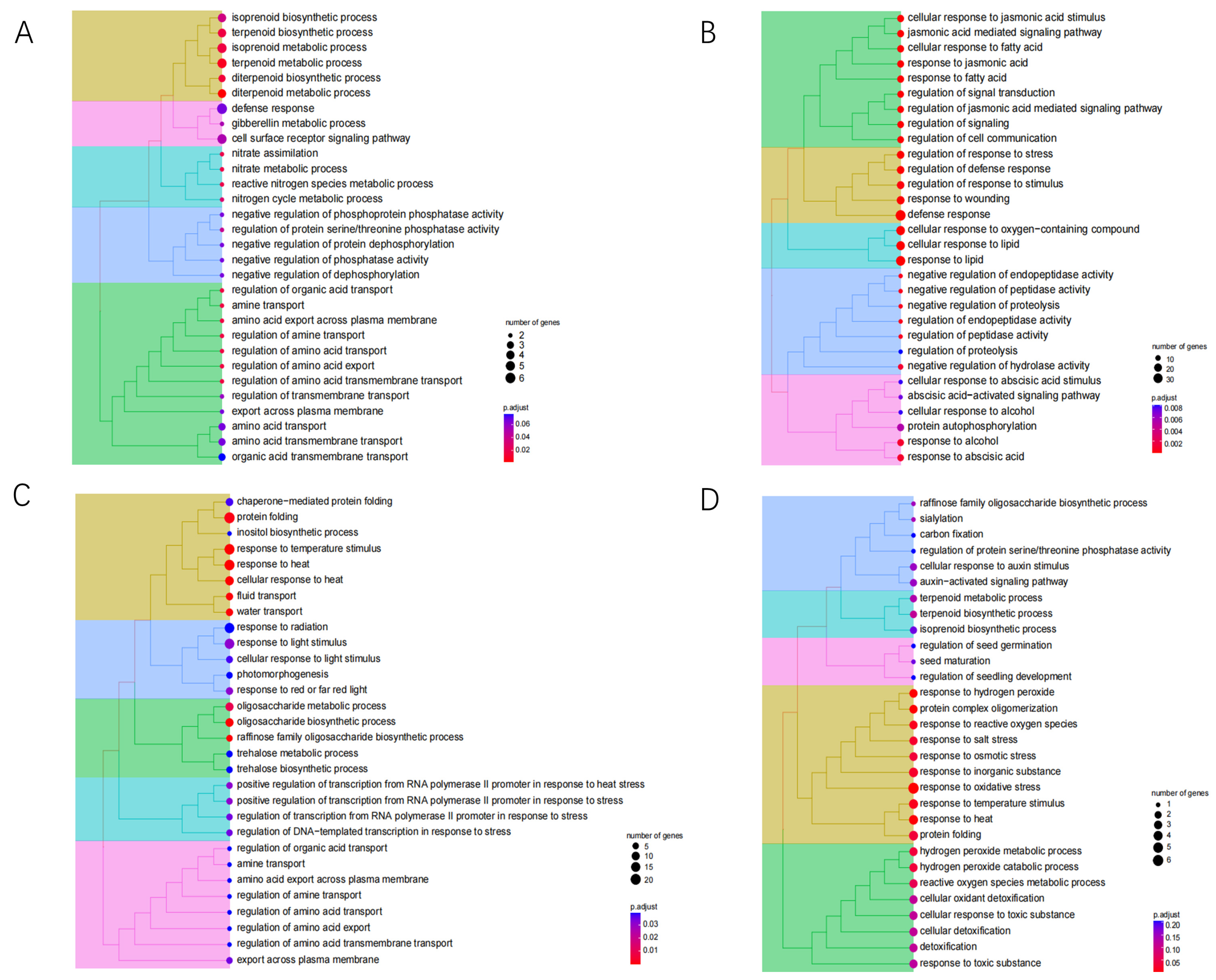
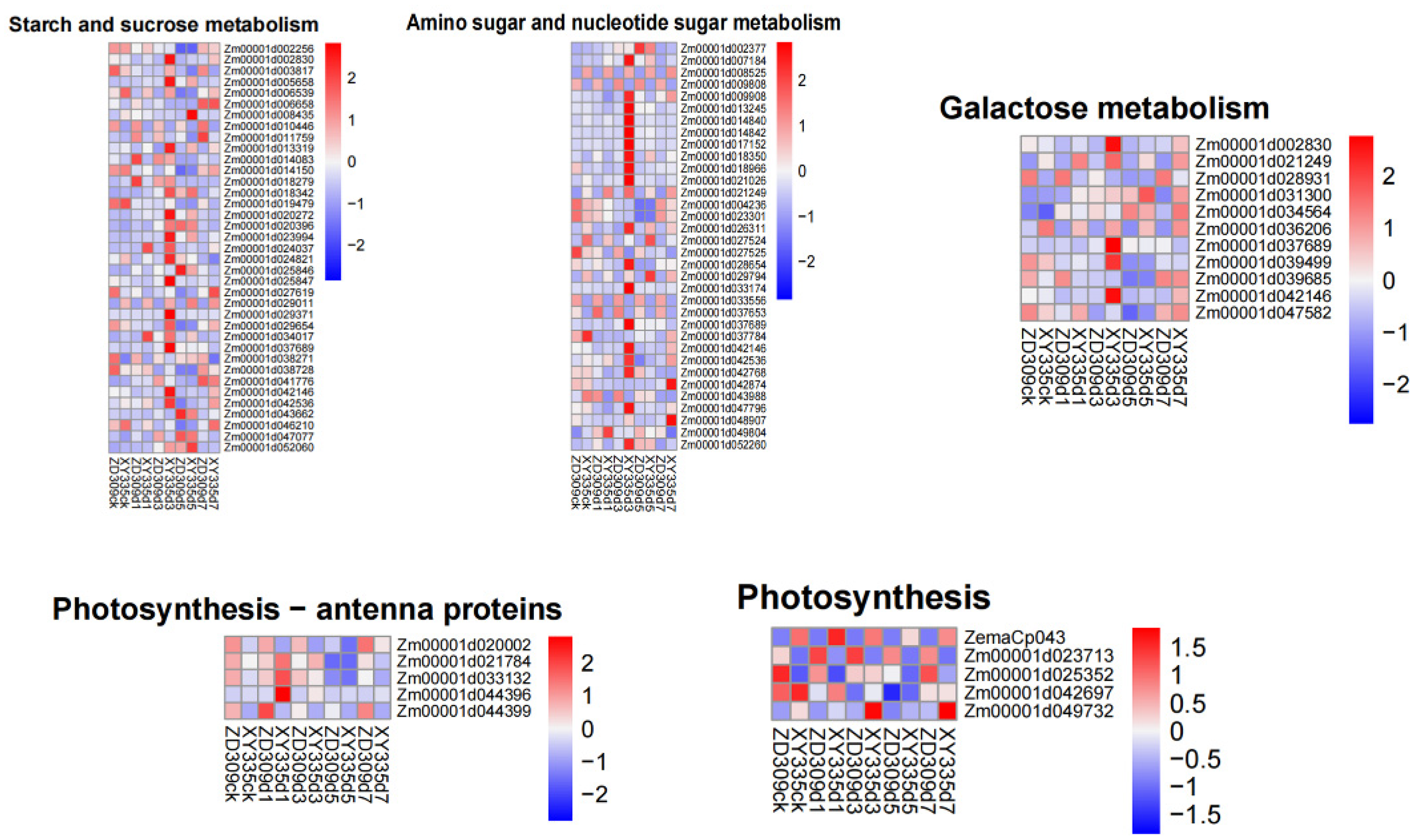
3.2. Transcriptome Analysis of Flowering Stage in Maize Hybrids Under Heat Stress
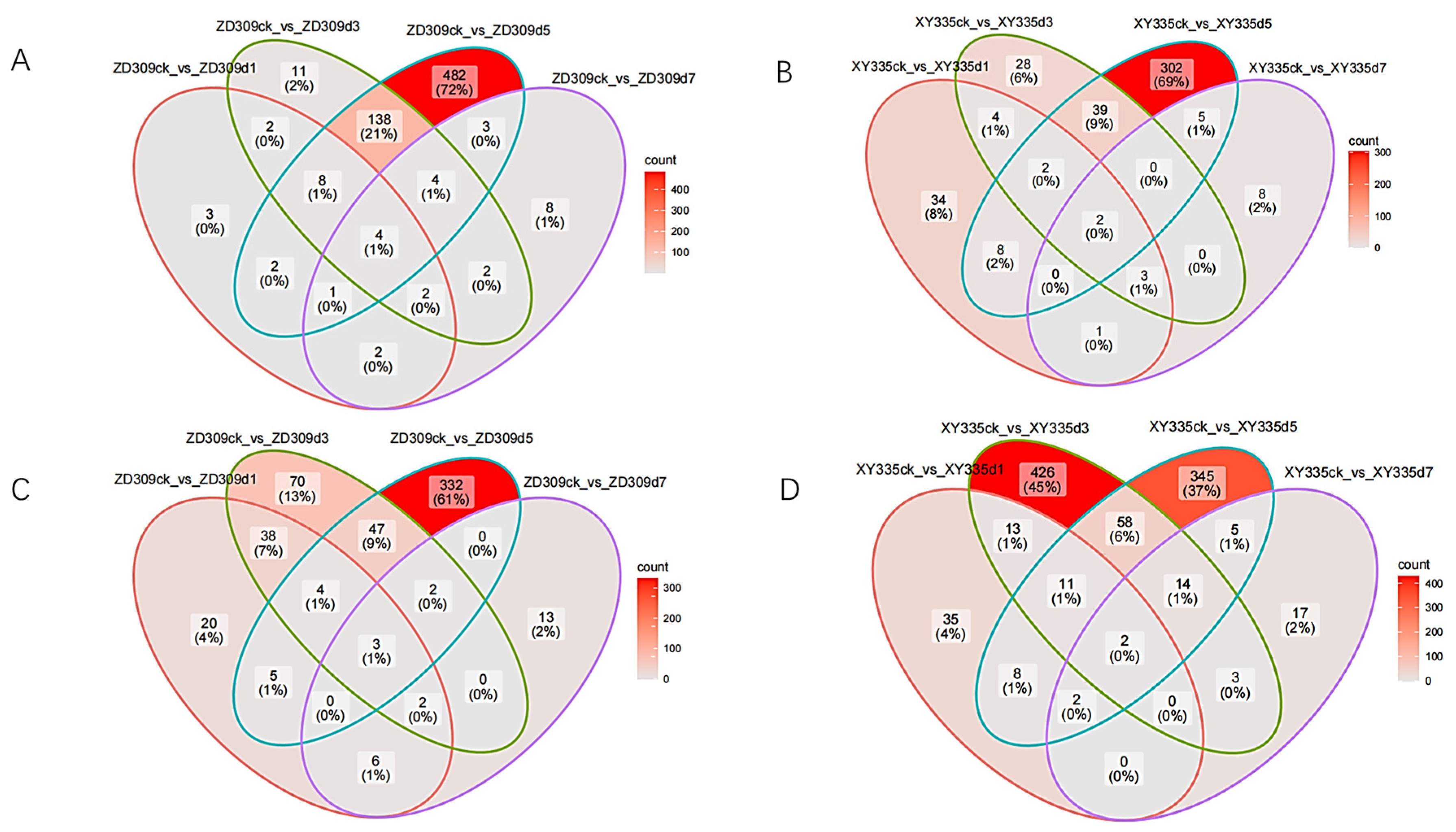
3.3. Identification of DEGs for XY335 and ZD309 Under Heat Stress During Flowering Stage
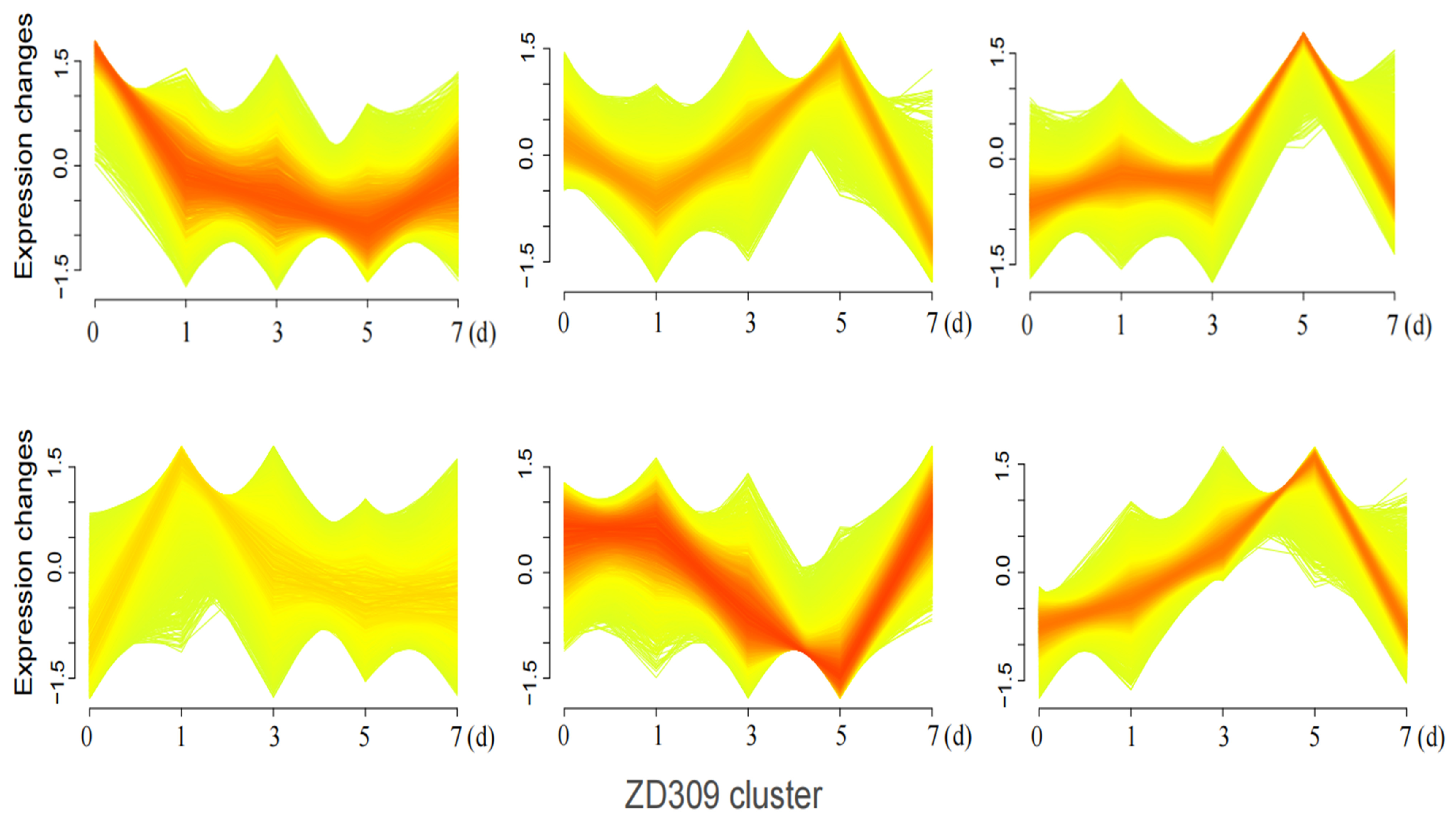
3.4. Mfuzz Time-Series Analysis
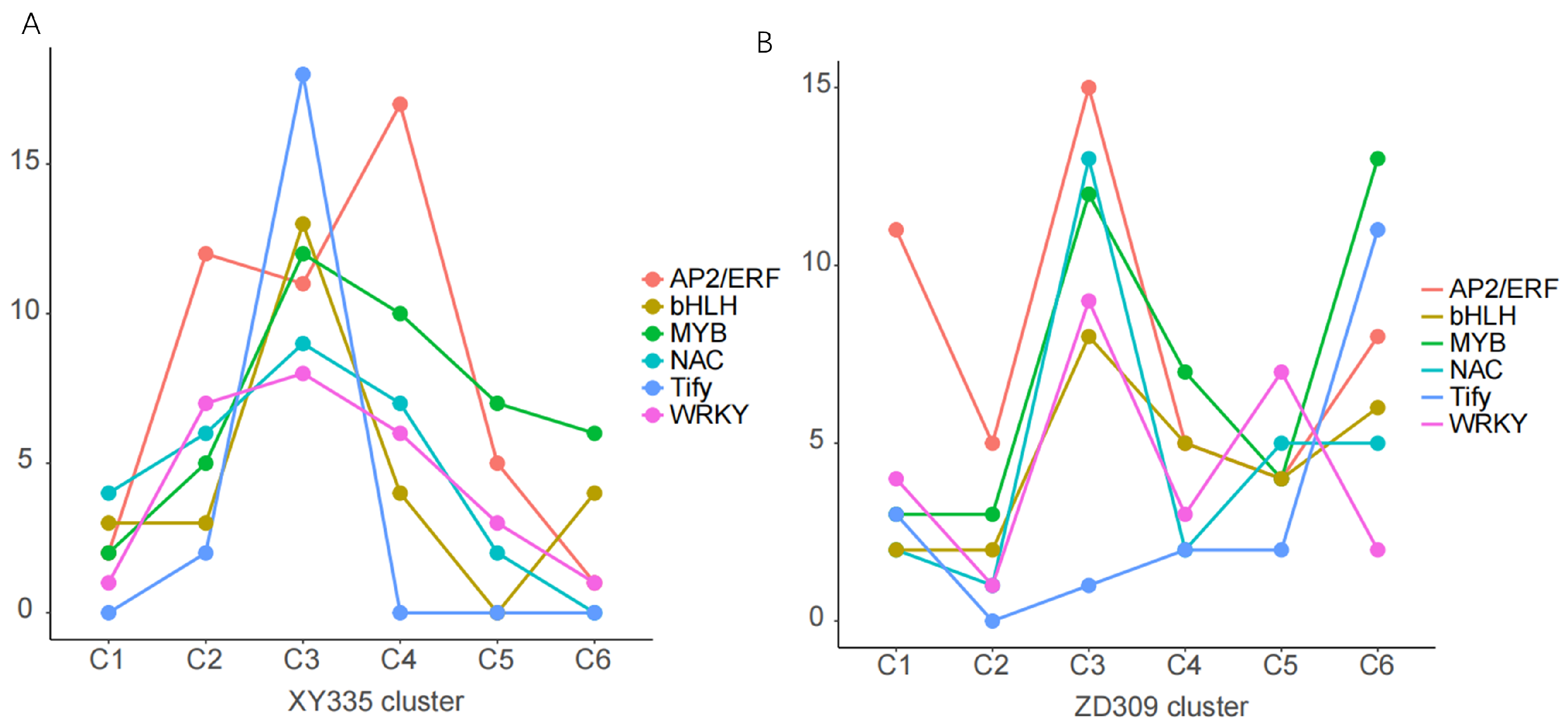
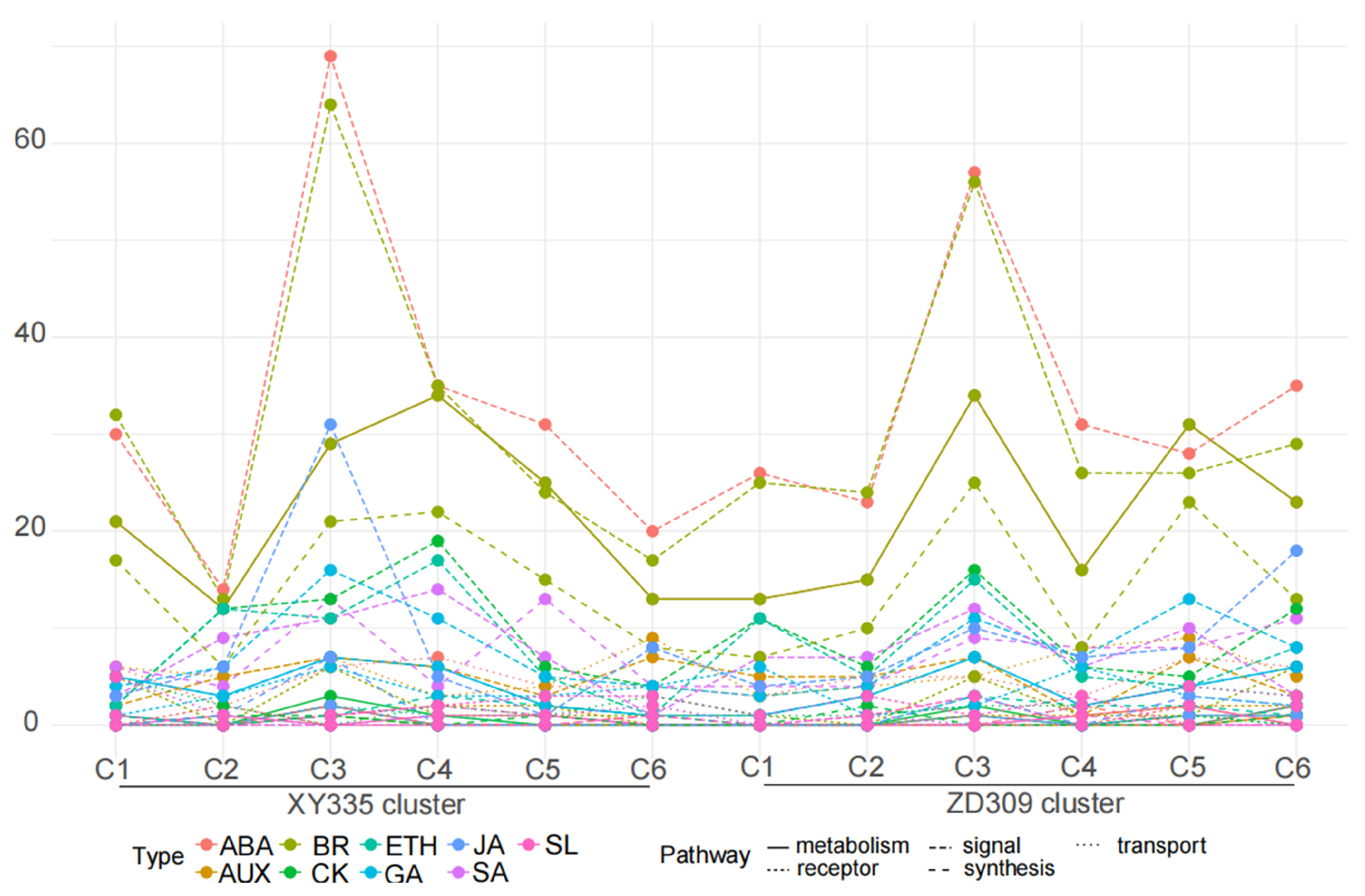
3.5. Transcription Factor (TF) and Hormone Analysis
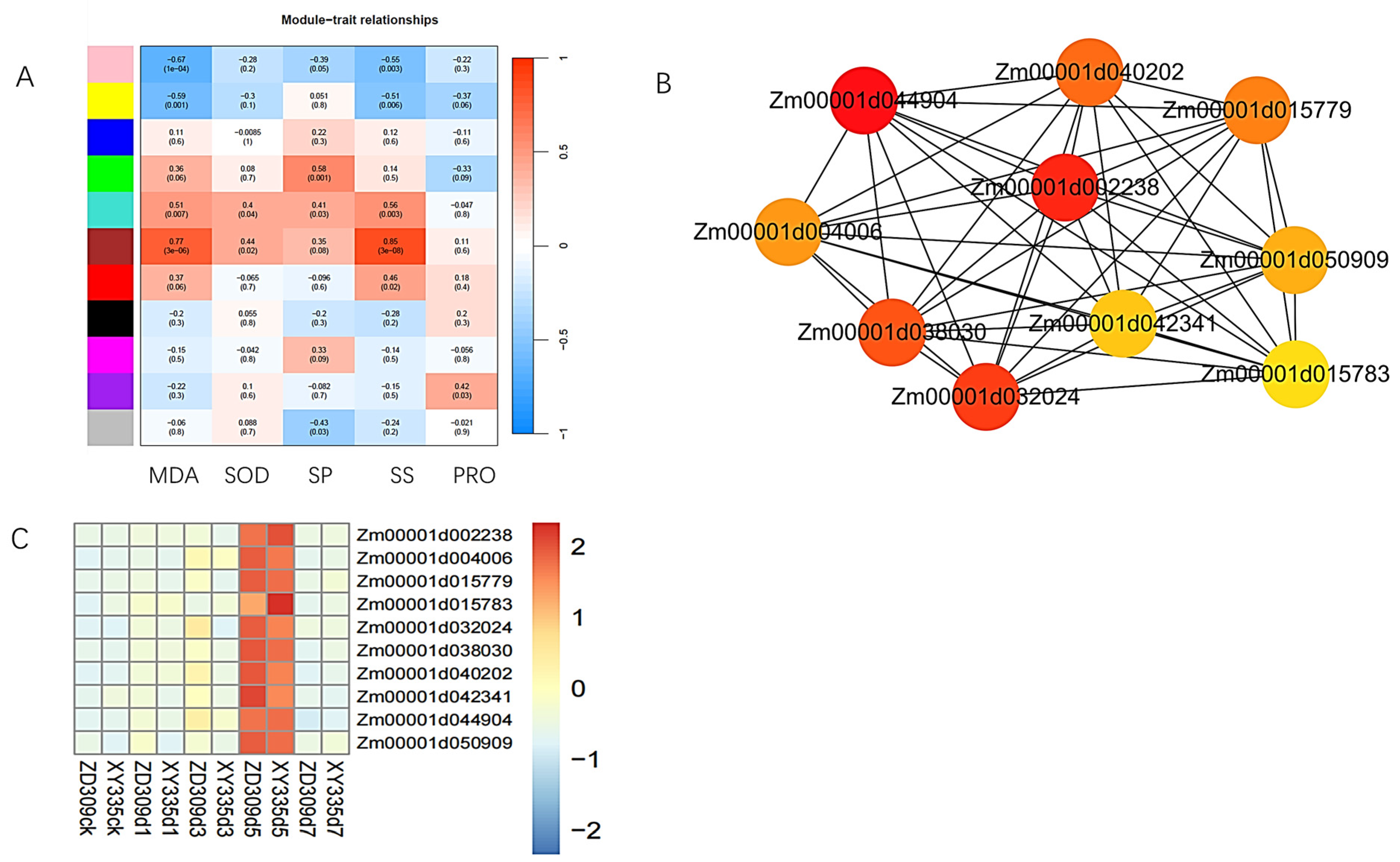
3.6. WGCNA Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
References
- Zhang, M.; An, P.; Li, H.; Wang, X.; Zhou, J.; Dong, P.; Zhao, Y.; Wang, Q.; Li, C. The miRNA-mediated post-transcriptional regulation of maize in response to high temperature. Int. J. Mol. Sci. 2019, 20, 1754. [Google Scholar] [CrossRef]
- Awasthi, R.; Bhandari, K.; Nayyar, H. Temperature stress and redox homeostasis in agricultural crops. Front. Environ. Sci. 2015, 3, 11. [Google Scholar] [CrossRef]
- Lobell, D.B.; Bänziger, M.; Magorokosho, C.; Vivek, B. Nonlinear heat effects on African maize as evidenced by historical yield trials. Nat. Clim. Chang. 2011, 1, 42–45. [Google Scholar] [CrossRef]
- Hawkins, E.; Fricker, T.E.; Challinor, A.J.; Ferro, C.A.; Ho, C.K.; Osborne, T.M. Increasing influence of heat stress on French maize yields from the 1960s to the 2030s. Glob. Change Biol. 2013, 19, 937–947. [Google Scholar] [CrossRef]
- Lobell, D.B.; Hammer, G.L.; McLean, G.; Messina, C.; Roberts, M.J.; Schlenker, W. The critical role of extreme heat for maize production in the United States. Nat. Clim. Chang. 2013, 3, 497–501. [Google Scholar] [CrossRef]
- Teng, L.; Zhang, X.-P.; Qing, L.; Jin, L.; Chen, Y.-Q.; Peng, S. Yield penalty of maize (Zea mays L.) under heat stress in different growth stages: A review. J. Integr. Agric. 2022, 21, 2465–2476. [Google Scholar]
