
Dynamic Changes in Prokaryotic and Eukaryotic Communities and Networks in Minimally Managed Cabbage-Cultivated Field Soils

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Soil Sampling Site
2.2. Soil DNA Purification and Amplicon Sequencing
2.3. Sequence Data Analysis
2.4. Soil Chemical Parameters
3. Results
3.1. Prokaryotic and Eukaryotic Phylum Compositions at Control and Cabbage-Cultivated Sites During Cultivation
3.2. α- and β-Diversities of Samples in Taxonomic Groups
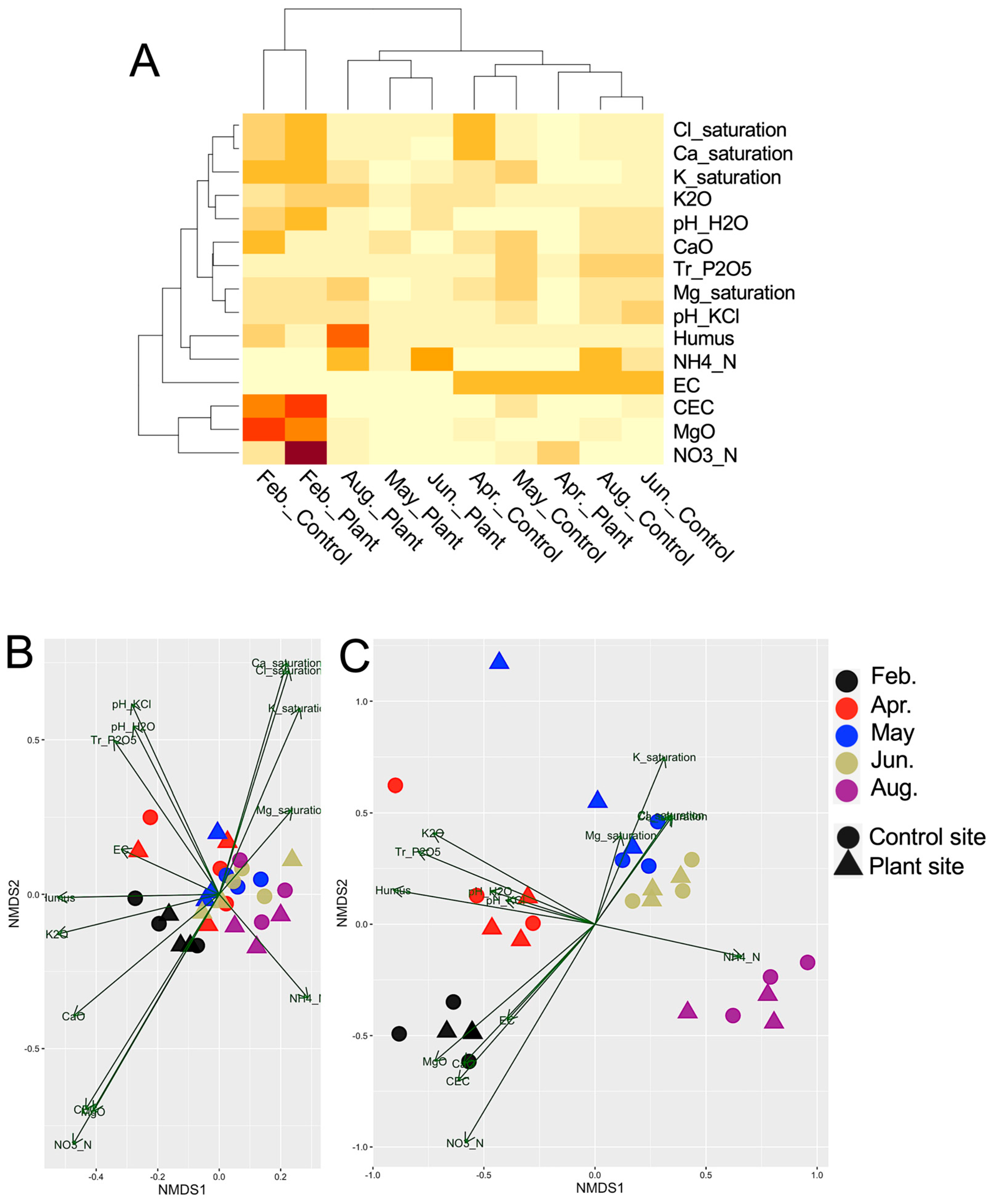
3.3. Redundancy Analysis (RDA) of the Sample Groups and Soil Chemical Parameters
3.4. Changes in Relative Abundances of SVs in Major Eukaryotic Groups and Abundant SVs in Phyla During Cultivation
3.5. Changes in Prokaryotic and Eukaryotic Co-Occurrence Networks During Cultivation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SV | Sequence variant |
| cp value | Colonizer–persister value |
| NMDS | Nonmetric multidimensional scaling |
| RDA | Redundancy analysis |
References
- Bhattacharyya, S.S.; Ros, G.H.; Furtak, K.; Iqbal, H.M.; Parra-Saldívar, R. Soil carbon sequestration–An interplay between soil microbial community and soil organic matter dynamics. Sci. Total Environ. 2022, 815, 152928. [Google Scholar] [CrossRef] [PubMed]
- Bardgett, R.D.; Caruso, T. Soil microbial community responses to climate extremes: Resistance, resilience and transitions to alternative states. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2020, 375, 20190112. [Google Scholar] [CrossRef]
- Bergmann, J.; Verbruggen, E.; Heinze, J.; Xiang, D.; Chen, B.; Joshi, J.; Rillig, M.C. The interplay between soil structure, roots, and microbiota as a determinant of plant–soil feedback. Ecol. Evol. 2016, 6, 7633–7644. [Google Scholar] [CrossRef]
- Thiele-Bruhn, S.; Bloem, J.; de Vries, F.T.; Kalbitz, K.; Wagg, C. Linking soil biodiversity and agricultural soil management. Curr. Opin. Environ. Sustain. 2012, 4, 523–528. [Google Scholar] [CrossRef]
- Philippot, L.; Chenu, C.; Kappler, A.; Rillig, M.C.; Fierer, N. The interplay between microbial communities and soil properties. Nat. Rev. Microbiol. 2024, 22, 226–239. [Google Scholar] [CrossRef]
- Neher, D.A. Soil community composition and ecosystem processes: Comparing agricultural ecosystems with natural ecosystems. Agrofor. Syst. 1999, 45, 159–185. [Google Scholar] [CrossRef]
- Larkin, R.P. Soil health paradigms and implications for disease management. Annu. Rev. Phytopathol. 2015, 53, 199–221. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhang, J.; Liu, H.; Wang, M.; Pan, L.; Chen, N.; Wang, T.; Jing, Y.; Chi, X.; Du, B. Long-term continuously monocropped peanut significantly disturbed the balance of soil fungal communities. J. Microbiol. 2020, 58, 563–573. [Google Scholar] [CrossRef]
