The Role of microRNAs in Mitochondria: Small Players Acting Wide




Abstract
:1. Introduction
2. Mitochondria
3. Translocation of miRNAs to Mitochondria
4. Mitochondrial Modulation through miRNAs
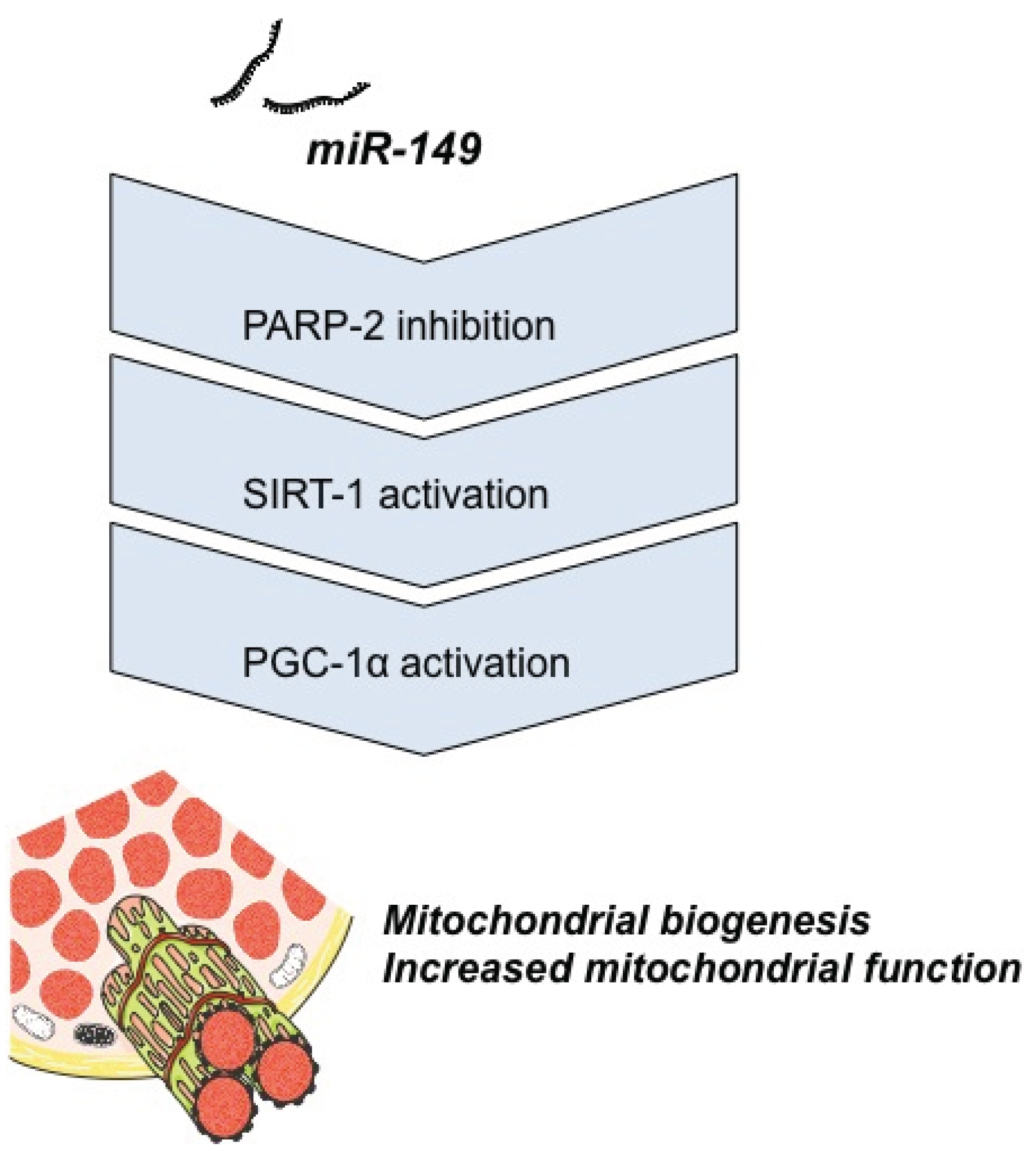
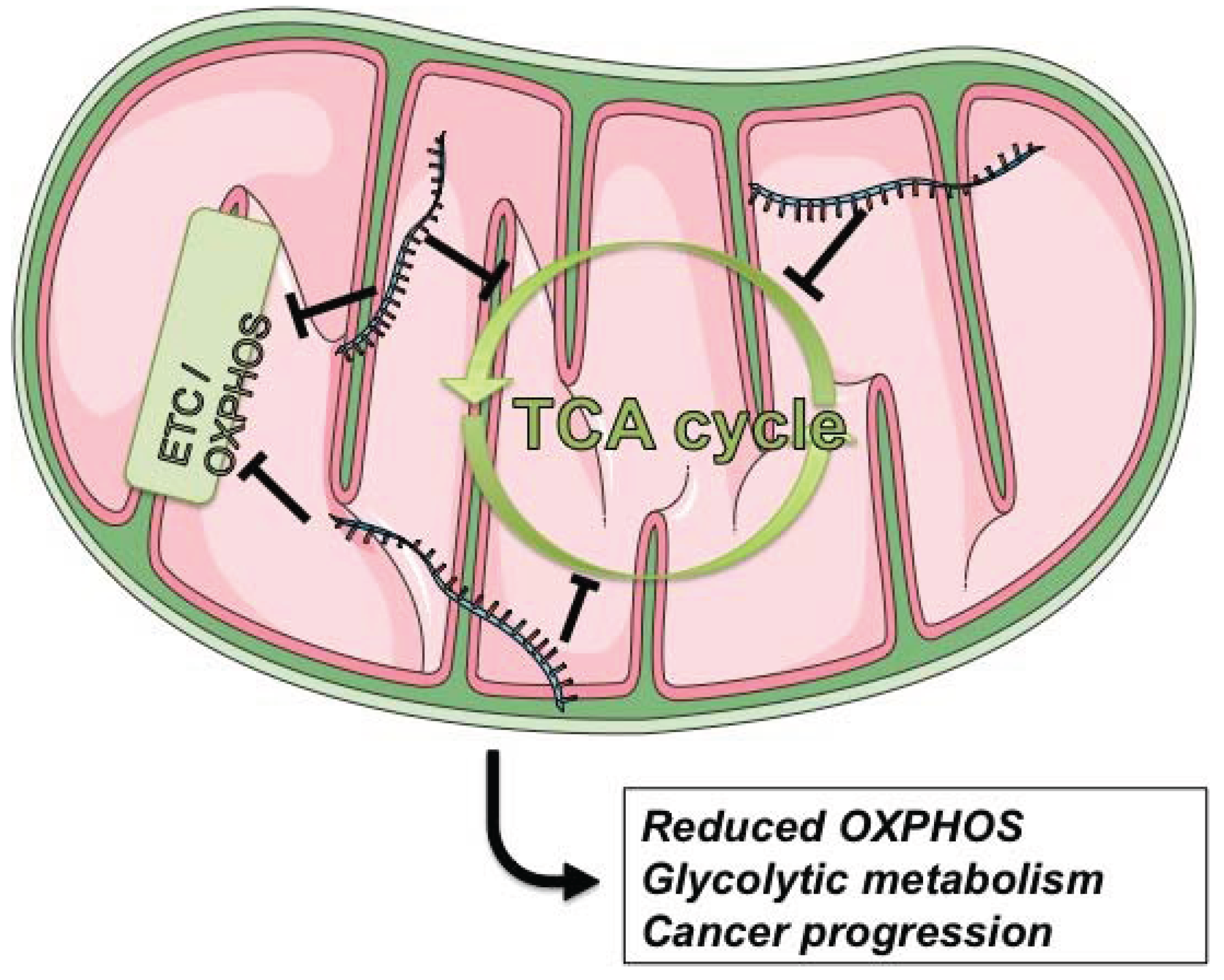
4.1. Energy Metabolism


{kind=link}
{kind=link}
{kind=link}
| miRNA | Target | Model | Reference |
|---|---|---|---|
| miR-181c | mt-COX1 | Rat, cardiomyocytes | [25] |
| miR-338 | COX IV | Rat, neurons | [50] |
| miR-210 | ISCU1/2; COX 10 | Human, placenta and fibroblasts | [54] |
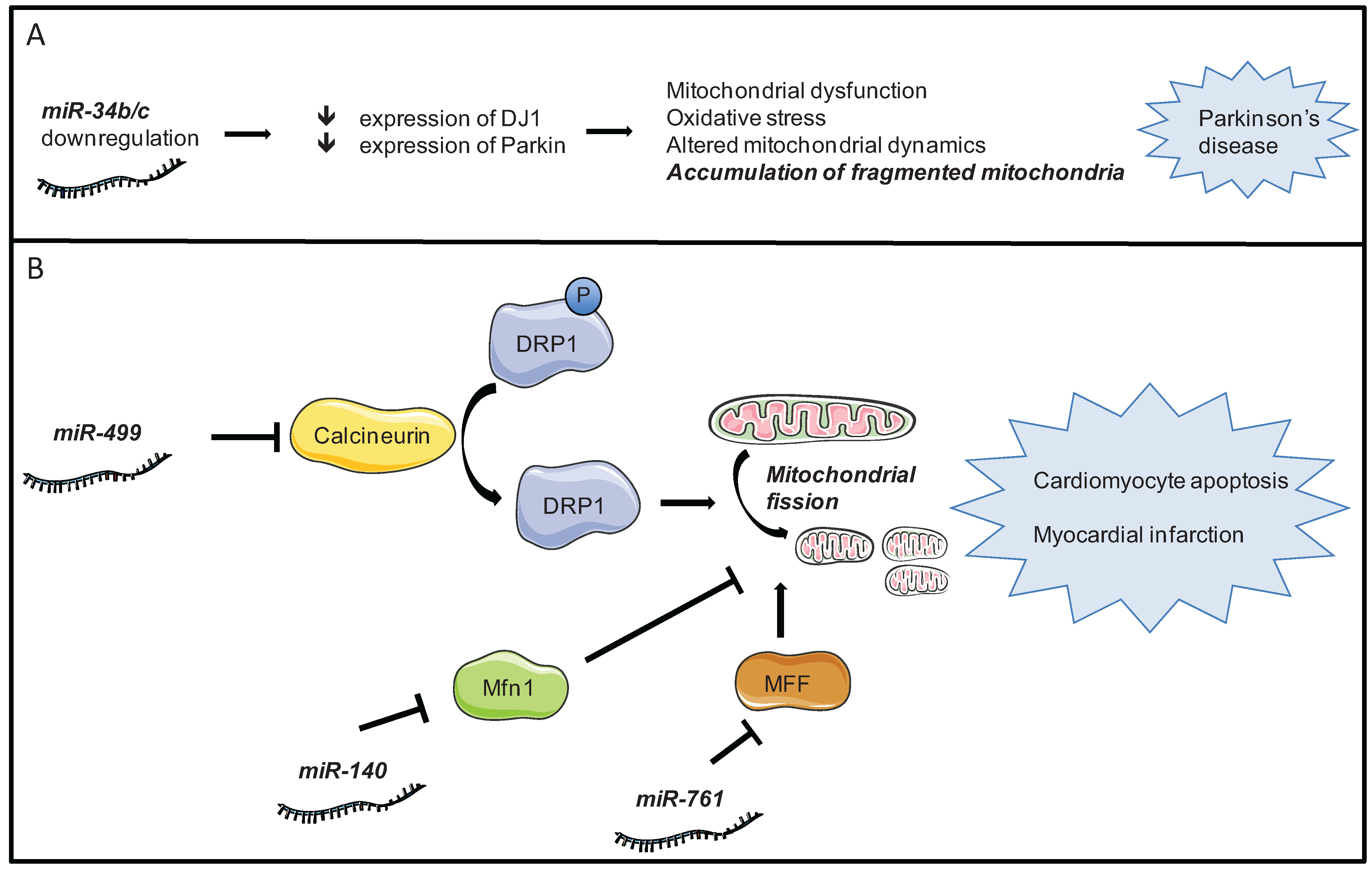
4.2. Mitochondrial Dynamics

| miRNA | Target | Effect on autophagy | Reference |
|---|---|---|---|
| miR-101 | STMN1, RAB5A and ATG4D | Inhibitor | [70] |
| miR-204 | LC3 | Inhibitor | [71] |
| miR-30a | Beclin 1 | Inhibitor | [72,76] |
| miR-137 | NIX, FUNDC1 | Inhibitor (mitophagy) | [77] |
4.3. Cancer

5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.S.; Ali, S.; Ahmad, A.; Bao, B.; Philip, P.A.; Sarkar, F.H. Expression of microRNAs: Potential molecular link between obesity, diabetes and cancer. Obes. Rev. 2011, 12, 1050–1062. [Google Scholar] [CrossRef] [PubMed]
- Tétreault, N.; de Guire, V. miRNAs: Their discovery, biogenesis and mechanism of action. Clin. Biochem. 2013, 46, 842–845. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, J.D.; Chevillet, J.R.; Kroh, E.M.; Ruf, I.K.; Pritchard, C.C.; Gibson, D.F.; Mitchell, P.S.; Bennett, C.F.; Pogosova-Agadjanyan, E.L.; Stirewalt, D.L.; et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc. Natl. Acad. Sci. USA 2011, 108, 5003–5008. [Google Scholar]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Kushnareva, Y.; Newmeyer, D.D. Bioenergetics and cell death. Ann. N. Y. Acad. Sci. 2010, 1201, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Michel, S.; Wanet, A.; de Pauw, A.; Rommelaere, G.; Arnould, T.; Renard, P. Crosstalk between mitochondrial (dys)function and mitochondrial abundance. J. Cell. Physiol. 2012, 227, 2297–2310. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S. A history of mitochondrial diseases. J. Inherit. Metab. Dis. 2011, 34, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Zorzano, A.; Hernández-Alvarez, M.I.; Palacín, M.; Mingrone, G. Alterations in the mitochondrial regulatory pathways constituted by the nuclear co-factors PGC-1alpha or PGC-1beta and mitofusin 2 in skeletal muscle in type 2 diabetes. Biochim. Biophys. Acta 2010, 1797, 1028–1033. [Google Scholar] [CrossRef] [PubMed]
- Peralta, S.; Wang, X.; Moraes, C.T. Mitochondrial transcription: Lessons from mouse models. BBA-Gene Regul. Mech. 2012, 1819, 961–969. [Google Scholar]
- Krishnan, K.J.; Greaves, L.C.; Reeve, A.K.; Turnbull, D. The ageing mitochondrial genome. Nucleic Acids Res. 2007, 35, 7399–7405. [Google Scholar] [CrossRef] [PubMed]