- Mu, X.; Ma, Z.; Lu, L.; Lyu, S.; Liu, T.; Hu, X.; Li, S.; Jiang, H.; Fan, Y.; Zhao, X. Effects of high temperature stress during pollination on plant morphology, leaf photosynthetic characteristics, and yield of summer maize. Chin. J. Eco-Agric. 2024, 32, 106–118. [Google Scholar]
- Szymańska, R.; Ślesak, I.; Orzechowska, A.; Kruk, J. Physiological and biochemical responses to high light and temperature stress in plants. Environ. Exp. Bot. 2017, 139, 165–177. [Google Scholar] [CrossRef]
- Wang, J.-Q.; Xiang, R.-H.; Li, Z.-G. The essential role of H2S-ABA crosstalk in maize thermotolerance through the ROS-scavenging system. Int. J. Mol. Sci. 2023, 24, 12264. [Google Scholar] [CrossRef]
- Long, Y.; Qin, Q.; Zhang, J.; Zhu, Z.; Liu, Y.; Gu, L.; Jiang, H.; Si, W. Transcriptomic and weighted gene co-expression network analysis of tropic and temperate maize inbred lines recovering from heat stress. Plant Sci. 2023, 327, 111538. [Google Scholar] [CrossRef]
- Li, Z.-G.; Ye, X.-Y. Transcriptome response of maize (Zea mays L.) seedlings to heat stress. Protoplasma 2022, 259, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Jacob, P.; Hirt, H.; Bendahmane, A. The heat-shock protein/chaperone network and multiple stress resistance. Plant Biotechnol. J. 2017, 15, 405–414. [Google Scholar] [CrossRef]
- Li, Y.; Huang, Y.; Sun, H.; Wang, T.; Ru, W.; Pan, L.; Zhao, X.; Dong, Z.; Huang, W.; Jin, W. Heat shock protein 101 contributes to the thermotolerance of male meiosis in maize. Plant Cell 2022, 34, 3702–3717. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, Z.; Ji, Y.; Wang, C.; Wang, S.; Shi, Y.; Le, J.; Zhang, M. The heat shock factor 20-HSF4-cellulose synthase A2 module regulates heat stress tolerance in maize. Plant Cell 2024, 36, 2652–2667. [Google Scholar] [CrossRef]