- Chen, M.; Liu, H.; Yu, S.; Wang, M.; Pan, L.; Chen, N.; Wang, T.; Chi, X.; Du, B. Long-term continuously monocropped peanut significantly changed the abundance and composition of soil bacterial communities. PeerJ 2020, 8, e9024. [Google Scholar] [CrossRef]
- Francioli, D.; Lentendu, G.; Lewin, S.; Kolb, S. DNA Metabarcoding for the Characterization of Terrestrial Microbiota-Pitfalls and Solutions. Microorganisms 2021, 9, 361. [Google Scholar] [CrossRef]
- Hassan, S.; Sabreena; Poczai, P.; Ganai, B.A.; Almalki, W.H.; Gafur, A.; Sayyed, R.Z. Environmental DNA Metabarcoding: A Novel Contrivance for Documenting Terrestrial Biodiversity. Biology 2022, 11, 1297. [Google Scholar] [CrossRef]
- Kestel, J.H.; Field, D.L.; Bateman, P.W.; White, N.E.; Allentoft, M.E.; Hopkins, A.J.M.; Gibberd, M.; Nevill, P. Applications of environmental DNA (eDNA) in agricultural systems: Current uses, limitations and future prospects. Sci. Total Environ. 2022, 847, 157556. [Google Scholar] [CrossRef] [PubMed]
- Kenmotsu, H.; Takabayashi, E.; Takase, A.; Hirose, Y.; Eki, T. Use of universal primers for the 18S ribosomal RNA gene and whole soil DNAs to reveal the taxonomic structures of soil nematodes by high-throughput amplicon sequencing. PLoS ONE 2021, 16, e0259842. [Google Scholar] [CrossRef] [PubMed]
- Kenmotsu, H.; Masuma, T.; Murakami, J.; Hirose, Y.; Eki, T. Distinct prokaryotic and eukaryotic communities and networks in two agricultural fields of central Japan with different histories of maize-cabbage rotation. Sci. Rep. 2023, 13, 15435. [Google Scholar] [CrossRef]
- Tscharntke, T.; Grass, I.; Wanger, T.C.; Westphal, C.; Batary, P. Beyond organic farming-harnessing biodiversity-friendly landscapes. Trends Ecol. Evol. 2021, 36, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahe, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Arbizu, P.M. pairwiseAdonis: Pairwise Multilevel Comparison Using Adonis. R Package Version 0.4. Available online: https://github.com/pmartinezarbizu/pairwiseAdonis (accessed on 3 March 2025).
- Wen, T.; Xie, P.; Yang, S.; Niu, G.; Liu, X.; Ding, Z.; Xue, C.; Liu, Y.X.; Shen, Q.; Yuan, J. ggClusterNet: An R package for microbiome network analysis and modularity-based multiple network layouts. Imeta 2022, 1, e32. [Google Scholar] [CrossRef]
- Yeates, G.W.; Bongers, T.; De Goede, R.G.; Freckman, D.W.; Georgieva, S.S. Feeding habits in soil nematode families and genera-an outline for soil ecologists. J. Nematol. 1993, 25, 315–331. [Google Scholar] [PubMed]