- Larsson, N.-G. Somatic mitochondrial DNA mutations in mammalian aging. Annu. Rev. Biochem. 2010, 79, 683–706. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-T.; Wu, S.-B.; Lee, W.-Y.; Wei, Y.-H. Mitochondrial respiratory dysfunction-elicited oxidative stress and posttranslational protein modification in mitochondrial diseases. Ann. N. Y. Acad. Sci. 2010, 1201, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Rolo, A.P.; Palmeira, C.M. Diabetes and mitochondrial function: Role of hyperglycemia and oxidative stress. Toxicol. Appl. Pharmacol. 2006, 212, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, C.M.; Rolo, A.P.; Berthiaume, J.; Bjork, J.A.; Wallace, K.B. Hyperglycemia decreases mitochondrial function: the regulatory role of mitochondrial biogenesis. Toxicol. Appl. Pharmacol. 2007, 225, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Rolo, A.P.; Gomes, A.P.; Palmeira, C.M. Regulation of mitochondrial biogenesis in metabolic syndrome. Curr. Drug Targets 2011, 12, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.P.; Duarte, F.V.; Nunes, P.; Hubbard, B.P.; Teodoro, J.S.; Varela, A.T.; Jones, J.G.; Sinclair, D.A.; Palmeira, C.M.; Rolo, A.P. Berberine protects against high fat diet-induced dysfunction in muscle mitochondria by inducing SIRT1-dependent mitochondrial biogenesis. Biochim. Biophys. Acta 2012, 1822, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Perier, C.; Vila, M. Mitochondrial biology and Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Physiological functions of mitochondrial fusion. Ann. N. Y. Acad. Sci. 2010, 1201, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev.Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Bandiera, S.; Matégot, R.; Girard, M.; Demongeot, J.; Henrion-Caude, A. MitomiRs delineating the intracellular localization of microRNAs at mitochondria. Free Radic. Biol. Med. 2013, 64, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Sripada, L.; Tomar, D.; Singh, R. Mitochondria: One of the destinations of miRNAs. Mitochondrion 2012, 12, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Bienertova-Vasku, J.; Sana, J.; Slaby, O. The role of microRNAs in mitochondria in cancer. Cancer Lett. 2013, 336, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Ferlito, M.; Kent, O.A.; Fox-Talbot, K.; Wang, R.; Liu, D.; Raghavachari, N.; Yang, Y.; Wheelan, S.J.; Murphy, E.; et al. Nuclear miRNA regulates the mitochondrial genome in the heart. Circ. Res. 2012, 110, 1596–1603. [Google Scholar] [CrossRef] [PubMed]
- Kren, B.T.; Wong, P.Y.P.; Sarver, A.; Zhang, X.; Zeng, Y.; Steer, C.J. MicroRNAs identified in highly purified liver-derived mitochondria may play a role in apoptosis. RNA Biol. 2009, 6, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Latronico, M.V.G.; Condorelli, G. The might of microRNA in mitochondria. Circ. Res. 2012, 110, 1540–1542. [Google Scholar] [CrossRef] [PubMed]
- Bandiera, S.; Rüberg, S.; Girard, M.; Cagnard, N.; Hanein, S.; Chrétien, D.; Munnich, A.; Lyonnet, S.; Henrion-Caude, A. Nuclear outsourcing of RNA interference components to human mitochondria. PLoS One 2011, 6, e20746. [Google Scholar] [CrossRef] [PubMed]