- Zhao, Y.; Du, H.; Wang, Y.; Wang, H.; Yang, S.; Li, C.; Chen, N.; Yang, H.; Zhang, Y.; Zhu, Y. The calcium-dependent protein kinase ZmCDPK7 functions in heat-stress tolerance in maize. J. Integr. Plant Biol. 2021, 63, 510–527. [Google Scholar] [CrossRef]
- Wang, N.; Liu, Q.; Ming, B.; Shang, W.; Zhao, X.; Wang, X.; Wang, J.; Zhang, J.; Luo, Z.; Liao, Y. Impacts of heat stress around flowering on growth and development dynamic of maize (Zea mays L.) ear and yield formation. Plants 2022, 11, 3515. [Google Scholar] [CrossRef]
- Begcy, K.; Nosenko, T.; Zhou, L.-Z.; Fragner, L.; Weckwerth, W.; Dresselhaus, T. Male sterility in maize after transient heat stress during the tetrad stage of pollen development. Plant Physiol. 2019, 181, 683–700. [Google Scholar] [CrossRef]
- Chen, Y.; Du, T.; Zhang, J.; Chen, S.; Fu, J.; Li, H.; Yang, Q. Genes and pathways correlated with heat stress responses and heat tolerance in maize kernels. Front. Plant Sci. 2023, 14, 1228213. [Google Scholar] [CrossRef]
- Zhao, P.; Sun, L.; Zhang, S.; Jiao, B.; Wang, J.; Ma, C. Integrated Transcriptomics and Metabolomics Analysis of Two Maize Hybrids (ZD309 and XY335) under Heat Stress at the Flowering Stage. Genes 2024, 15, 189. [Google Scholar] [CrossRef]
- Ko, D.K.; Brandizzi, F. Network-based approaches for understanding gene regulation and function in plants. Plant J. 2020, 104, 302–317. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Tan, M.; Cheng, D.; Yang, Y.; Zhang, G.; Qin, M.; Chen, J.; Chen, Y.; Jiang, M. Co-expression network analysis of the transcriptomes of rice roots exposed to various cadmium stresses reveals universal cadmium-responsive genes. BMC Plant Biol. 2017, 17, 194. [Google Scholar] [CrossRef]
- Zou, X.; Liu, A.; Zhang, Z.; Ge, Q.; Fan, S.; Gong, W.; Li, J.; Gong, J.; Shi, Y.; Tian, B. Co-expression network analysis and hub gene selection for high-quality fiber in upland cotton (Gossypium hirsutum) using RNA sequencing analysis. Genes 2019, 10, 119. [Google Scholar] [CrossRef]
