
Single Amino Acid Supplementation in Inherited Metabolic Disorders: An Evidence-Based Review of Interventions



Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Sources and Search Strategies
2.2. Identification of Relevant Studies
2.3. Study Selection and Evaluation
3. Results
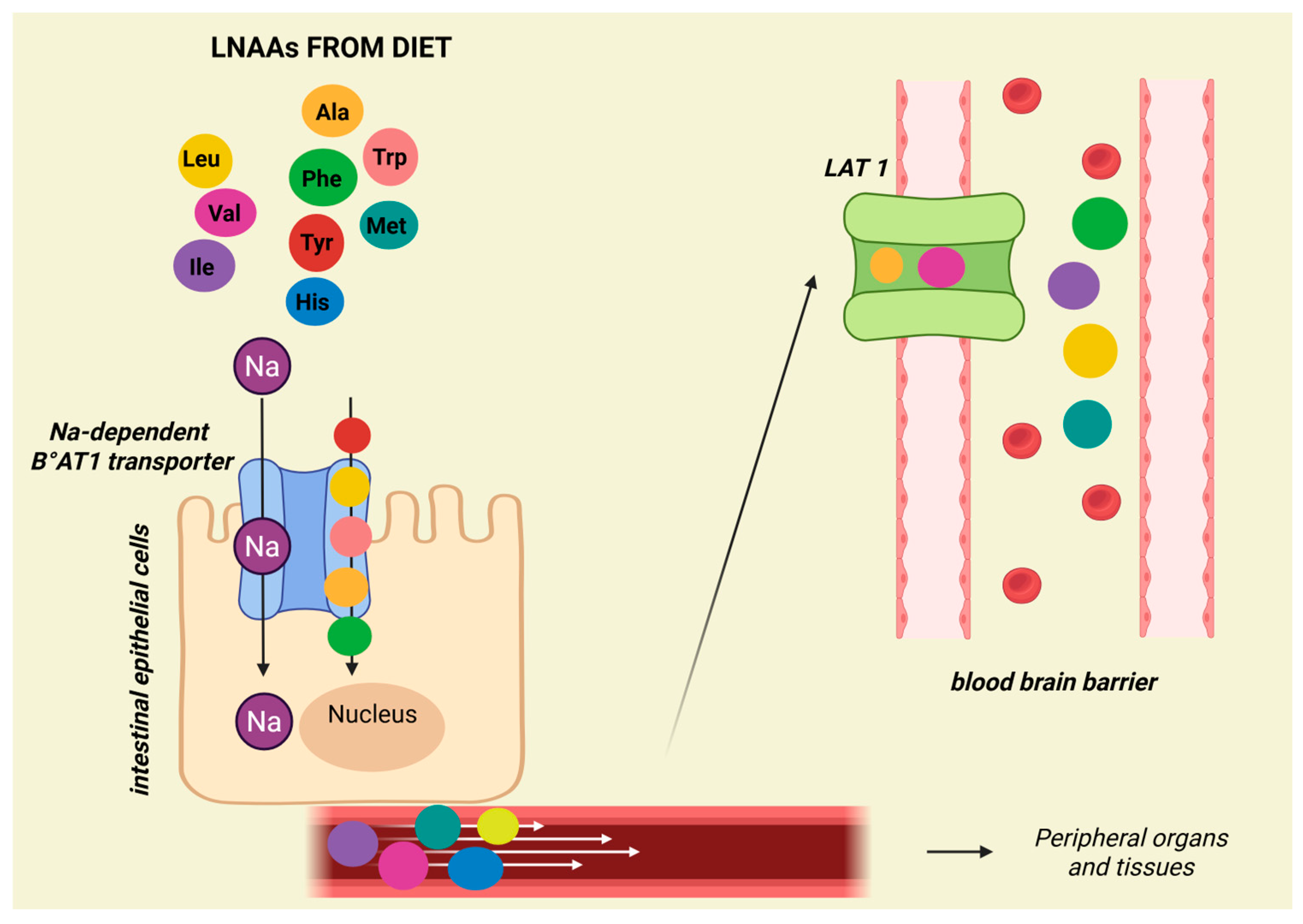
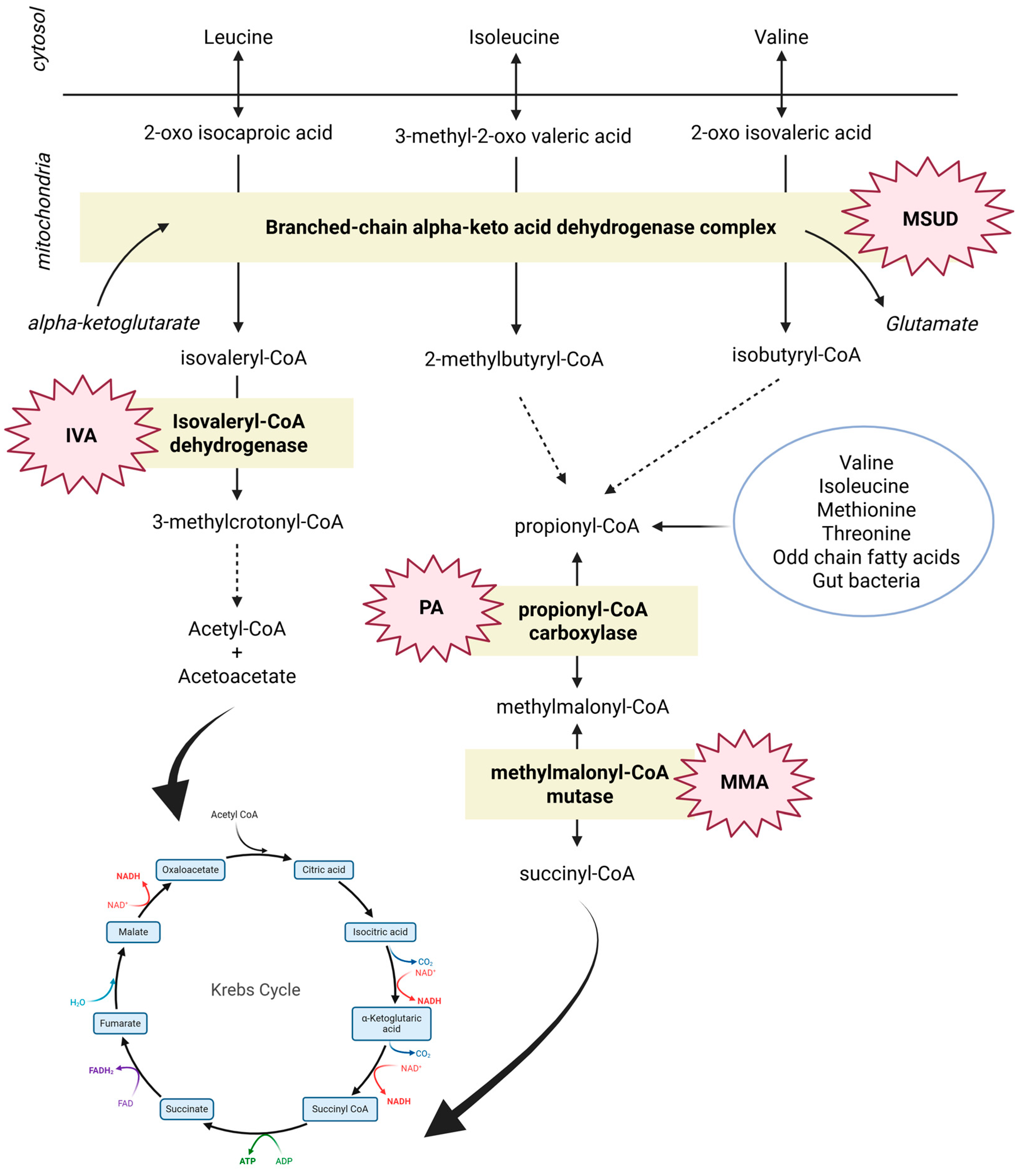
3.1. Branched Chain Amino Acids (BCAAs): Leucine, Isoleucine, and Valine
- Urea cycle disorders (UCDs);