- Ferris, H. Nematode Ecophysiological Parameter Search. Available online: http://nemaplex.ucdavis.edu/Ecology/EcophysiologyParms/EcoParameterMenu.html (accessed on 14 April 2025).
- Bongers, T.; Bongers, M. Functional diversity of nematodes. Appl. Soil. Ecol. 1998, 10, 239–251. [Google Scholar] [CrossRef]
- Bongers, T. The maturity index: An ecological measure of environmental disturbance based on nematode species composition. Oecologia 1990, 83, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Hodda, M.; Peters, L.; Traunspurger, W. Nematode diversity in terresterial, freshwater aquatic and marine systems. In Nematodes as Environmental Indicators; Wilson, M.J., Kakouli-Duarte, T., Eds.; CABI International: Oxfordshire, UK, 2009; pp. 45–93. [Google Scholar]
- Singh, J.S.; Kumar, A.; Rai, A.N.; Singh, D.P. Cyanobacteria: A Precious Bio-resource in Agriculture, Ecosystem, and Environmental Sustainability. Front. Microbiol. 2016, 7, 529. [Google Scholar] [CrossRef]
- Wang, H.; Weil, M.; Dumack, K.; Zak, D.; Münch, D.; Günther, A.; Jurasinski, G.; Blume-Werry, G.; Kreyling, J.; Urich, T. Eukaryotic rather than prokaryotic microbiomes change over seasons in rewetted fen peatlands. FEMS Microbiol. Ecol. 2021, 97, fiab121. [Google Scholar] [CrossRef]
- Redecker, D.; Schüßler, A.; Stockinger, H.; Stürmer, S.L.; Morton, J.B.; Walker, C. An evidence-based consensus for the classification of arbuscular mycorrhizal fungi (Glomeromycota). Mycorrhiza 2013, 23, 515–531. [Google Scholar] [CrossRef]
- Corsaro, D.; Köhsler, M.; Wylezich, C.; Venditti, D.; Walochnik, J.; Michel, R. New insights from molecular phylogenetics of amoebophagous fungi (Zoopagomycota, Zoopagales). Parasitol. Res. 2018, 117, 157–167. [Google Scholar] [CrossRef]
- Arcate, J.M.; Karp, M.A.; Nelson, E.B. Diversity of peronosporomycete (oomycete) communities associated with the rhizosphere of different plant species. Microb. Ecol. 2006, 51, 36–50. [Google Scholar] [CrossRef]
- Del Campo, J.; Heger, T.J.; Rodríguez-Martínez, R.; Worden, A.Z.; Richards, T.A.; Massana, R.; Keeling, P.J. Assessing the diversity and distribution of apicomplexans in host and free-living environments using high-throughput amplicon data and a phylogenetically informed reference framework. Front. Microbiol. 2019, 10, 2373. [Google Scholar] [CrossRef] [PubMed]
- Esteban, G.F.; Clarke, K.J.; Olmo, J.L.; Finlay, B.J. Soil protozoa—An intensive study of population dynamics and community structure in an upland grassland. Appl. Soil. Ecol. 2006, 33, 137–151. [Google Scholar] [CrossRef]
- Buonanno, F. The changes in the predatory behavior of the microturbellarian Stenostomum sphagnetorum on two species of toxin-secreting ciliates of the genus Spirostomum. Biologia 2011, 66, 648–653. [Google Scholar] [CrossRef]