- Yan, Y.X.; Sang, Z.; Xu, C.; Dai, W.S.; Zou, C. Identification of maize flowering gene co-expression modules by WGCNA. Acta Agron. Sin. 2019, 45, 161–174. [Google Scholar]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J. iTAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, Z. Impact of extreme heat on corn yield in main summer corn cultivating area of China at present and under future climate change. Int. J. Plant Prod. 2019, 13, 267–274. [Google Scholar] [CrossRef]
- Li, Z.; Howell, S.H. Heat stress responses and thermotolerance in maize. Int. J. Mol. Sci. 2021, 22, 948. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Lu, Z.; Wang, L.; Jin, B. Plant responses to heat stress: Physiology, transcription, noncoding RNAs, and epigenetics. Int. J. Mol. Sci. 2020, 22, 117. [Google Scholar] [CrossRef]
- Li, H.; Tiwari, M.; Tang, Y.; Wang, L.; Yang, S.; Long, H.; Guo, J.; Wang, Y.; Wang, H.; Yang, Q. Metabolomic and transcriptomic analyses reveal that sucrose synthase regulates maize pollen viability under heat and drought stress. Ecotoxicol. Environ. Saf. 2022, 246, 114191. [Google Scholar] [CrossRef]
- Jagtap, A.B.; Yadav, I.S.; Vikal, Y.; Praba, U.P.; Kaur, N.; Gill, A.S.; Johal, G.S. Transcriptional dynamics of maize leaves, pollens and ovules to gain insights into heat stress-related responses. Front. Plant Sci. 2023, 14, 1117136. [Google Scholar] [CrossRef]
- Smith, L.M. The heat is on: Maize pollen development after a heat wave. Plant Physiol. 2019, 181, 387–388. [Google Scholar] [CrossRef]
- Xue, M.; Han, X.; Zhang, L.; Chen, S. Heat-resistant inbred lines coordinate the heat response gene expression remarkably in maize (Zea mays L.). Genes 2024, 15, 289. [Google Scholar] [CrossRef]