- Maple Syrup Urine Disease (MSUD);
- Methylmalonic (MMA) and Propionic (PA) acidemia;
- Glycogen storage disease due to acid maltase deficiency (Pompe Disease);
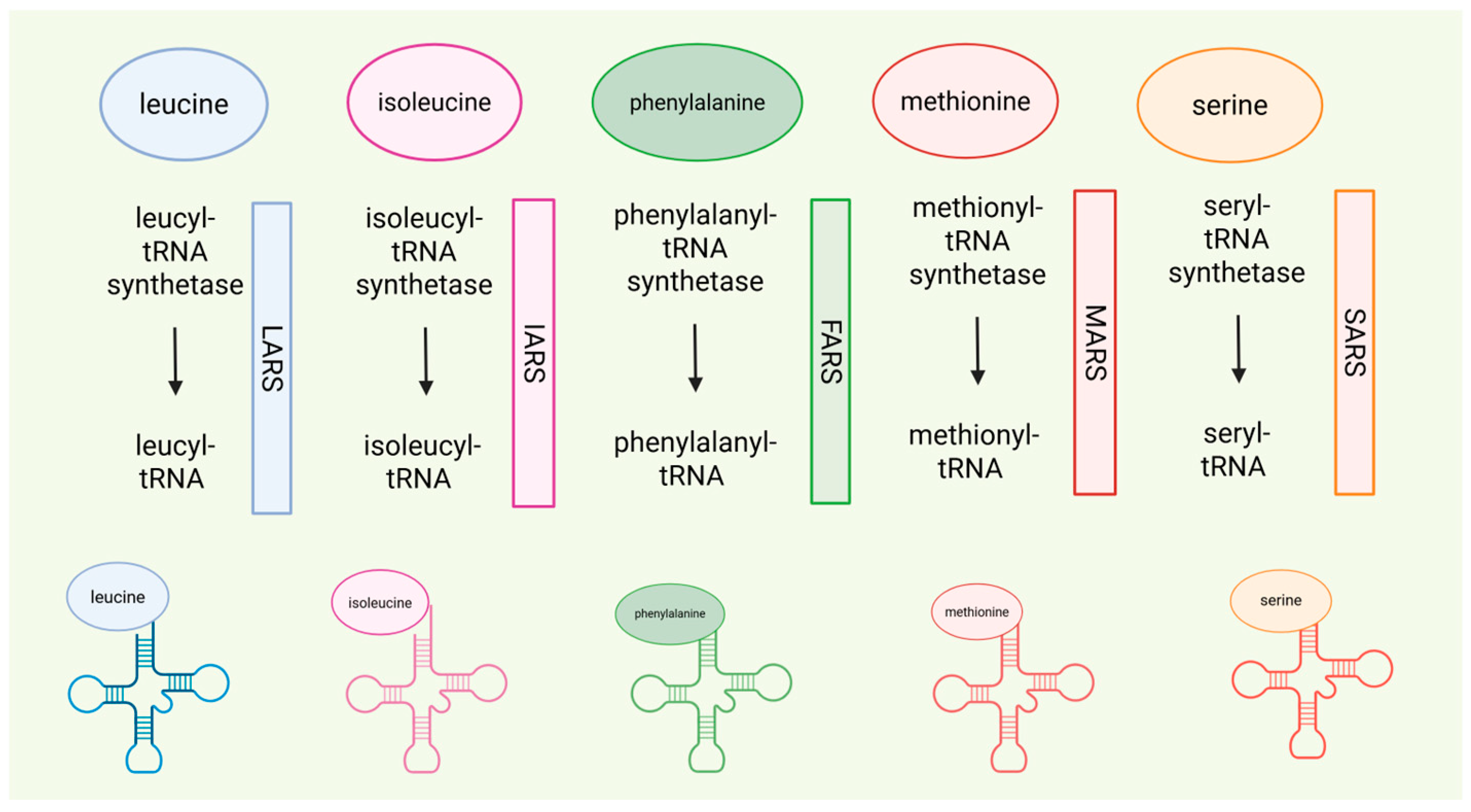
- t-RNA synthetase defects Isoleucyl-tRNA synthetase (IARS) and Leucyl-tRNA synthetase (LARS).
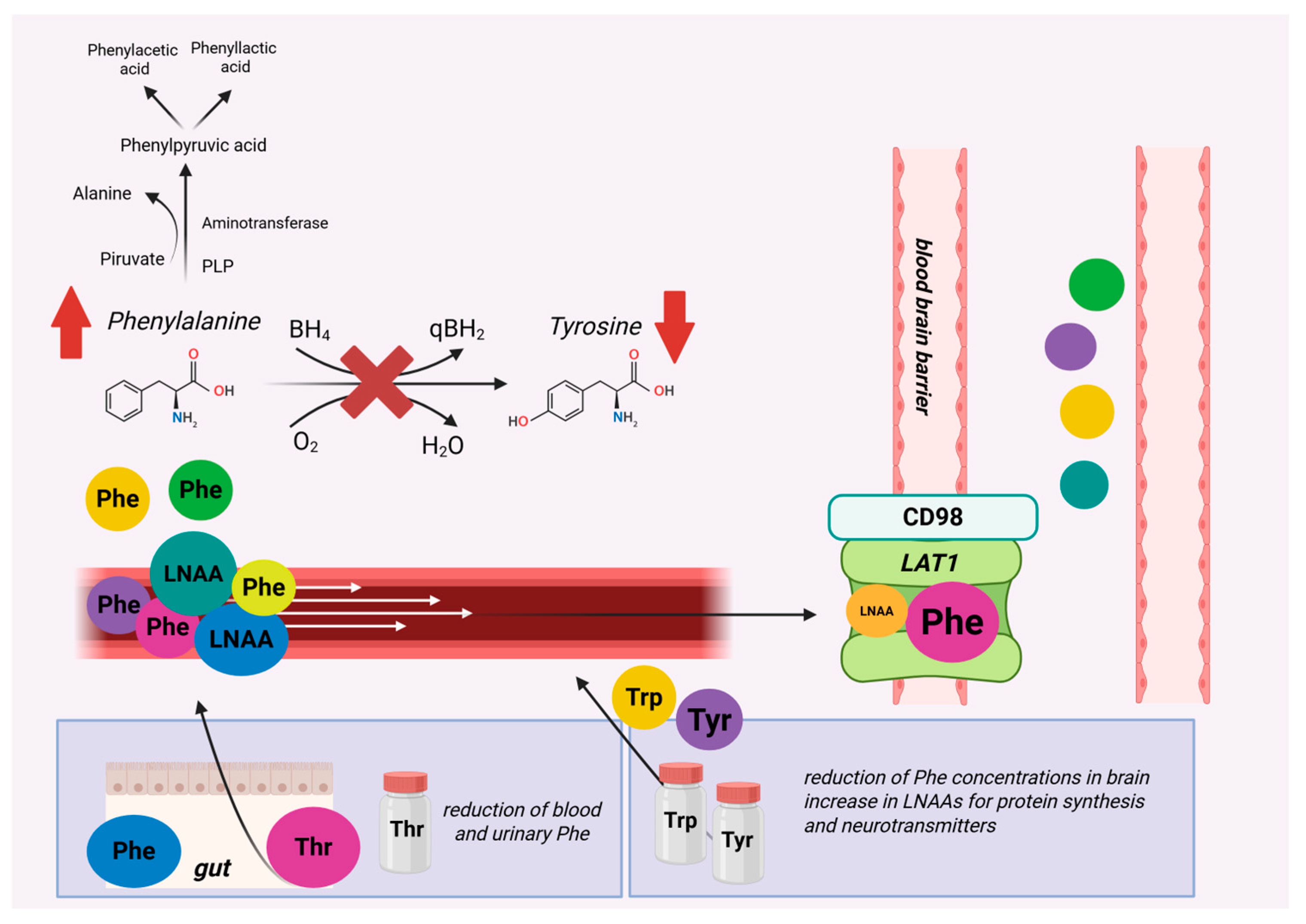
3.2. Aromatic Amino Acids (AAAs): Phenylalanine, Tryptophan, and Tyrosine
- Phenylketonuria (PKU);
- Phenylalanyl-tRNA synthetase (FARS).