- Barrey, E.; Saint-Auret, G.; Bonnamy, B.; Damas, D.; Boyer, O.; Gidrol, X. Pre-microRNA and mature microRNA in human mitochondria. PLoS One 2011, 6, e20220. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Lowe, S.W. The microcosmos of cancer. Nature 2012, 482, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Guay, C.; Regazzi, R. Circulating microRNAs as novel biomarkers for diabetes mellitus. Nat. Rev. Endocrinol. 2013, 9, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Raitoharju, E.; Oksala, N.; Lehtimäki, T. MicroRNAs in the atherosclerotic plaque. Clin. Chem. 2013, 59, 1708–1721. [Google Scholar] [CrossRef] [PubMed]
- Rayner, K.J.; Esau, C.C.; Hussain, F.N.; McDaniel, A.L.; Marshall, S.M.; van Gils, J.M.; Ray, T.D.; Sheedy, F.J.; Goedeke, L.; Liu, X.; et al. Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature 2011, 478, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Zhao, X.; Xu, K.; Li, Q.; Cheng, J.; Gao, Y.; Du, J.; Shi, H.; Zhou, L. Polymorphisms in lipid metabolism related miRNA binding sites and risk of metabolic syndrome. Gene 2013, 528, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Liu, C.; Ye, Z. Deep sequencing of small RNA repertoires in mice reveals metabolic disorders-associated hepatic miRNAs. PLoS One 2013, 8, e80774. [Google Scholar] [CrossRef] [PubMed]
- Civelek, M.; Hagopian, R.; Pan, C.; Che, N.; Yang, W.-P.; Kayne, P.S.; Saleem, N.K.; Cederberg, H.; Kuusisto, J.; Gargalovic, P.S.; et al. Genetic regulation of human adipose microRNA expression and its consequences for metabolic traits. Hum. Mol. Genet. 2013, 22, 3023–3037. [Google Scholar] [CrossRef] [PubMed]
- Rottiers, V.; Näär, A.M. MicroRNAs in metabolism and metabolic disorders. Nat. Rev. Mol. Cell Biol. 2012, 13, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Koch, L. Paediatrics: Circulating microRNAs-predictors of obesity? Nat. Rev. Endocrinol. 2013, 9, 565. [Google Scholar] [CrossRef]
- Pescador, N.; Pérez-Barba, M.; Ibarra, J.M.; Corbatón, A.; Martínez-Larrad, M.T.; Serrano-Ríos, M. Serum circulating microRNA profiling for identification of potential type 2 diabetes and obesity biomarkers. PLoS One 2013, 8, e77251. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, J.S.; Hajira, A.; Pardo, P.S.; Boriek, A.M. MicroRNA-149 inhibits PARP-2 and promotes mitochondrial biogenesis via SIRT-1/PGC-1α network in skeletal muscle. Diabetes 2014, 63, 1546–1559. [Google Scholar] [CrossRef] [PubMed]
- Nezami, B.G.; Mwangi, S.M.; Lee, J.E.; Jeppsson, S.; Anitha, M.; Yarandi, S.S.; Farris, A.B.; Srinivasan, S. MicroRNA 375 mediates palmitate-induced enteric neuronal damage and high-fat diet-induced delayed intestinal transit in mice. Gastroenterology 2014, 146, 473–483.e3. [Google Scholar]
- Stranahan, A.M.; Cutler, R.G.; Button, C.; Telljohann, R.; Mattson, M.P. Diet-induced elevations in serum cholesterol are associated with alterations in hippocampal lipid metabolism and increased oxidative stress. J. Neurochem. 2011, 118, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Schrauwen, P.; Schrauwen-Hinderling, V.; Hoeks, J.; Hesselink, M.K.C. Mitochondrial dysfunction and lipotoxicity. Biochim. Biophys. Acta 2010, 1801, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Wegner, M.; Neddermann, D.; Piorunska-Stolzmann, M.; Jagodzinski, P.P. Role of epigenetic mechanisms in the development of chronic complications of diabetes. Diabetes Res. Clin. Pract. 