- Wahid, A.; Gelani, S.; Ashraf, M.; Foolad, M.R. Heat tolerance in plants: An overview. Environ. Exp. Bot. 2007, 61, 199–223. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, L.; Huang, L.; Yang, T.; Ma, J.; Yu, T.; Zhu, W.; Zhang, Z.; Tang, J. Uncovering the gene regulatory network of maize hybrid ZD309 under heat stress by transcriptomic and metabolomic analysis. Plants 2022, 11, 677. [Google Scholar] [CrossRef]
- Liu, J.; Xu, L.; Shang, J.; Hu, X.; Yu, H.; Wu, H.; Lv, W.; Zhao, Y. Genome-wide analysis of the maize superoxide dismutase (SOD) gene family reveals important roles in drought and salt responses. Genet. Mol. Biol. 2021, 44, e20210035. [Google Scholar] [CrossRef]
- Shi, J.; Yan, B.; Lou, X.; Ma, H.; Ruan, S. Comparative transcriptome analysis reveals the transcriptional alterations in heat-resistant and heat-sensitive sweet maize (Zea mays L.) varieties under heat stress. BMC Plant Biol. 2017, 17, 26. [Google Scholar] [CrossRef]
- Wang, K.; Liu, Y.; Tian, J.; Huang, K.; Shi, T.; Dai, X.; Zhang, W. Transcriptional profiling and identification of heat-responsive genes in perennial ryegrass by RNA-sequencing. Front. Plant Sci. 2017, 8, 1032. [Google Scholar] [CrossRef]
- Yan, J.; Yu, L.; Xuan, J.; Lu, Y.; Lu, S.; Zhu, W. De novo transcriptome sequencing and gene expression profiling of spinach (Spinacia oleracea L.) leaves under heat stress. Sci. Rep. 2016, 6, 19473. [Google Scholar] [CrossRef]
- Akula, R.; Ravishankar, G.A. Influence of abiotic stress signals on secondary metabolites in plants. Plant Signal. Behav. 2011, 6, 1720–1731. [Google Scholar] [CrossRef]
- Qian, Y.; Ren, Q.; Zhang, J.; Chen, L. Transcriptomic analysis of the maize (Zea mays L.) inbred line B73 response to heat stress at the seedling stage. Gene 2019, 692, 68–78. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, B.; Zhang, P.; Han, Q.; Zhao, G.; Zhao, F. Comparative Transcriptome Analysis Reveals the Underlying Response Mechanism to Salt Stress in Maize Seedling Roots. Metabolites 2023, 13, 1155. [Google Scholar] [CrossRef]