3.3. Sulfur Amino Acids (S-AAs): Cysteine and Methionine
- Cystathionine β-synthase-deficient homocystinuria;
- Methylmalonic acidemia with homocystinuria, type cblC;
- Methylcobalamin deficiency type cblG;
- Methionyl-tRNA synthetase (MARS).
3.4. Urea Cycle Amino Acids (UCD-AAs): Arginine, Citrulline, and Ornithine
- Urea Cycle Disorders (UCDs);
- Citrin deficiency;
- Lysinuric protein intolerance (LPI);
- Glutaric Acidemia type 1;
- Pyridoxine-dependent epilepsy (PDE) and antiquitin deficiency (ATQ);
- X-linked creatine transporter deficiency (CRTR-D);
- Guanidinoacetate methyltransferase deficiency (GAMT);
- ALDH18A1-related De Barsy syndrome (P5CS deficiency);
- Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes syndrome (MELAS).
3.5. Other Essential Amino Acids (EAAs): Threonine and Lysine
- Ornithine aminotransferase deficiency (OAT);
- Phenylketonuria (PKU);
- Lysinuric protein intolerance.
3.6. Other Non-Essential Amino Acids (Non-EAAs): Alanine, Glycine, Glutamine, Proline, and Serine
- Isovaleric acidemia (IVA);
- X-linked creatine transporter deficiency (CRTR-D);
- Glutamine synthetase deficiency;
- Ornithine aminotransferase deficiency (OAT);
- ALDH18A1-related De Barsy syndrome (P5CS deficit);
- Seryl-tRNA synthetase 1 (SARS);
- Neurometabolic disorder due to serine deficiency;
- 3-Phosphoglycerate dehydrogenase deficiency (3-PGDH deficiency);
- Deficiency of phosphoserine aminotransferase (PSAT);
- GRIN-related disorders;
- Glycogen storage disease due to acid maltase deficiency (Pompe Disease).
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferreira, C.R.; Rahman, S.; Keller, M.; Zschocke, J.; ICIMD Advisory Group. An International Classification of Inherited Metabolic Disorders (ICIMD). J. Inherit. Metab. Dis. 2021, 44, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Saudubray, J.-M.; García-Cazorla, Á. Clinical Approach to Inborn Errors of Metabolism in Paediatrics. In Inborn Metabolic Diseases: Diagnosis and Treatment; Saudubray, J.-M., Baumgartner, M.R., García-Cazorla, Á., Walter, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2022; pp. 3–123. ISBN 978-3-662-63123-2. [Google Scholar]
- Saudubray, J.-M.; Sedel, F.; Walter, J.H. Clinical Approach to Treatable Inborn Metabolic Diseases: An Introduction. J. Inherit. Metab. Dis. 2006, 29, 261–274. [Google Scholar] [CrossRef]
- Ling, Z.-N.; Jiang, Y.-F.; Ru, J.-N.; Lu, J.-H.; Ding, B.; Wu, J. Amino Acid Metabolism in Health and Disease. Signal Transduct. Target. Ther. 2023, 8, 345. [Google Scholar] [CrossRef]
- Agnoli, C.; Baroni, L.; Bertini, I.; Ciappellano, S.; Fabbri, A.; Papa, M.; Pellegrini, N.; Sbarbati, R.; Scarino, M.L.; Siani, V.; et al. Position Paper on Vegetarian Diets from the Working Group of the Italian Society of Human Nutrition. Nutr. Metab. Cardiovasc. Dis. NMCD 2017, 27, 1037–1052. [Google Scholar] [CrossRef] [PubMed]
- Holeček, M. Branched-Chain Amino Acids in Health and Disease: Metabolism, Alterations in Blood Plasma, and as Supplements. Nutr. Metab. 2018, 15, 33. [Google Scholar] [CrossRef]
- de la O, V.; Zazpe, I.; Ruiz-Canela, M. Effect of Branched-Chain Amino Acid Supplementation, Dietary Intake and Circulating Levels in Cardiometabolic Diseases: An Updated Review. Curr. Opin. Clin. Nutr. Metab. Care 2020, 23, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Kuhara, T.; Ikeda, S.; Ohneda, A.; Sasaki, Y. Effects of Intravenous Infusion of 17 Amino Acids on the Secretion of GH, Glucagon, and Insulin in Sheep. Am. J. Physiol. 1991, 260, E21–E26. [Google Scholar] [CrossRef]
- Goichon, A.; Chan, P.; Lecleire, S.; Coquard, A.; Cailleux, A.-F.; Walrand, S.; Lerebours, E.; Vaudry, D.; Déchelotte, P.; Coëffier, M. An Enteral Leucine Supply Modulates Human Duodenal Mucosal Proteome and Decreases the Expression of Enzymes Involved in Fatty Acid β-Oxidation. J. Proteom. 2013, 78, 535–544. [Google Scholar] [CrossRef]
- van Vliet, D.; Derks, T.G.J.; van Rijn, M.; de Groot, M.J.; MacDonald, A.; Heiner-Fokkema, M.R.; van Spronsen, F.J. Single Amino Acid Supplementation in Aminoacidopathies: A Systematic Review. Orphanet J. Rare Dis. 2014, 9, 7. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G. PRISMA Group Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef]
- OCEBM Levels of Evidence. Available online: https://www.cebm.ox.ac.uk/resources/levels-of-evidence/ocebm-levels-of-evidence (accessed on 27 October 2024).