2014, 105, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.L.; Distefano, J.K. The role of non-coding RNAs in diabetic nephropathy: Potential applications as biomarkers for disease development and progression. Diabetes Res. Clin. Pract. 2013, 99, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zhang, Y.; Wang, Z.; Wang, L.; Wei, X.; Zhang, B.; Wen, Z.; Fang, H.; Pang, Q.; Yi, F. Regulation of NADPH oxidase activity is associated with miRNA-25-mediated NOX4 expression in experimental diabetic nephropathy. Am. J. Nephrol. 2010, 32, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Peng, H.; Chen, J.; Chen, X.; Han, F.; Xu, X.; He, X.; Yan, N. MicroRNA-21 protects from mesangial cell proliferation induced by diabetic nephropathy in db/db mice. FEBS Lett. 2009, 583, 2009–2014. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, M.; Neuzil, J.; Dong, L. MicroRNAs as regulators of mitochondrial function: Role in cancer suppression. Biochim. Biophys. Acta 2014, 1840, 1441–1453. [Google Scholar] [CrossRef]
- Nishi, H.; Ono, K.; Iwanaga, Y.; Horie, T.; Nagao, K.; Takemura, G.; Kinoshita, M.; Kuwabara, Y.; Mori, R.T.; Hasegawa, K.; et al. MicroRNA-15b modulates cellular ATP levels and degenerates mitochondria via Arl2 in neonatal rat cardiac myocytes. J. Biol. Chem. 2010, 285, 4920–4930. [Google Scholar] [CrossRef] [PubMed]
- Aschrafi, A.; Schwechter, A.D.; Mameza, M.G.; Natera-Naranjo, O.; Gioio, A.E.; Kaplan, B.B. MicroRNA-338 regulates local cytochrome c oxidase IV mRNA levels and oxidative phosphorylation in the axons of sympathetic neurons. J. Neurosci. 2008, 28, 12581–12590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Bedja, D.; Campbell, N.; Dunkerly, B.; Chenna, V.; Maitra, A.; Steenbergen, C. miR-181c regulates the mitochondrial genome, bioenergetics, and propensity for heart failure in vivo. PLoS One 2014, 9, e96820. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Huang, H.; Fan, Y.; Kong, B.; Hu, H.; Hu, K.; Guo, J.; Mei, Y.; Liu, W.-L. Effects of downregulation of microRNA-181a on H2O2-induced H9c2 cell apoptosis via the mitochondrial apoptotic pathway. Oxid. Med. Cell Longev. 2014, 2014, 960362. [Google Scholar] [PubMed]
- Colleoni, F.; Padmanabhan, N.; Yung, H.-W.; Watson, E.D.; Cetin, I.; Tissot van Patot, M.C.; Burton, G.J.; Murray, A.J. Suppression of mitochondrial electron transport chain function in the hypoxic human placenta: A role for miRNA-210 and protein synthesis inhibition. PLoS One 2013, 8, e55194. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J. Oxygen, the Janus gas; its effects on human placental development and function. J. Anat. 2009, 215, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Karbiener, M.; Pisani, D.F.; Frontini, A.; Oberreiter, L.M.; Lang, E.; Vegiopoulos, A.; Mössenböck, K.; Bernhardt, G.A.; Mayr, T.; Hildner, F.; et al. MicroRNA-26 family is required for human adipogenesis and drives characteristics of brown adipocytes. Stem Cells 2014, 32, 1578–1590. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.; Lu, W.; Xu, W.; Anderson, L.; Bacanamwo, M.; Thompson, W.; Chen, Y.E.; Liu, D. MicroRNA-27 (miR-27) targets prohibitin and impairs adipocyte differentiation and mitochondrial function in human adipose-derived stem cells. J. Biol. Chem. 2013, 288, 34394–34402. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef] [PubMed]