- Wu, L.; Zu, X.; Zhang, H.; Wu, L.; Xi, Z.; Chen, Y. Overexpression of ZmMAPK1 enhances drought and heat stress in transgenic Arabidopsis thaliana. Plant Mol. Biol. 2015, 88, 429–443. [Google Scholar] [CrossRef]
- Yu, Y.; He, L.; Wu, Y. Wheat WRKY transcription factor TaWRKY24 confers drought and salt tolerance in transgenic plants. Plant Physiol. Biochem. 2023, 205, 108137. [Google Scholar] [CrossRef]
- Mahmud, S.; Ullah, C.; Kortz, A.; Bhattacharyya, S.; Yu, P.; Gershenzon, J.; Vothknecht, U.C. Constitutive expression of JASMONATE RESISTANT 1 induces molecular changes that prime the plants to better withstand drought. Plant Cell Environ. 2022, 45, 2906–2922. [Google Scholar] [CrossRef]
- Lim, C.W.; Han, S.-W.; Hwang, I.S.; Kim, D.S.; Hwang, B.K.; Lee, S.C. The pepper lipoxygenase CaLOX1 plays a role in osmotic, drought and high salinity stress response. Plant Cell Physiol. 2015, 56, 930–942. [Google Scholar] [CrossRef]
- Yan, L.; Zhai, Q.; Wei, J.; Li, S.; Wang, B.; Huang, T.; Du, M.; Sun, J.; Kang, L.; Li, C.-B. Role of tomato lipoxygenase D in wound-induced jasmonate biosynthesis and plant immunity to insect herbivores. PLoS Genet. 2013, 9, e1003964. [Google Scholar] [CrossRef]
- Liu, X.; Ji, P.; Yang, H.; Jiang, C.; Liang, Z.; Chen, Q.; Lu, F.; Chen, X.; Yang, Y.; Zhang, X. Priming effect of exogenous ABA on heat stress tolerance in rice seedlings is associated with the upregulation of antioxidative defense capability and heat shock-related genes. Plant Growth Regul. 2022, 98, 23–38. [Google Scholar] [CrossRef]
- Sun, X.; Sun, C.; Li, Z.; Hu, Q.; Han, L.; Luo, H. AsHSP17, a creeping bentgrass small heat shock protein modulates plant photosynthesis and ABA-dependent and independent signalling to attenuate plant response to abiotic stress. Plant Cell Environ. 2016, 39, 1320–1337. [Google Scholar] [CrossRef]
- Mazorra, L.M.; Holton, N.; Bishop, G.J.; Núñez, M. Heat shock response in tomato brassinosteroid mutants indicates that thermotolerance is independent of brassinosteroid homeostasis. Plant Physiol. Biochem. 2011, 49, 1420–1428. [Google Scholar] [CrossRef]