- Häberle, J.; Burlina, A.; Chakrapani, A.; Dixon, M.; Karall, D.; Lindner, M.; Mandel, H.; Martinelli, D.; Pintos-Morell, G.; Santer, R.; et al. Suggested Guidelines for the Diagnosis and Management of Urea Cycle Disorders: First Revision. J. Inherit. Metab. Dis. 2019, 42, 1192–1230. [Google Scholar] [CrossRef] [PubMed]
- Adam, S.; Almeida, M.F.; Assoun, M.; Baruteau, J.; Bernabei, S.M.; Bigot, S.; Champion, H.; Daly, A.; Dassy, M.; Dawson, S.; et al. Dietary Management of Urea Cycle Disorders: European Practice. Mol. Genet. Metab. 2013, 110, 439–445. [Google Scholar] [CrossRef]
- Scaglia, F. New Insights in Nutritional Management and Amino Acid Supplementation in Urea Cycle Disorders. Mol. Genet. Metab. 2010, 100 (Suppl. S1), S72–S76. [Google Scholar] [CrossRef]
- Brunetti-Pierri, N.; Lanpher, B.; Erez, A.; Ananieva, E.A.; Islam, M.; Marini, J.C.; Sun, Q.; Yu, C.; Hegde, M.; Li, J.; et al. Phenylbutyrate Therapy for Maple Syrup Urine Disease. Hum. Mol. Genet. 2011, 20, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Strauss, K.A.; Puffenberger, E.G.; Carson, V.J. Maple Syrup Urine Disease. In GeneReviews; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Strauss, K.A.; Wardley, B.; Robinson, D.; Hendrickson, C.; Rider, N.L.; Puffenberger, E.G.; Shellmer, D.; Moser, A.B.; Morton, D.H. Classical Maple Syrup Urine Disease and Brain Development: Principles of Management and Formula Design. Mol. Genet. Metab. 2010, 99, 333–345. [Google Scholar] [CrossRef]
- Koch, S.E.; Packman, S.; Koch, T.K.; Williams, M.L. Dermatitis in Treated Maple Syrup Urine Disease. J. Am. Acad. Dermatol. 1993, 28, 289–292. [Google Scholar] [CrossRef]
- Tornqvist, K.; Tornqvist, H. Corneal deepithelialization caused by acute deficiency of isoleucine during treatment of a patient with maple syrup urine disease. Acta Ophthalmol. Scand. Suppl. 1996, 74, 48–49. [Google Scholar] [CrossRef]
- Oztürk, Y. Acrodermatitis Enteropathica-like Syndrome Secondary to Branched-Chain Amino Acid Deficiency in Inborn Errors of Metabolism. Pediatr. Dermatol. 2008, 25, 415. [Google Scholar] [CrossRef] [PubMed]
- Morton, D.H.; Strauss, K.A.; Robinson, D.L.; Puffenberger, E.G.; Kelley, R.I. Diagnosis and Treatment of Maple Syrup Disease: A Study of 36 Patients. Pediatrics 2002, 109, 999–1008. [Google Scholar] [CrossRef]
- Parini, R.; Sereni, L.P.; Bagozzi, D.C.; Corbetta, C.; Rabier, D.; Narcy, C.; Hubert, P.; Saudubray, J.M. Nasogastric Drip Feeding as the Only Treatment of Neonatal Maple Syrup Urine Disease. Pediatrics 1993, 92, 280–283. [Google Scholar] [CrossRef]
- Nyhan, W.L.; Rice-Kelts, M.; Klein, J.; Barshop, B.A. Treatment of the Acute Crisis in Maple Syrup Urine Disease. Arch. Pediatr. Adolesc. Med. 1998, 152, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Frazier, D.M.; Allgeier, C.; Homer, C.; Marriage, B.J.; Ogata, B.; Rohr, F.; Splett, P.L.; Stembridge, A.; Singh, R.H. Nutrition Management Guideline for Maple Syrup Urine Disease: An Evidence- and Consensus-Based Approach. Mol. Genet. Metab. 2014, 112, 210–217. [Google Scholar] [CrossRef]
- Forny, P.; Hörster, F.; Ballhausen, D.; Chakrapani, A.; Chapman, K.A.; Dionisi-Vici, C.; Dixon, M.; Grünert, S.C.; Grunewald, S.; Haliloglu, G.; et al. Guidelines for the Diagnosis and Management of Methylmalonic Acidaemia and Propionic Acidaemia: First Revision. J. Inherit. Metab. Dis. 2021, 44, 566–592. [Google Scholar] [CrossRef]
- Manoli, I.; Myles, J.G.; Sloan, J.L.; Shchelochkov, O.A.; Venditti, C.P. A Critical Reappraisal of Dietary Practices in Methylmalonic Acidemia Raises Concerns about the Safety of Medical Foods. Part 1: Isolated Methylmalonic Acidemias. Genet. Med. 2016, 18, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Fraga, S.; Pinho, M.J.; Soares-da-Silva, P. Expression of LAT1 and LAT2 Amino Acid Transporters in Human and Rat Intestinal Epithelial Cells. Amino Acids 2005, 29, 229–233. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, A.; Singh, R.H.; Rocha, J.C.; van Spronsen, F.J. Optimising Amino Acid Absorption: Essential to Improve Nitrogen Balance and Metabolic Control in Phenylketonuria. Nutr. Res. Rev. 2019, 32, 70–78. [Google Scholar] [CrossRef]
- Molema, F.; Gleich, F.; Burgard, P.; van der Ploeg, A.T.; Summar, M.L.; Chapman, K.A.; Barić, I.; Lund, A.M.; Kölker, S.; Williams, M.; et al. Evaluation of Dietary Treatment and Amino Acid Supplementation in Organic Acidurias and Urea-Cycle Disorders: On the Basis of Information from a European Multicenter Registry. J. Inherit. Metab. Dis. 2019, 42, 1162–1175. [Google Scholar] [CrossRef]
- Gugelmo, G.; Lenzini, L.; Francini-Pesenti, F.; Fasan, I.; Spinella, P.; Valentini, R.; Miraval, A.; Avogaro, A.; Vitturi, N. Anthropometrics, Dietary Intake and Body Composition in Urea Cycle Disorders and Branched Chain Organic Acidemias: A Case Study of 18 Adults on Low-Protein Diets. Nutrients 2022, 14, 467. [Google Scholar] [CrossRef]
- Hauser, N.S.; Manoli, I.; Graf, J.C.; Sloan, J.; Venditti, C.P. Variable Dietary Management of Methylmalonic Acidemia: Metabolic and Energetic Correlations. Am. J. Clin. Nutr. 2011, 93, 47–56. [Google Scholar] [CrossRef]
- Jurecki, E.; Ueda, K.; Frazier, D.; Rohr, F.; Thompson, A.; Hussa, C.; Obernolte, L.; Reineking, B.; Roberts, A.M.; Yannicelli, S.; et al. Nutrition Management Guideline for Propionic Acidemia: An Evidence- and Consensus-Based Approach. Mol. Genet. Metab. 2019, 126, 341–354. [Google Scholar] [CrossRef]
- Myles, J.G.; Manoli, I.; Venditti, C.P. Effects of Medical Food Leucine Content in the Management of Methylmalonic and Propionic Acidemias. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Fecarotta, S.; Alagia, M.; Attaianese, F.; Verde, A.; Tarallo, A.; Gragnaniello, V.; Ziagaki, A.; Guimaraes, M.J.; Aguiar, P.; et al. The European Reference Network for Metabolic Diseases (MetabERN) Clinical Pathway Recommendations for Pompe Disease (Acid Maltase Deficiency, Glycogen Storage Disease Type II). Orphanet J. Rare Dis. 2024, 19, 408. [Google Scholar] [CrossRef]