- Pereira e Silva, M.C.; Dias, A.C.; van Elsas, J.D.; Salles, J.F. Spatial and temporal variation of archaeal, bacterial and fungal communities in agricultural soils. PLoS ONE 2012, 7, e51554. [Google Scholar] [CrossRef] [PubMed]
- Dózsa-Farkas, K.; Felföldi, T.; Hong, Y. New enchytraeid species (Enchytraeidae, Oligochaeta) from Korea. Zootaxa 2015, 4006, 171–197. [Google Scholar] [CrossRef]
- Degtyarev, M.I.; Lebedev, I.M.; Kuznetsova, K.G.; Gongalsky, K.B. A history of study and new records of terrestrial enchytraeids (Annelida, Clitellata, Enchytraeidae) from the Russian Far East. ZooKeys 2020, 955, 79–96. [Google Scholar] [CrossRef]
- Singh, J.; Schädler, M.; Demetrio, W.; Brown, G.G.; Eisenhauer, N. Climate change effects on earthworms—A review. Soil. Org. 2019, 91, 113–137. [Google Scholar] [CrossRef]
- Wu, W.; Ogawa, F.; Ochiai, M.; Yamada, K.; Fukui, H. Common strategies to control Pythium disease. Rev. Agric. Sci. 2020, 8, 58–69. [Google Scholar] [CrossRef]
- Erper, I.; Ozer, G.; Zholdoshbekova, S.; Turkkan, M. First report of garden cucumber root rot caused by Globisporangium ultimum var. ultimum in Kyrgyzstan. J. Plant Pathol. 2020, 102, 1363–1364. [Google Scholar] [CrossRef]
- Dixon, G.R. The occurrence and economic impact of Plasmodiophora brassicae and clubroot disease. J. Plant Growth Regul. 2009, 28, 194–202. [Google Scholar] [CrossRef]
- Hittorf, M.; Letsch-Praxmarer, S.; Windegger, A.; Bass, D.; Kirchmair, M.; Neuhauser, S. Revised taxonomy and expanded biodiversity of the Phytomyxea (Rhizaria, Endomyxa). J. Eukaryot. Microbiol. 2020, 67, 648–659. [Google Scholar] [CrossRef]
- Foets, J.; Wetzel, C.E.; Teuling, A.J.; Pfister, L. Temporal and spatial variability of terrestrial diatoms at the catchment scale: Controls on productivity and comparison with other soil algae. PeerJ 2020, 8, e9198. [Google Scholar] [CrossRef]
- Bedano, J.C.; Cantú, M.P.; Doucet, M.E. Abundance of soil mites (Arachnida: Acari) in a natural soil of central Argentina. Zool. Stud. 2005, 44, 505–512. [Google Scholar]
- Hess, S.; Suthaus, A. The Vampyrellid Amoebae (Vampyrellida, Rhizaria). Protist 2022, 173, 125854. [Google Scholar] [CrossRef] [PubMed]
- Sayre, R.M. Theratromyxa weberi, An Amoeba Predatory on Plant-Parasitic Nematodes. J. Nematol. 1973, 5, 258–264. [Google Scholar] [PubMed]
- Honna, Y.; Ishii, M. Perforation of hyphae and sclerotia of Rhizoctonia solani Kühn by mycophagous soil amoebae from vegetable field soils in Japan. Jpn. J. Phytopathol. 1984, 50, 229–240. [Google Scholar] [CrossRef]
- Suthaus, A.; Hess, S. Pseudovampyrella gen. nov.: A genus of Vampyrella-like protoplast extractors finds its place in the Leptophryidae. J. Eukaryot. Microbiol. 2024, 71, e13002. [Google Scholar] [CrossRef]