- Long, B.; Wang, K.; Li, N.; Murtaza, I.; Xiao, J.-Y.; Fan, Y.-Y.; Liu, C.-Y.; Li, W.-H.; Cheng, Z.; Li, P. miR-761 regulates the mitochondrial network by targeting mitochondrial fission factor. Free Radic. Biol. Med. 2013, 65C, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Donath, S.; Li, Y.; Qin, D.; Prabhakar, B.S.; Li, P. miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet. 2010, 6, e1000795. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Long, B.; Jiao, J.-Q.; Wang, J.-X.; Liu, J.-P.; Li, Q.; Li, P.-F. miR-484 regulates mitochondrial network through targeting Fis1. Nat. Commun. 2012, 3, 781. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.; Galloway, C.A.; Jhun, B.S.; Yu, T. Mitochondrial dynamics in diabetes. Antioxid. Redox. Signal. 2011, 14, 439–457. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, Y.; Jiao, J.; Wang, J.; Li, Y.; Qin, D.; Li, P. Mitofusin 1 is negatively regulated by microRNA 140 in cardiomyocyte apoptosis. Mol. Cell. Biol. 2014, 34, 1788–1799. [Google Scholar] [CrossRef] [PubMed]
- Van Empel, V.P.M.; Bertrand, A.T.; van der Nagel, R.; Kostin, S.; Doevendans, P.A.; Crijns, H.J.; de Wit, E.; Sluiter, W.; Ackerman, S.L.; de Windt, L.J. Downregulation of apoptosis-inducing factor in harlequin mutant mice sensitizes the myocardium to oxidative stress-related cell death and pressure overload-induced decompensation. Circ. Res. 2005, 96, e92–e101. [Google Scholar] [CrossRef] [PubMed]
- Webster, K.A.; Discher, D.J.; Kaiser, S.; Hernandez, O.; Sato, B.; Bishopric, N.H. Hypoxia-activated apoptosis of cardiac myocytes requires reoxygenation or a pH shift and is independent of p53. J. Clin. Invest. 1999, 104, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Kitsis, R.N.; Mann, D.L. Apoptosis and the heart: A decade of progress. J. Mol. Cell. Cardiol. 2005, 38, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Barsoum, M.J.; Yuan, H.; Gerencser, A.A.; Liot, G.; Kushnareva, Y.; Gräber, S.; Kovacs, I.; Lee, W.D.; Waggoner, J.; Cui, J.; et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006, 25, 3900–3911. [Google Scholar] [CrossRef] [PubMed]
- Kundu, M.; Lindsten, T.; Yang, C.-Y.; Wu, J.; Zhao, F.; Zhang, J.; Selak, M.A.; Ney, P.A.; Thompson, C.B. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 2008, 112, 1493–1502. [Google Scholar] [CrossRef] [PubMed]