- Mobarhan, S.; Pintozzi, R.L.; Damle, P.; Friedman, H. Treatment of Acid Maltase Deficiency with a Diet High in Branched-Chain Amino Acids. JPEN J. Parenter. Enteral. Nutr. 1990, 14, 210–212. [Google Scholar] [CrossRef]
- Umpleby, A.M.; Trend, P.S.; Chubb, D.; Conaglen, J.V.; Williams, C.D.; Hesp, R.; Scobie, I.N.; Wiles, C.M.; Spencer, G.; Sönksen, P.H. The Effect of a High Protein Diet on Leucine and Alanine Turnover in Acid Maltase Deficiency. J. Neurol. Neurosurg. Psychiatry 1989, 52, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Kok, G.; Tseng, L.; Schene, I.F.; Dijsselhof, M.E.; Salomons, G.; Mendes, M.I.; Smith, D.E.C.; Wiedemann, A.; Canton, M.; Feillet, F.; et al. Treatment of ARS Deficiencies with Specific Amino Acids. Genet. Med. 2021, 23, 2202–2207. [Google Scholar] [CrossRef]
- Hoytema van Konijnenburg, E.M.M.; Rohof, J.; Kok, G.; van Hasselt, P.M.; van Karnebeek, C.D.; Muffels, I.J.J.; Fuchs, S.A. Setting the Stage for Treatment of Aminoacyl-tRNA Synthetase (ARS)1-Deficiencies: Phenotypic Characterization and a Review of Treatment Effects. J. Inherit. Metab. Dis. 2025, 48, e70017. [Google Scholar] [CrossRef]
- van Wegberg, A.M.J.; MacDonald, A.; Ahring, K.; Bélanger-Quintana, A.; Blau, N.; Bosch, A.M.; Burlina, A.; Campistol, J.; Feillet, F.; Giżewska, M.; et al. The Complete European Guidelines on Phenylketonuria: Diagnosis and Treatment. Orphanet J. Rare Dis. 2017, 12, 162. [Google Scholar] [CrossRef] [PubMed]
- Remmington, T.; Smith, S. Tyrosine Supplementation for Phenylketonuria. Cochrane Database Syst. Rev. 2013, 2021, CD001507. [Google Scholar] [CrossRef]
- Pietz, J.; Kreis, R.; Rupp, A.; Mayatepek, E.; Rating, D.; Boesch, C.; Bremer, H.J. Large Neutral Amino Acids Block Phenylalanine Transport into Brain Tissue in Patients with Phenylketonuria. J. Clin Investig. 1999, 103, 1169–1178. [Google Scholar] [CrossRef]
- van Spronsen, F.J.; de Groot, M.J.; Hoeksma, M.; Reijngoud, D.-J.; van Rijn, M. Large Neutral Amino Acids in the Treatment of PKU: From Theory to Practice. J. Inherit. Metab. Dis. 2010, 33, 671–676. [Google Scholar] [CrossRef]
- de Groot, M.J.; Sijens, P.E.; Reijngoud, D.-J.; Paans, A.M.; van Spronsen, F.J. Phenylketonuria: Brain Phenylalanine Concentrations Relate Inversely to Cerebral Protein Synthesis. J. Cereb. Blood Flow Metab. 2015, 35, 200–205. [Google Scholar] [CrossRef]
- de Groot, M.J.; Hoeksma, M.; Reijngoud, D.-J.; de Valk, H.W.; Paans, A.M.J.; Sauer, P.J.J.; van Spronsen, F.J. Phenylketonuria: Reduced Tyrosine Brain Influx Relates to Reduced Cerebral Protein Synthesis. Orphanet J. Rare Dis. 2013, 8, 133. [Google Scholar] [CrossRef] [PubMed]
- Sanjurjo, P.; Aldamiz, L.; Georgi, G.; Jelinek, J.; Ruiz, J.I.; Boehm, G. Dietary Threonine Reduces Plasma Phenylalanine Levels in Patients with Hyperphenylalaninemia. J. Pediatr. Gastroenterol. Nutr. 2003, 36, 23–26. [Google Scholar] [CrossRef]
- Yano, S.; Moseley, K.; Azen, C. Melatonin and Dopamine as Biomarkers to Optimize Treatment in Phenylketonuria: Effects of Tryptophan and Tyrosine Supplementation. J. Pediatr. 2014, 165, 184–189.e1. [Google Scholar] [CrossRef] [PubMed]
- Oswald, S.L.; Steinbrücker, K.; Achleitner, M.T.; Göschl, E.; Bittner, R.E.; Schmidt, W.M.; Tiefenthaler, E.; Hammerl, E.; Eisl, A.; Mayr, D.; et al. Treatment of Mitochondrial Phenylalanyl-tRNa-Synthetase Deficiency (FARS2) with Oral Phenylalanine. Neuropediatrics 2023, 54, 351–355. [Google Scholar] [CrossRef]
- Morris, A.A.M.; Kožich, V.; Santra, S.; Andria, G.; Ben-Omran, T.I.M.; Chakrapani, A.B.; Crushell, E.; Henderson, M.J.; Hochuli, M.; Huemer, M.; et al. Guidelines for the Diagnosis and Management of Cystathionine β-Synthase Deficiency. J. Inherit. Metab. Dis. 2017, 40, 49–74. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.J.; Briddon, A. A Rationale for Cystine Supplementation in Severe Homocystinuria. J. Inherit. Metab. Dis. 2007, 30, 35–38. [Google Scholar] [CrossRef]
- Huemer, M.; Diodato, D.; Schwahn, B.; Schiff, M.; Bandeira, A.; Benoist, J.-F.; Burlina, A.; Cerone, R.; Couce, M.L.; Garcia-Cazorla, A.; et al. Guidelines for Diagnosis and Management of the Cobalamin-Related Remethylation Disorders cblC, cblD, cblE, cblF, cblG, cblJ and MTHFR Deficiency. J. Inherit. Metab. Dis. 2017, 40, 21–48. [Google Scholar] [CrossRef]
- Arhip, L.; Brox-Torrecilla, N.; Romero, I.; Motilla, M.; Serrano-Moreno, C.; Miguélez, M.; Cuerda, C. Late-Onset Methylmalonic Acidemia and Homocysteinemia (cblC Disease): Systematic Review. Orphanet J. Rare Dis. 2024, 19, 20. [Google Scholar] [CrossRef]
- Broomfield, A.; Abulhoul, L.; Pitt, W.; Jameson, E.; Cleary, M. Reversal of Respiratory Failure in Both Neonatal and Late Onset Isolated Remethylation Disorders. JIMD Rep. 2014, 16, 51–56. [Google Scholar] [CrossRef]
- Kripps, K.A.; Sremba, L.; Larson, A.A.; Van Hove, J.L.K.; Nguyen, H.; Wright, E.L.; Mirsky, D.M.; Watkins, D.; Rosenblatt, D.S.; Ketteridge, D.; et al. Methionine Synthase Deficiency: Variable Clinical Presentation and Benefit of Early Diagnosis and Treatment. J. Inherit. Metab. Dis. 2022, 45, 157–168. [Google Scholar] [CrossRef]
- Lenz, D.; Stahl, M.; Seidl, E.; Schöndorf, D.; Brennenstuhl, H.; Gesenhues, F.; Heinzmann, T.; Longerich, T.; Mendes, M.I.; Prokisch, H.; et al. Rescue of Respiratory Failure in Pulmonary Alveolar Proteinosis Due to Pathogenic MARS1 Variants. Pediatr. Pulmonol. 2020, 55, 3057–3066. [Google Scholar] [CrossRef] [PubMed]
- Hadchouel, A.; Drummond, D.; Pontoizeau, C.; Aoust, L.; Hurtado Nedelec, M.-M.; El Benna, J.; Gachelin, E.; Perisson, C.; Vigier, C.; Schiff, M.; et al. Methionine Supplementation for Multi-Organ Dysfunction in MetRS-Related Pulmonary Alveolar Proteinosis. Eur. Respir. J. 2022, 59, 2101554. [Google Scholar] [CrossRef]