- Foissner, W. An updated compilation of world soil ciliates (Protozoa, Ciliophora), with ecological notes, new records, and descriptions of new species. Eur. J. Protistol. 1998, 34, 195–235. [Google Scholar] [CrossRef]
- Andrássy, I. On the Genera Mononchus Bastían, 1865 and Prionchuhis (Cobb, 1916) Wu & Hoeppli, 1929 (Nematode: Mononchidae). Opusc. Zool. Bp. 1985, 21, 9–22. [Google Scholar]
- Murchie, A.K.; Gordon, A.W. The impact of the ‘New Zealand flatworm’, Arthurdendyus triangulatus, on earthworm populations in the field. Biol. Invasions 2013, 15, 569–586. [Google Scholar] [CrossRef]
- Lay, C.Y.; Hamel, C.; St-Arnaud, M. Taxonomy and pathogenicity of Olpidium brassicae and its allied species. Fungal Biol. 2018, 122, 837–846. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Profile of Change | Group | Abundant SV | Phylum | The Closest Hit Species | Enhanced Changes at Plant Site |
|---|---|---|---|---|---|
| Increased | Fungi | SV_165 | Basidiomycota | Alloexidiopsis calcea | Yes |
| SV_90 | Mucoromycota | Cetraspora gilmorei | |||
| SV_407 | Zoopagomycota | Ramicandelaber sp. | Yes | ||
| Protists | SV_36 | Apicomplexa | Eimeria tropidura | Yes | |
| SV_614 | Bicosoecida | Bicosoeca petiolata | |||
| SV_169 | Peronosporomycetes | Achlya bisexualis/Thraustotheca clavata | Yes | ||
| Animals | SV_28 | Arthropoda | Stigmalychus spp. | Yes | |
| SV_32 | Nematozoa | Dorylaimellus parvulus | |||
| SV_39 | Nematozoa | Prismatolaimus spp. | |||
| SV_51 | Gastrotricha | Neogossea spp. | |||
| SV_71 | Platyhelminthes | Stenostomum spp. | |||
| SV_99 | Platyhelminthes | Stenostomum spp. | |||
| SV_100 | Platyhelminthes | Stenostomum leucops | |||
| Decreased | Fungi | SV_121 | Aphelidea | Aphelidium parallelum | |
| SV_8 | Ascomycota | Chaetomium globosum/Humicola sp. | |||
| SV_16 | Ascomycota | C. globosum/Humicola sp. | |||
| SV_43 | Ascomycota | Ascobolus spp. | |||
| SV_37 | Basidiomycota | Lyophyllum sp. etc. | |||
| SV_13 | Chytridiomycota | Chytridium polysiphoniae | |||
| SV_65 | Chytridiomycota | Rhizophydium patellarium/Betamyces sp. | |||
| SV_94 | Chytridiomycota | Rhizophydium sp. | |||
| SV_102 | Chytridiomycota | Catenomyces persicinus | |||
| SV_161 | Cryptomycota | Rozella spp. | |||
| SV_327 | Cryptomycota | Vinositunica sp./Bullera formosensis | |||
| SV_421 | Cryptomycota | Rozella spp. | |||
| Protists | SV_57 | Peronosporomycetes | Pythium spp./Globisporangium spp. | ||
| Animals | SV_1 | Annelida | Enchytraeus dichaetus | Yes | |
| SV_5 | Annelida | Enchytraeus bulbosus | |||
| Increased and then decreased | Fungi | SV_91 | Aphelidea | A. parallelum | |