- Shutt, T.E.; McBride, H.M. Staying cool in difficult times: mitochondrial dynamics, quality control and the stress response. Biochim. Biophys. Acta 2013, 1833, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Frankel, L.B.; Wen, J.; Lees, M.; Høyer-Hansen, M.; Farkas, T.; Krogh, A.; Jäättelä, M.; Lund, A.H. microRNA-101 is a potent inhibitor of autophagy. EMBO J. 2011, 30, 4628–4641. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Zhu, X.; He, B.; Zhang, Y.; Kang, B.; Wang, Z.; Ni, X. MiR-204 regulates cardiomyocyte autophagy induced by ischemia-reperfusion through LC3-II. J. Biomed. Sci. 2011, 18, 35. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wu, H.; Liu, X.; Li, B.; Chen, Y.; Ren, X.; Liu, C.-G.; Yang, J.-M. Regulation of autophagy by a beclin 1-targeted microRNA, miR-30a, in cancer cells. Autophagy 2009, 5, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Frankel, L.B.; Lund, A.H. MicroRNA regulation of autophagy. Carcinogenesis 2012, 33, 2018–2025. [Google Scholar] [CrossRef] [PubMed]
- Minones-Moyano, E.; Porta, S.; Escaramis, G.; Rabionet, R.; Iraola, S.; Kagerbauer, B.; Espinosa-Parrilla, Y.; Ferrer, I.; Estivill, X.; Marti, E. MicroRNA profiling of Parkinson’s disease brains identifies early downregulation of miR-34b/c which modulate mitochondrial function. Hum. Mol. Genet. 2011, 20, 3067–3078. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Liang, J.; Li, Y.; Li, J.; Yang, X.; Zhang, X.; Han, S.; Li, S.; Li, J. Down-regulation of miRNA-30a alleviates cerebral ischemic injury through enhancing beclin 1-mediated autophagy. Neurochem. Res. 2014, 39, 1279–1291. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, X.; Zhuang, H.; Chen, H.-G.; Chen, Y.; Tian, W.; Wu, W.; Li, Y.; Wang, S.; Zhang, L.; et al. MicroRNA-137 is a novel hypoxia-responsive microRNA that inhibits mitophagy via regulation of two mitophagy receptors FUNDC1 and NIX. J. Biol. Chem. 2014, 289, 10691–10701. [Google Scholar] [CrossRef] [PubMed]
- Coskun, P.; Wyrembak, J.; Schriner, S.E.; Chen, H.-W.; Marciniack, C.; Laferla, F.; Wallace, D.C. A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim. Biophys. Acta 2012, 1820, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Gelino, S.; Hansen, M. Autophagy—An emerging anti-aging mechanism. J Clin Exp Pathol. 2012, Suppl 4, 006. [Google Scholar]
- Bender, C.E.; Fitzgerald, P.; Tait, S.W.G.; Llambi, F.; McStay, G.P.; Tupper, D.O.; Pellettieri, J.; Sánchez Alvarado, A.; Salvesen, G.S.; Green, D.R. Mitochondrial pathway of apoptosis is ancestral in metazoans. Proc. Natl. Acad. Sci. USA 2012, 109, 4904–4909. [Google Scholar] [CrossRef] [PubMed]
- Rippo, M.R.; Olivieri, F.; Monsurrò, V.; Prattichizzo, F.; Albertini, M.C.; Procopio, A.D. MitomiRs in human inflamm-aging: A hypothesis involving miR-181a, miR-34a and miR-146a. Exp. Gerontol. 2014, 56, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [PubMed]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [PubMed]
- Fogg, V.C.; Lanning, N.J.; Mackeigan, J.P. Mitochondria in cancer: At the crossroads of life and death. Chin. J. Cancer 2011, 30, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Krell, J.; Frampton, A.E.; Stebbing, J. MicroRNAs in the cancer clinic. Front. Biosci. (Elite Ed.) 2013, 5, 204–213. [Google Scholar]