- Roy, C.; Allou, N.; Coulomb, A.; Grenet, D.; Borie, R.; Zuber, B.; Hamid, A.; Glorion, M.; Brun, A.-L.; Longchamps, E.; et al. Successful Lung Transplantation in Genetic Methionyl-tRNA Synthetase-Related Alveolar Proteinosis/Lung Fibrosis without Recurrence under Methionine Supplementation: Medium-Term Outcome in 4 Cases. Am. J. Transplant. 2024, 24, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Imbard, A.; Bouchereau, J.; Arnoux, J.-B.; Brassier, A.; Schiff, M.; Bérat, C.-M.; Pontoizeau, C.; Benoist, J.-F.; Josse, C.; Montestruc, F.; et al. Citrulline in the Management of Patients with Urea Cycle Disorders. Orphanet J. Rare Dis. 2023, 18, 207. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, D.; Diodato, D.; Ponzi, E.; Monné, M.; Boenzi, S.; Bertini, E.; Fiermonte, G.; Dionisi-Vici, C. The Hyperornithinemia-Hyperammonemia-Homocitrullinuria Syndrome. Orphanet J. Rare Dis. 2015, 10, 29. [Google Scholar] [CrossRef]
- Vuković, T.; Kuek, L.E.; Yu, B.; Makris, G.; Häberle, J. The Therapeutic Landscape of Citrin Deficiency. J. Inherit. Metab. Dis. 2024, 47, 1157–1174. [Google Scholar] [CrossRef]
- Pinto, A.; Ashmore, C.; Batzios, S.; Daly, A.; Dawson, C.; Dixon, M.; Evans, S.; Green, D.; Gribben, J.; Hunjan, I.; et al. Dietary Management, Clinical Status and Outcome of Patients with Citrin Deficiency in the UK. Nutrients 2020, 12, 3313. [Google Scholar] [CrossRef]
- Imamura, Y.; Kobayashi, K.; Shibatou, T.; Aburada, S.; Tahara, K.; Kubozono, O.; Saheki, T. Effectiveness of Carbohydrate-Restricted Diet and Arginine Granules Therapy for Adult-Onset Type II Citrullinemia: A Case Report of Siblings Showing Homozygous SLC25A13 Mutation with and without the Disease. Hepatol. Res. 2003, 26, 68–72. [Google Scholar] [CrossRef]
- Ogier de Baulny, H.; Schiff, M.; Dionisi-Vici, C. Lysinuric Protein Intolerance (LPI): A Multi Organ Disease by Far More Complex than a Classic Urea Cycle Disorder. Mol. Genet. Metab. 2012, 106, 12–17. [Google Scholar] [CrossRef]
- Ziegler, S.G.; Kim, J.; Ehmsen, J.T.; Vernon, H.J. Inborn Errors of Amino Acid Metabolism—From Underlying Pathophysiology to Therapeutic Advances. Dis. Model. Mech. 2023, 16, dmm050233. [Google Scholar] [CrossRef]
- Mizutani, N.; Kato, T.; Maehara, M.; Watanabe, K.; Ban, M. Oral Administration of Arginine and Citrulline in the Treatment of Lysinuric Protein Intolerance. Tohoku J. Exp. Med. 1984, 142, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, T.O.; Levy, H.L.; Holtrop, M.E.; Shih, V.E.; Anast, C.S. Lysinuric Protein Intolerance Presenting as Childhood Osteoporosis. Clinical and Skeletal Response to Citrulline Therapy. N. Engl. J. Med. 1985, 312, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Rajantie, J.; Simell, O.; Rapola, J.; Perheentupa, J. Lysinuric Protein Intolerance: A Two-Year Trial of Dietary Supplementation Therapy with Citrulline and Lysine. J. Pediatr. 1980, 97, 927–932. [Google Scholar] [CrossRef]
- Kang, D.-W.; Adams, J.B.; Coleman, D.M.; Pollard, E.L.; Maldonado, J.; McDonough-Means, S.; Caporaso, J.G.; Krajmalnik-Brown, R. Long-Term Benefit of Microbiota Transfer Therapy on Autism Symptoms and Gut Microbiota. Sci. Rep. 2019, 9, 5821. [Google Scholar] [CrossRef]
- Nunes, V.; Niinikoski, H. Lysinuric Protein Intolerance. In GeneReviews; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Kölker, S.; Christensen, E.; Leonard, J.V.; Greenberg, C.R.; Boneh, A.; Burlina, A.B.; Burlina, A.P.; Dixon, M.; Duran, M.; García Cazorla, A.; et al. Diagnosis and Management of Glutaric Aciduria Type I—Revised Recommendations. J. Inherit. Metab. Dis. 2011, 34, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.-M. Update Current Understanding of Neurometabolic Disorders Related to Lysine Metabolism. Epilepsy Behav. 2023, 146, 109363. [Google Scholar] [CrossRef]
- Mercimek-Mahmutoglu, S.; Cordeiro, D.; Cruz, V.; Hyland, K.; Struys, E.A.; Kyriakopoulou, L.; Mamak, E. Novel Therapy for Pyridoxine Dependent Epilepsy Due to ALDH7A1 Genetic Defect: L-Arginine Supplementation Alternative to Lysine-Restricted Diet. Eur. J. Paediatr. Neurol. 2014, 18, 741–746. [Google Scholar] [CrossRef]
- Coughlin, C.R.; van Karnebeek, C.D.M.; Al-Hertani, W.; Shuen, A.Y.; Jaggumantri, S.; Jack, R.M.; Gaughan, S.; Burns, C.; Mirsky, D.M.; Gallagher, R.C.; et al. Triple Therapy with Pyridoxine, Arginine Supplementation and Dietary Lysine Restriction in Pyridoxine-Dependent Epilepsy: Neurodevelopmental Outcome. Mol. Genet. Metab. 2015, 116, 35–43. [Google Scholar] [CrossRef]
- Kim, J.; Pipitone Dempsey, A.; Kim, S.Y.; Gunay-Aygun, M.; Vernon, H.J. An Atypical Presentation of Pyridoxine-Dependent Epilepsy Diagnosed with Whole Exome Sequencing and Treated with Lysine Restriction and Supplementation with Arginine and Pyridoxine. Case Rep. Genet. 2022, 2022, 7138435. [Google Scholar] [CrossRef]
- Tseng, L.A.; Abdenur, J.E.; Andrews, A.; Aziz, V.G.; Bok, L.A.; Boyer, M.; Buhas, D.; Hartmann, H.; Footitt, E.J.; Grønborg, S.; et al. Timing of Therapy and Neurodevelopmental Outcomes in 18 Families with Pyridoxine-Dependent Epilepsy. Mol. Genet. Metab. 2022, 135, 350–356. [Google Scholar] [CrossRef]
- Tauer, K.; Theile, C.; Owens, J.W.; Cecil, K.M.; Shillington, A. Arginine, Glycine, and Creatine Supplementation Improves Symptoms in a Female with Creatine Transporter Deficiency. Psychiatr. Genet. 2024, 34, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Mejdahl Nielsen, M.; Petersen, E.T.; Fenger, C.D.; Ørngreen, M.C.; Siebner, H.R.; Boer, V.O.; Považan, M.; Lund, A.; Grønborg, S.W.; Hammer, T.B. X-Linked Creatine Transporter (SLC6A8) Deficiency in Females: Difficult to Recognize, but a Potentially Treatable Disease. Mol. Genet. Metab. 2023, 140, 107694. [Google Scholar] [CrossRef]
- Li, J.; Xu, S. Diagnosis and Treatment of X-Linked Creatine Transporter Deficiency: Case Report and Literature Review. Brain Sci. 2023, 13, 1382. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, D.; Häberle, J.; Rubio, V.; Giunta, C.; Hausser, I.; Carrozzo, R.; Gougeard, N.; Marco-Marín, C.; Goffredo, B.M.; Meschini, M.C.; et al. Understanding Pyrroline-5-Carboxylate Synthetase Deficiency: Clinical, Molecular, Functional, and Expression Studies, Structure-Based Analysis, and Novel Therapy with Arginine. J. Inherit. Metab. Dis. 2012, 35, 761–776. [Google Scholar] [CrossRef] [PubMed]