- Mazorra, L.M.; Nunez, M.; Hechavarria, M.; Coll, F.; Sánchez-Blanco, M.J. Influence of brassinosteroids on antioxidant enzymes activity in tomato under different temperatures. Biol. Plant. 2002, 45, 593–596. [Google Scholar] [CrossRef]
- Gao, F.; Dubos, C. The arabidopsis bHLH transcription factor family. Trends Plant Sci. 2024, 29, 668–680. [Google Scholar] [CrossRef]
- Ma, Z.; Hu, L.; Jiang, W. Understanding AP2/ERF transcription factor responses and tolerance to various abiotic stresses in plants: A comprehensive review. Int. J. Mol. Sci. 2024, 25, 893. [Google Scholar] [CrossRef]
- Ma, Z.; Hu, L. WRKY transcription factor responses and tolerance to abiotic stresses in plants. Int. J. Mol. Sci. 2024, 25, 6845. [Google Scholar] [CrossRef]
- Guo, Z.; Dzinyela, R.; Yang, L.; Hwarari, D. bZIP transcription factors: Structure, modification, abiotic stress responses and application in plant improvement. Plants 2024, 13, 2058. [Google Scholar] [CrossRef]
- Li, S.; Fu, Q.; Chen, L.; Huang, W.; Yu, D. Arabidopsis thaliana WRKY25, WRKY26, and WRKY33 coordinate induction of plant thermotolerance. Planta 2011, 233, 1237–1252. [Google Scholar] [CrossRef]
- Sakuraba, Y.; Kim, Y.-S.; Han, S.-H.; Lee, B.-D.; Paek, N.-C. The Arabidopsis transcription factor NAC016 promotes drought stress responses by repressing AREB1 transcription through a trifurcate feed-forward regulatory loop involving NAP. Plant Cell 2015, 27, 1771–1787. [Google Scholar] [CrossRef]
- Lee, S.; Seo, P.J.; Lee, H.J.; Park, C.M. A NAC transcription factor NTL4 promotes reactive oxygen species production during drought-induced leaf senescence in Arabidopsis. Plant J. 2012, 70, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Humbert, S.; Liu, J.-X.; Srivastava, R.; Rothstein, S.J.; Howell, S.H. Heat induces the splicing by IRE1 of a mRNA encoding a transcription factor involved in the unfolded protein response in Arabidopsis. Proc. Natl. Acad. Sci. USA 2011, 108, 7247–7252. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GeneID | Description |