- Radojicic, J.; Zaravinos, A.; Vrekoussis, T.; Kafousi, M.; Spandidos, D.A.; Stathopoulos, E.N. MicroRNA expression analysis in triple-negative (ER, PR and Her2/neu) breast cancer. Cell Cycle 2011, 10, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Zhou, E.; Wang, Y.; Xu, F.; Zhang, D.; Zhong, D. microRNA-200a inhibits cell proliferation by targeting mitochondrial transcription factor A in breast cancer. DNA Cell Biol. 2014, 33, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, F.; Yuan, J.-H.; Yuan, S.-X.; Zhou, W.-P.; Huo, X.-S.; Xu, D.; Bi, H.-S.; Wang, F.; Sun, S.-H. Epigenetic activation of the MiR-200 family contributes to H19-mediated metastasis suppression in hepatocellular carcinoma. Carcinogenesis 2013, 34, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Roybal, J.D.; Zang, Y.; Ahn, Y.-H.; Yang, Y.; Gibbons, D.L.; Baird, B.N.; Alvarez, C.; Thilaganathan, N.; Liu, D.D.; Saintigny, P.; et al. miR-200 inhibits lung adenocarcinoma cell invasion and metastasis by targeting Flt1/VEGFR1. Mol. Cancer Res. 2011, 9, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, S.A.K.; Teo, C.R.; Beillard, E.J.; Voorhoeve, P.M.; Casey, P.J. MicroRNA-182 and microRNA-200a control G-protein subunit α-13 (GNA13) expression and cell invasion synergistically in prostate cancer cells. J. Biol. Chem. 2013, 288, 7986–7995. [Google Scholar] [CrossRef] [PubMed]
- Bonawitz, N.D.; Clayton, D.A.; Shadel, G.S. Initiation and beyond: Multiple functions of the human mitochondrial transcription machinery. Mol. Cell 2006, 24, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-C.; Wei, Y.-H. Mitochondrial DNA instability and metabolic shift in human cancers. Int. J. Mol. Sci. 2009, 10, 674–701. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Izumi, H.; Yasuniwa, Y.; Akiyama, M.; Yamaguchi, T.; Fujimoto, N.; Matsumoto, T.; Wu, B.; Tanimoto, A.; Sasaguri, Y.; et al. Human mitochondrial transcription factor A functions in both nuclei and mitochondria and regulates cancer cell growth. Biochem. Biophys. Res. Commun. 2011, 408, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Morino, K.; Nishio, Y.; Ugi, S.; Yoshizaki, T.; Kashiwagi, A.; Maegawa, H. MicroRNA-494 regulates mitochondrial biogenesis in skeletal muscle through mitochondrial transcription factor A and Forkhead box j3. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E1419–E1427. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Mazumdar, M.; Mukherjee, S.; Bhattacharjee, P.; Adhikary, A.; Manna, A.; Chakraborty, S.; Khan, P.; Sen, A.; Das, T. Restoration of p53/miR-34a regulatory axis decreases survival advantage and ensures Bax-dependent apoptosis of non-small cell lung carcinoma cells. FEBS Lett. 2014, 588, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Le Bras, M.; Rouy, I.; Brenner, C. The modulation of inter-organelle cross-talk to control apoptosis. Med. Chem. 2006, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Liu, X.; Xu, X.; Fan, X.; Liu, M.; Li, X.; Zhong, Q.; Tang, H. MicroRNA-373, a new regulator of protein phosphatase 6, functions as an oncogene in hepatocellular carcinoma. FEBS J. 2011, 278, 2044–2054. [Google Scholar] [CrossRef] [PubMed]
- Annibali, D.; Gioia, U.; Savino, M.; Laneve, P.; Caffarelli, E.; Nasi, S. A new module in neural differentiation control: Two microRNAs upregulated by retinoic acid, miR-9 and -103, target the differentiation inhibitor ID2. PLoS One 2012, 7, e40269. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.A.; Krichevsky, A.M.; Kosik, K.S. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005, 65, 6029–6033. [Google Scholar] [CrossRef] [PubMed]