- Koenig, M.K.; Emrick, L.; Karaa, A.; Korson, M.; Scaglia, F.; Parikh, S.; Goldstein, A. Recommendations for the Management of Strokelike Episodes in Patients With Mitochondrial Encephalomyopathy, Lactic Acidosis, and Strokelike Episodes. JAMA Neurol. 2016, 73, 591–594. [Google Scholar] [CrossRef]
- Barros, C.D.S.; Coutinho, A.; Tengan, C.H. Arginine Supplementation in MELAS Syndrome: What Do We Know about the Mechanisms? Int. J. Mol. Sci. 2024, 25, 3629. [Google Scholar] [CrossRef]
- Argudo, J.M.; Astudillo Moncayo, O.M.; Insuasti, W.; Garofalo, G.; Aguirre, A.S.; Encalada, S.; Villamarin, J.; Oña, S.; Tenemaza, M.G.; Eissa-Garcés, A.; et al. Arginine for the Treatment of Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-Like Episodes: A Systematic Review. Cureus 2022, 14, e32709. [Google Scholar] [CrossRef]
- Stefanetti, R.J.; Ng, Y.S.; Errington, L.; Blain, A.P.; McFarland, R.; Gorman, G.S. L-Arginine in Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like Episodes: A Systematic Review. Neurology 2022, 98, e2318–e2328. [Google Scholar] [CrossRef]
- Peltola, K.; Heinonen, O.J.; Näntö-Salonen, K.; Pulkki, K.; Simell, O. Oral Lysine Feeding in Gyrate Atrophy with Hyperornithinaemia--a Pilot Study. J. Inherit. Metab. Dis. 2000, 23, 305–307. [Google Scholar] [CrossRef]
- Elpeleg, N.; Korman, S.H. Sustained Oral Lysine Supplementation in Ornithine Delta-Aminotransferase Deficiency. J. Inherit. Metab. Dis. 2001, 24, 423–424. [Google Scholar] [CrossRef]
- Brands, M.; Balfoort, B.; Acharya, K.; Bergen, A.; Brunetti-Pierri, N.; Buijs, M.; Cellini, B.; Schultink, P.; Singh, M.; Schulze, A.; et al. A Mini-Review on the International Gyrate Atrophy Symposium 2023: More than Meets the Eye. Focus on Outstanding Research Questions. Mol. Genet. Metab. 2024, 143, 108609. [Google Scholar] [CrossRef]
- Hjelm, M.; Seakins, J.; Antoshechkin, A. Indications of Changed Amino Acid Homeostasis in Untreated and Treated PKU. Acta Paediatr. Suppl. 1994, 407, 57–59. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Ye, J.; Han, L.; Qiu, W.; Zhang, H.; Yu, Y.; Zhu, T.; Xu, F.; Zhan, X.; Bao, P.; et al. Examining the Blood Amino Acid Status in Pretherapeutic Patients with Hyperphenylalaninemia. J. Clin. Lab. Anal. 2020, 34, e23106. [Google Scholar] [CrossRef] [PubMed]
- Maenz, D.D.; Patience, J.F. L-Threonine Transport in Pig Jejunal Brush Border Membrane Vesicles. Functional Characterization of the Unique System B in the Intestinal Epithelium. J. Biol. Chem. 1992, 267, 22079–22086. [Google Scholar] [CrossRef] [PubMed]
- Mütze, U.; Reischl-Hajiabadi, A.; Kölker, S. Classic Isovaleric Acidemia. In GeneReviews; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Roe, C.R.; Millington, D.S.; Maltby, D.A.; Kahler, S.G.; Bohan, T.P. L-Carnitine Therapy in Isovaleric Acidemia. J. Clin. Investig. 1984, 74, 2290–2295. [Google Scholar] [CrossRef]
- Krieger, I.; Tanaka, K. Therapeutic Effects of Glycine in Isovaleric Acidemia. Pediatr. Res. 1976, 10, 25–29. [Google Scholar] [CrossRef]
- Yudkoff, M.; Cohn, R.M.; Puschak, R.; Rothman, R.; Segal, S. Glycine Therapy in Isovaleric Acidemia. J. Pediatr. 1978, 92, 813–817. [Google Scholar] [CrossRef]
- Shigematsu, Y.; Sudo, M.; Momoi, T.; Inoue, Y.; Suzuki, Y.; Kameyama, J. Changing Plasma and Urinary Organic Acid Levels in a Patient with Isovaleric Acidemia during an Attack. Pediatr. Res. 1982, 16, 771–775. [Google Scholar] [CrossRef]
- Mütze, U.; Henze, L.; Gleich, F.; Lindner, M.; Grünert, S.C.; Spiekerkoetter, U.; Santer, R.; Blessing, H.; Thimm, E.; Ensenauer, R.; et al. Newborn Screening and Disease Variants Predict Neurological Outcome in Isovaleric Aciduria. J. Inherit. Metab. Dis. 2021, 44, 857–870. [Google Scholar] [CrossRef]
- Spodenkiewicz, M.; Diez-Fernandez, C.; Rüfenacht, V.; Gemperle-Britschgi, C.; Häberle, J. Minireview on Glutamine Synthetase Deficiency, an Ultra-Rare Inborn Error of Amino Acid Biosynthesis. Biology 2016, 5, 40. [Google Scholar] [CrossRef]
- Häberle, J.; Shahbeck, N.; Ibrahim, K.; Schmitt, B.; Scheer, I.; O’Gorman, R.; Chaudhry, F.A.; Ben-Omran, T. Glutamine Supplementation in a Child with Inherited GS Deficiency Improves the Clinical Status and Partially Corrects the Peripheral and Central Amino Acid Imbalance. Orphanet J. Rare Dis. 2012, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Hayasaka, S.; Saito, T.; Nakajima, H.; Takahashi, O.; Mizuno, K.; Tada, K. Clinical Trials of Vitamin B6 and Proline Supplementation for Gyrate Atrophy of the Choroid and Retina. Br. J. Ophthalmol. 1985, 69, 283–290. [Google Scholar] [CrossRef]
- van der Crabben, S.N.; de Koning, T.J. Serine Deficiency Disorders. In GeneReviews; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Tabatabaie, L.; Klomp, L.W.J.; Rubio-Gozalbo, M.E.; Spaapen, L.J.M.; Haagen, A.a.M.; Dorland, L.; de Koning, T.J. Expanding the Clinical Spectrum of 3-Phosphoglycerate Dehydrogenase Deficiency. J. Inherit. Metab. Dis. 2011, 34, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Hart, C.E.; Race, V.; Achouri, Y.; Wiame, E.; Sharrard, M.; Olpin, S.E.; Watkinson, J.; Bonham, J.R.; Jaeken, J.; Matthijs, G.; et al. Phosphoserine Aminotransferase Deficiency: A Novel Disorder of the Serine Biosynthesis Pathway. Am. J. Hum. Genet. 2007, 80, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Montanucci, L.; Brünger, T.; Bhattarai, N.; Boßelmann, C.M.; Kim, S.; Allen, J.P.; Zhang, J.; Klöckner, C.; Krey, I.; Fariselli, P.; et al. Ligand Distances as Key Predictors of Pathogenicity and Function in NMDA Receptors. Hum. Mol. Genet. 2025, 34, 128–139. [Google Scholar] [CrossRef]
- Wolosker, H. The Neurobiology of D-Serine Signaling. Adv. Pharmacol. 2018, 82, 325–348. [Google Scholar] [CrossRef]
- MacKay, M.-A.B.; Kravtsenyuk, M.; Thomas, R.; Mitchell, N.D.; Dursun, S.M.; Baker, G.B. D-Serine: Potential Therapeutic Agent and/or Biomarker in Schizophrenia and Depression? Front. Psychiatry 2019, 10, 25. [Google Scholar] [CrossRef]