|---|---|
| Zm00001d002238 | Ubiquitin thiolesterase |
| Zm00001d044904 | - PPR repeat (PPR) |
| Zm00001d042341 | RNA modification |
| Zm00001d015779 | 14-3-3 protein |
| Zm00001d032024 | MYB-like DNA-binding protein |
| Zm00001d040202 | A.THALIANA mRNA (ORF19) from chromosome III |
| Zm00001d015783 | Glucan endo-1,3-β-D-glucosidase/Laminarinase |
| Zm00001d050909 | - |
| Zm00001d004006 | - AMP-activated protein kinase |
| Zm00001d038030 | Protein of unknown function (DUF2985) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Sun, L.; Ma, C.; Xu, D.; Jiao, B.; Wang, J.; Dong, F.; Yang, F.; Zhou, S.; Yang, Q.; et al. Transcriptomic Analysis of Leaves from Two Maize Hybrids Under Heat Stress During the Early Generative Stage. Genes 2025, 16, 480. https://doi.org/10.3390/genes16050480
Zhang S, Sun L, Ma C, Xu D, Jiao B, Wang J, Dong F, Yang F, Zhou S, Yang Q, et al. Transcriptomic Analysis of Leaves from Two Maize Hybrids Under Heat Stress During the Early Generative Stage. Genes. 2025; 16(5):480. https://doi.org/10.3390/genes16050480
Chicago/Turabian StyleZhang, Siqi, Lei Sun, Chunhong Ma, Dajin Xu, Bo Jiao, Jiao Wang, Fushuang Dong, Fan Yang, Shuo Zhou, Qing Yang, and et al. 2025. "Transcriptomic Analysis of Leaves from Two Maize Hybrids Under Heat Stress During the Early Generative Stage" Genes 16, no. 5: 480. https://doi.org/10.3390/genes16050480
APA StyleZhang, S., Sun, L., Ma, C., Xu, D., Jiao, B., Wang, J., Dong, F., Yang, F., Zhou, S., Yang, Q., & Zhao, P. (2025). Transcriptomic Analysis of Leaves from Two Maize Hybrids Under Heat Stress During the Early Generative Stage. Genes, 16(5), 480. https://doi.org/10.3390/genes16050480