- He, L.; He, X.; Lim, L.P.; de Stanchina, E.; Xuan, Z.; Liang, Y.; Xue, W.; Zender, L.; Magnus, J.; Ridzon, D.; et al. microRNA component of the p53 tumour suppressor network. Nature 2007, 447, 1130–1134. [Google Scholar] [CrossRef] [PubMed]
- Yamakuchi, M.; Lowenstein, C.J. MiR-34, SIRT1 and p53: The feedback loop. Cell Cycle 2009, 8, 712–715. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-G.; Wang, J.-J.; Zhao, F.; Liu, Q.; Jiang, K.; Yang, G.-H. MicroRNA-21 (miR-21) represses tumor suppressor PTEN and promotes growth and invasion in non-small cell lung cancer (NSCLC). Clin. Chim. Acta 2010, 411, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wu, H.; Ling, T. Suppressive effect of microRNA-126 on oral squamous cell carcinoma in vitro. Mol. Med. Rep. 2014, 10, 125–130. [Google Scholar] [PubMed]
- Jia, A.Y.; Castillo-Martin, M.; Bonal, D.M.; Sánchez-Carbayo, M.; Silva, J.M.; Cordon-Cardo, C. MicroRNA-126 inhibits invasion in bladder cancer via regulation of ADAM9. Br. J. Cancer 2014, 110, 2945–2954. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Jung, S.B.; Kim, J.-S.; Roh, M.S.; Lee, J.H.; Lee, E.H.; Lee, H.W. Expression of microRNA miR-126 and miR-200c is associated with prognosis in patients with non-small cell lung cancer. Virchows Arch. 2014. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, Y.; Feng, X.; An, P.; Quan, X.; Wang, H.; Ye, S.; Yu, C.; He, Y.; Luo, H. MicroRNA-126 functions as a tumor suppressor in colorectal cancer cells by targeting CXCR4 via the AKT and ERK1/2 signaling pathways. Int. J. Oncol. 2014, 44, 203–210. [Google Scholar] [PubMed]
- Tomasetti, M.; Nocchi, L.; Staffolani, S.; Manzella, N.; Amati, M.; Goodwin, J.; Kluckova, K.; Nguyen, M.; Strafella, E.; Bajzikova, M.; et al. MicroRNA-126 suppresses mesothelioma malignancy by targeting IRS1 and interfering with the mitochondrial function. Antioxid. Redox. Signal. 2014. [Google Scholar] [CrossRef]
- Zhang, J.; Du, Y.-Y.; Lin, Y.-F.; Chen, Y.-T.; Yang, L.; Wang, H.-J.; Ma, D. The cell growth suppressor, mir-126, targets IRS-1. Biochem. Biophys. Res. Commun. 2008, 377, 136–140. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duarte, F.V.; Palmeira, C.M.; Rolo, A.P. The Role of microRNAs in Mitochondria: Small Players Acting Wide. Genes 2014, 5, 865-886. https://doi.org/10.3390/genes5040865
Duarte FV, Palmeira CM, Rolo AP. The Role of microRNAs in Mitochondria: Small Players Acting Wide. Genes. 2014; 5(4):865-886. https://doi.org/10.3390/genes5040865
Chicago/Turabian StyleDuarte, Filipe V., Carlos M. Palmeira, and Anabela P. Rolo. 2014. "The Role of microRNAs in Mitochondria: Small Players Acting Wide" Genes 5, no. 4: 865-886. https://doi.org/10.3390/genes5040865
APA StyleDuarte, F. V., Palmeira, C. M., & Rolo, A. P. (2014). The Role of microRNAs in Mitochondria: Small Players Acting Wide. Genes, 5(4), 865-886. https://doi.org/10.3390/genes5040865