- Soto, D.; Olivella, M.; Grau, C.; Armstrong, J.; Alcon, C.; Gasull, X.; Santos-Gómez, A.; Locubiche, S.; Gómez de Salazar, M.; García-Díaz, R.; et al. L-Serine Dietary Supplementation Is Associated with Clinical Improvement of Loss-of-Function GRIN2B-Related Pediatric Encephalopathy. Sci. Signal 2019, 12, eaaw0936. [Google Scholar] [CrossRef]
- Juliá-Palacios, N.; Olivella, M.; Sigatullina Bondarenko, M.; Ibáñez-Micó, S.; Muñoz-Cabello, B.; Alonso-Luengo, O.; Soto-Insuga, V.; García-Navas, D.; Cuesta-Herraiz, L.; Andreo-Lillo, P.; et al. L-Serine Treatment in Patients with GRIN-Related Encephalopathy: A Phase 2A, Non-Randomized Study. Brain 2024, 147, 1653–1666. [Google Scholar] [CrossRef]
- Kelts, D.G.; Ney, D.; Bay, C.; Saudubray, J.M.; Nyhan, W.L. Studies on Requirements for Amino Acids in Infants with Disorders of Amino Acid Metabolism. I. Effect of Alanine. Pediatr. Res. 1985, 19, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Mundy, H.R.; Williams, J.E.; Cousins, A.J.; Lee, P.J. The Effect of L-Alanine Therapy in a Patient with Adult Onset Glycogen Storage Disease Type II. J. Inherit. Metab. Dis. 2006, 29, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Bodamer, O.A.; Halliday, D.; Leonard, J.V. The Effects of L-Alanine Supplementation in Late-Onset Glycogen Storage Disease Type II. Neurology 2000, 55, 710–712. [Google Scholar] [CrossRef]
- Bodamer, O.A.; Haas, D.; Hermans, M.M.; Reuser, A.J.; Hoffmann, G.F. L-Alanine Supplementation in Late Infantile Glycogen Storage Disease Type II. Pediatr. Neurol. 2002, 27, 145–146. [Google Scholar] [CrossRef] [PubMed]
- Rovelli, V.; Zuvadelli, J.; Piotto, M.; Scopari, A.; Dionigi, A.R.; Ercoli, V.; Paci, S.; Cefalo, G.; Salvatici, E.; Banderali, G. L-Alanine Supplementation in Pompe Disease (IOPD): A Potential Therapeutic Implementation for Patients on ERT? A Case Report. Ital. J. Pediatr. 2022, 48, 48. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Level of Evidence | Type of Study |
|---|---|
| Level 1: High-Quality Evidence | 1a: Systematic Review (with homogeneity) of Randomized Controlled Trials (RCTs) 1b: Individual Randomized Controlled Trial (RCT) with a Narrow Confidence Interval 1c: “All or None” Study |
| Level 2: Individual RCTs or cohort studies with dramatic effects | 2a: Systematic Review (with homogeneity) of Cohort Studies 2b: Individual Cohort Study or Low-Quality RCT 2c: “Outcomes” Research or Ecological Studies |
| Level 3: Non-randomized controlled cohort/follow-up study | 3a: Systematic Review (with homogeneity) of Case-Control Studies 3b: Individual Case-Control Study |
| Level 4: Case Series or Poor-Quality Cohort and Case-Control Studies | |
| Level 5: Expert opinion or reasoning without supporting evidence from clinical studies | |
| IMDs | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| AAs | MSUD | PA | MMA | IVA | UCDs (Not Arg1) | IARS | LARS | SARS | FARS | MARS | POMPE |
| Leu | LoE: 1a | LoE: 4 D: 35–100 mg/kg/day | X | ||||||||
| Ile | LoE: 4 D: 10–30 mg/kg/day (routine) D: 20–120 mg/kg/day (acute episodes) | LoE: 4 D: 100 mg/day or 50–350 mg/kg/die | LoE: 4 D: 48–340 mg/day or 50–350 mg/kg/die | LoE: 1a | LoE: 4 D: 35–70 or 200 mg/kg/day | X | |||||
| Val | LoE: 4 D: 10–30 mg/kg/day (routine) D: 20–120 mg/kg/day (acute episodes) | LoE: 4 D: 70–170 mg/kg/day | LoE: 4 D: 70–170 mg/kg/day | LoE: 1a | X | ||||||
| Ala | LoE: 4 up to 600 mg/kg/day on ERT | ||||||||||
| Gly | LoE: 4 D: 150–300 mg/kg/day | ||||||||||
| Arg | LoE: 2b D: 200–300 mg/kg/day | ||||||||||
| Cit | LoE: 2b D: 150–400 mg/kg/day | ||||||||||
| Met | LoE: 4 D: 50–150 mg/kg/day | ||||||||||
| Phe | LoE: 4 D: 40–100 mg/kg/day | ||||||||||
| Ser | LoE: 4 85.7–97.5 mg/kg/day | ||||||||||
| IMDs | |||||||||||
| AAs | PKU | HCU | CblC | CblG | GA1 | GAMT | CRTR-D | CD | LPI | ||
| Gly | LoE: 4 D: 150–180 mg/kg/day | ||||||||||
| Arg | LoE: 4 D: 100–150 mg/kg/day | LoE: 4 D: 400 mg/kg/day | X | ||||||||
| Orn | LoE: 4 D: 100–800 mg/kg/day | ||||||||||
| Cit | LoE: 4 D: 0.5–1.1 mmol/kg/day (long term) | ||||||||||
| Lys | LoE: 4 D: 20–30 mg/kg/day | ||||||||||
| Cys | X | ||||||||||
| Met | X | LoE: 4 D: 10–12 mg/kg/day | |||||||||
| Phe | |||||||||||
| Tyr | X | ||||||||||
| Thr | LoE: 2b D: 50 mg/kg/day | ||||||||||
| Trp | LoE: 2b D: 100 mg/kg/day | ||||||||||
| IMDs | |||||||||||
| AAs | P5CSD | MELAS | PDE | OAT | GS def. | Serine def. | 3-PGDH | PSAT | GRIN 2B | ||
| Gly | LoE: 4 D: 200–300 mg/kg/day | LoE: 4 D: 200 mg/kg/day | |||||||||
| Arg | LoE: 4 D: 150 mg/kg/day | X | LoE: 3b | ||||||||
| Orn | X | ||||||||||
| Cit | X | X | |||||||||
| Lys | LoE: 4 D: 10–15 g/day | ||||||||||
| Gln | LoE: 4 From 17 mg/kg/day to 1020 mg/kg/day (enteral and parenteral) | ||||||||||
| Pro | X | 4 | |||||||||
| Ser | LoE: 4 D: From 200–400 to 500–700 mg/kg/day | LoE: 2b Treatment: 250–500 mg/kg/day (without exceeding 30 g/day for patients weighing ≥60 kg) | |||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verduci, E.; Tosi, M.; Dionisi Vici, C.; Spada, M. Single Amino Acid Supplementation in Inherited Metabolic Disorders: An Evidence-Based Review of Interventions. Genes 2025, 16, 502. https://doi.org/10.3390/genes16050502
Verduci E, Tosi M, Dionisi Vici C, Spada M. Single Amino Acid Supplementation in Inherited Metabolic Disorders: An Evidence-Based Review of Interventions. Genes. 2025; 16(5):502. https://doi.org/10.3390/genes16050502
Chicago/Turabian StyleVerduci, Elvira, Martina Tosi, Carlo Dionisi Vici, and Marco Spada. 2025. "Single Amino Acid Supplementation in Inherited Metabolic Disorders: An Evidence-Based Review of Interventions" Genes 16, no. 5: 502. https://doi.org/10.3390/genes16050502
APA StyleVerduci, E., Tosi, M., Dionisi Vici, C., & Spada, M. (2025). Single Amino Acid Supplementation in Inherited Metabolic Disorders: An Evidence-Based Review of Interventions. Genes, 16(5), 502. https://doi.org/10.3390/genes16050502