| SV_17 | Basidiomycota | Trechispora spp. | |||
| SV_259 | Blastocladiomycota | Paraphysoderma sedebokerense | Yes | ||
| SV_47 | Chytridiomycota | Olpidium brassicae | Yes | ||
| SV_147 | LKM15 | Podoscypha spp. | Yes | ||
| SV_247 | LKM15 | Morellospora saccamoebae | |||
| Protists | SV_307 | Bicosoecida | Trieres chinensis | ||
| SV_24 | Cercozoa (class Vampyrellidae) | Theratromyxa weberi/Arachnula impatiens | |||
| SV_38 | Cercozoa (class Vampyrellidae) | Vampyrella closterii | |||
| SV_41 | Cercozoa (class Vampyrellidae) | T. weberi/A. impatiens | |||
| SV_116 | Ciliophora | Obertrumia georgiana | |||
| SV_159 | Ciliophora | O. georgiana | |||
| SV_189 | Ciliophora | Colpoda spp. | Yes | ||
| SV_168 | Centrohelida | Triangulopteris lacunata | |||
| SV_18 | Diatomea | Craticula subminuscula/Stauroneis acuta | |||
| SV_30 | Diatomea | Hantzschia amphioxys | |||
| SV_103 | Diatomea | Halamphora montana/Amphora montana | |||
| SV_106 | Diatomea | Nitzschia palea | |||
| Animals | SV_10 | Annelida | Achaeta spp./Hemienchytraeus spp. | ||
| SV_14 | Arthropoda | Alicorhagia spp. | |||
| SV_29 | Arthropoda | Bourletiella hortensis | |||
| SV_35 | Arthropoda | Pergamasus spp. etc. | |||
| SV_42 | Nematozoa | Mononchus truncatus | |||
| SV_101 | Platyhelminthes | Arthurdendyus triangulatus | Yes | ||
| Decreased and then increased | Protists | SV_20 | Cercozoa | Rhogostoma bowseri | |
| SV_111 | Ciliophora | Kalometopia duplicata | |||
| Animals | SV_23 | Nematozoa | Plectus spp. | ||
| SV_26 | Nematozoa | Acrobeloides spp. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ito, S.; Murakami, J.; Suzuki, M.; Hirose, Y.; Yamauchi, T.; Eki, T. Dynamic Changes in Prokaryotic and Eukaryotic Communities and Networks in Minimally Managed Cabbage-Cultivated Field Soils. Genes 2025, 16, 482. https://doi.org/10.3390/genes16050482
Ito S, Murakami J, Suzuki M, Hirose Y, Yamauchi T, Eki T. Dynamic Changes in Prokaryotic and Eukaryotic Communities and Networks in Minimally Managed Cabbage-Cultivated Field Soils. Genes. 2025; 16(5):482. https://doi.org/10.3390/genes16050482
Chicago/Turabian StyleIto, Sentaro, Junya Murakami, Mio Suzuki, Yuu Hirose, Takahiro Yamauchi, and Toshihiko Eki. 2025. "Dynamic Changes in Prokaryotic and Eukaryotic Communities and Networks in Minimally Managed Cabbage-Cultivated Field Soils" Genes 16, no. 5: 482. https://doi.org/10.3390/genes16050482
APA StyleIto, S., Murakami, J., Suzuki, M., Hirose, Y., Yamauchi, T., & Eki, T. (2025). Dynamic Changes in Prokaryotic and Eukaryotic Communities and Networks in Minimally Managed Cabbage-Cultivated Field Soils. Genes, 16(5), 482. https://doi.org/10.3390/genes16050482