Non-Additive Transcriptomic Responses to Inoculation with Rhizobia in a Young Allopolyploid Compared with Its Diploid Progenitors


Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. RNA Sequencing
2.3. RNA-Seq Data Processing
2.4. Homoeologue Mapping
2.5. Expression-Level Analyses
2.6. Weighted Gene Co-Expression Network Analysis (WGCNA)
3. Results and Discussion
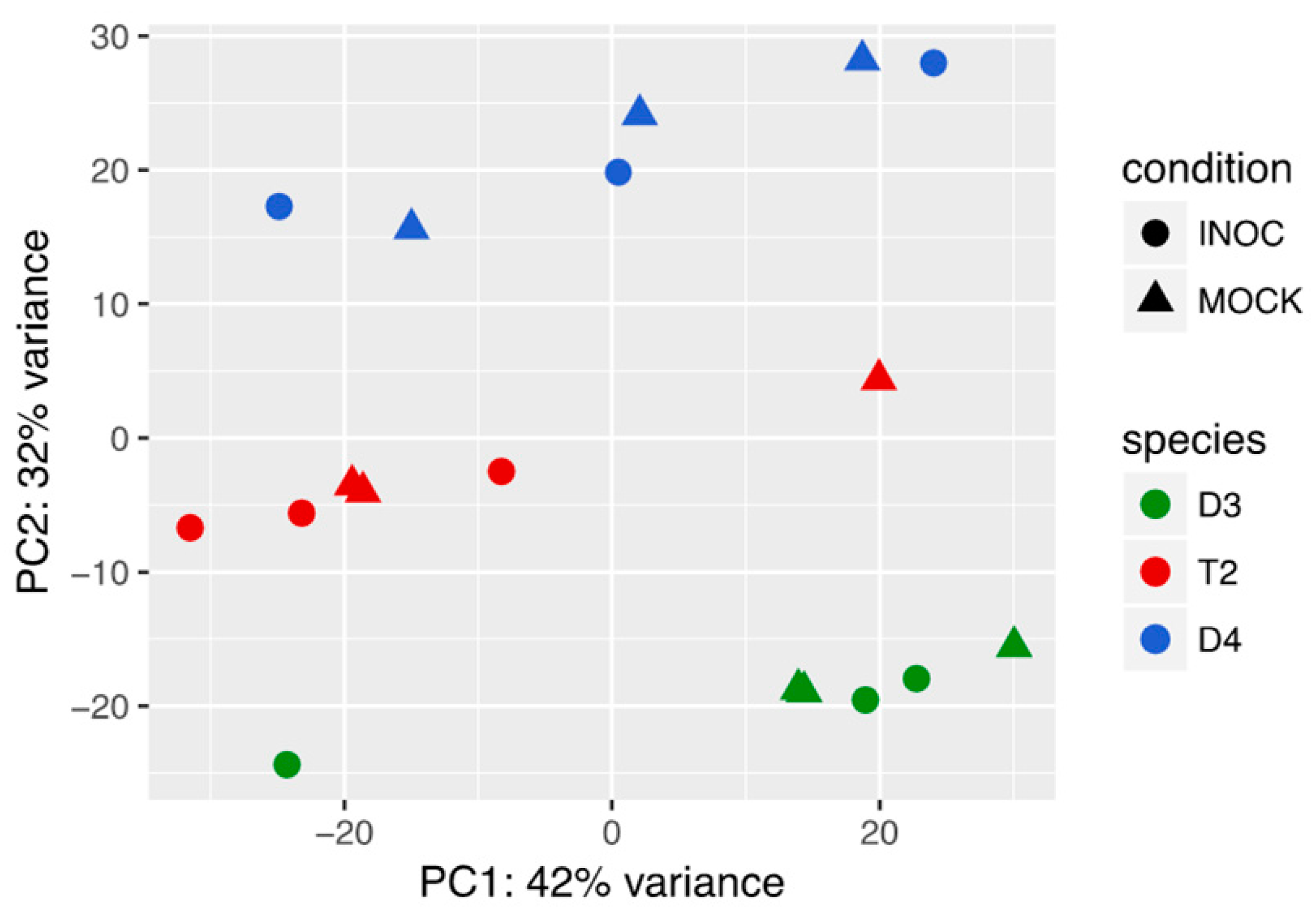
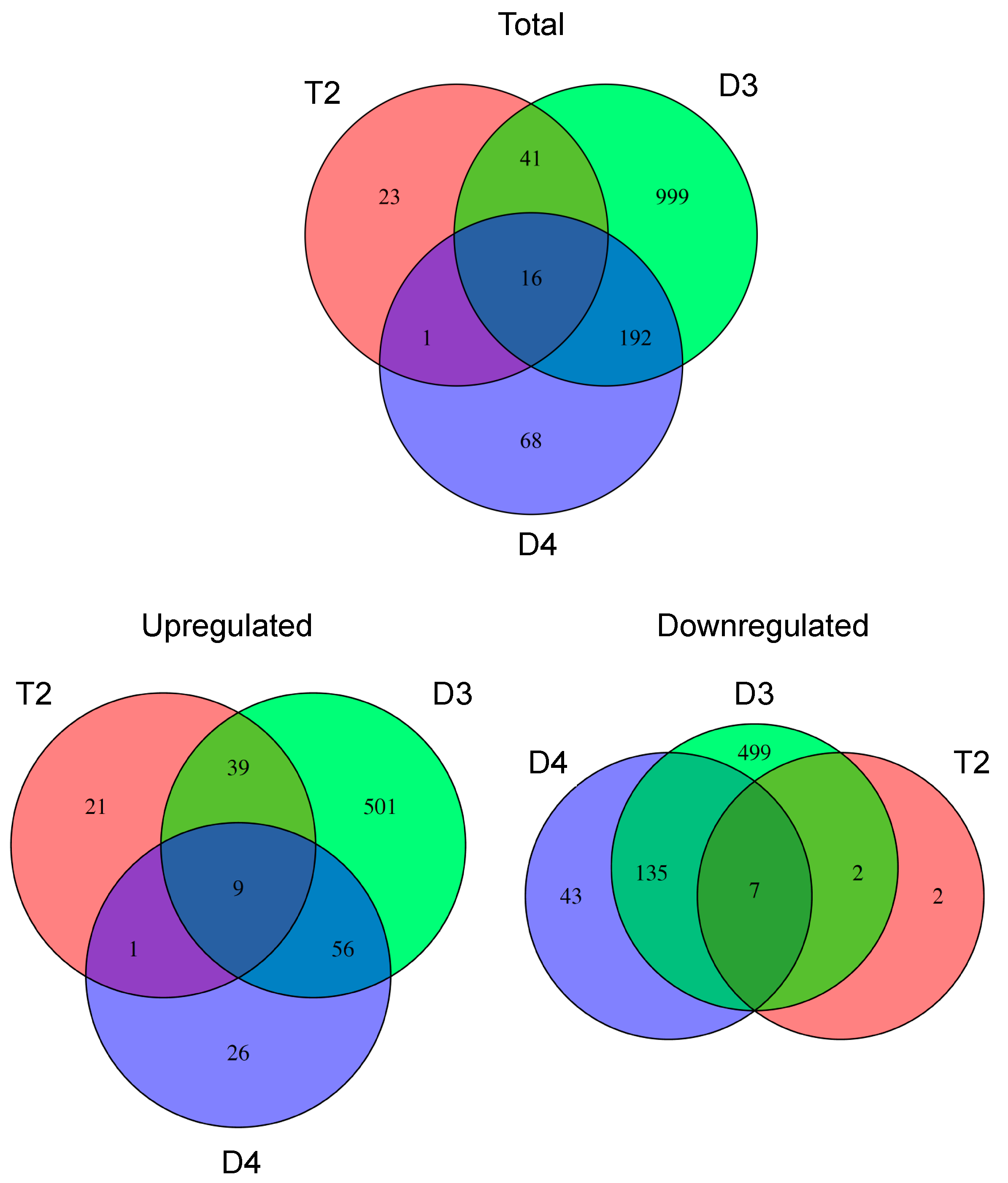
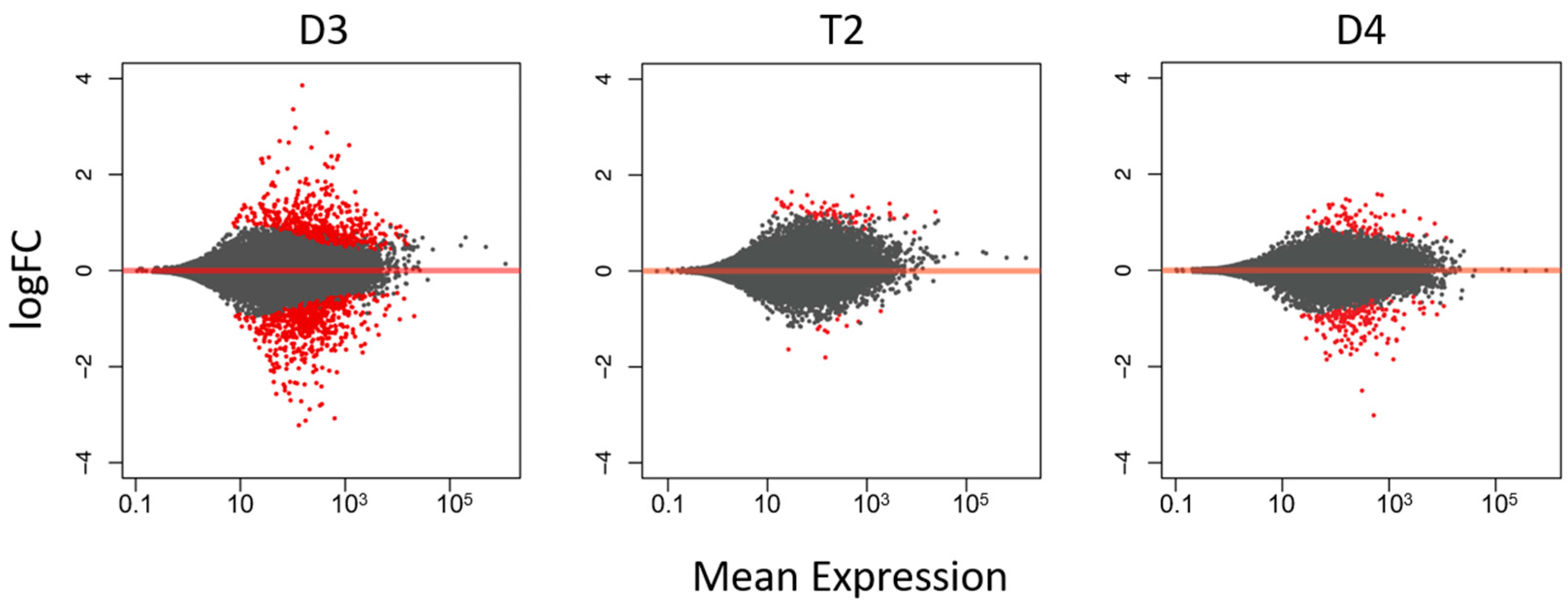
3.1. T2 Exhibited Overall Expression-Level Intermediacy, but Had Fewer Differentially Expressed Genes Compared to Diploids
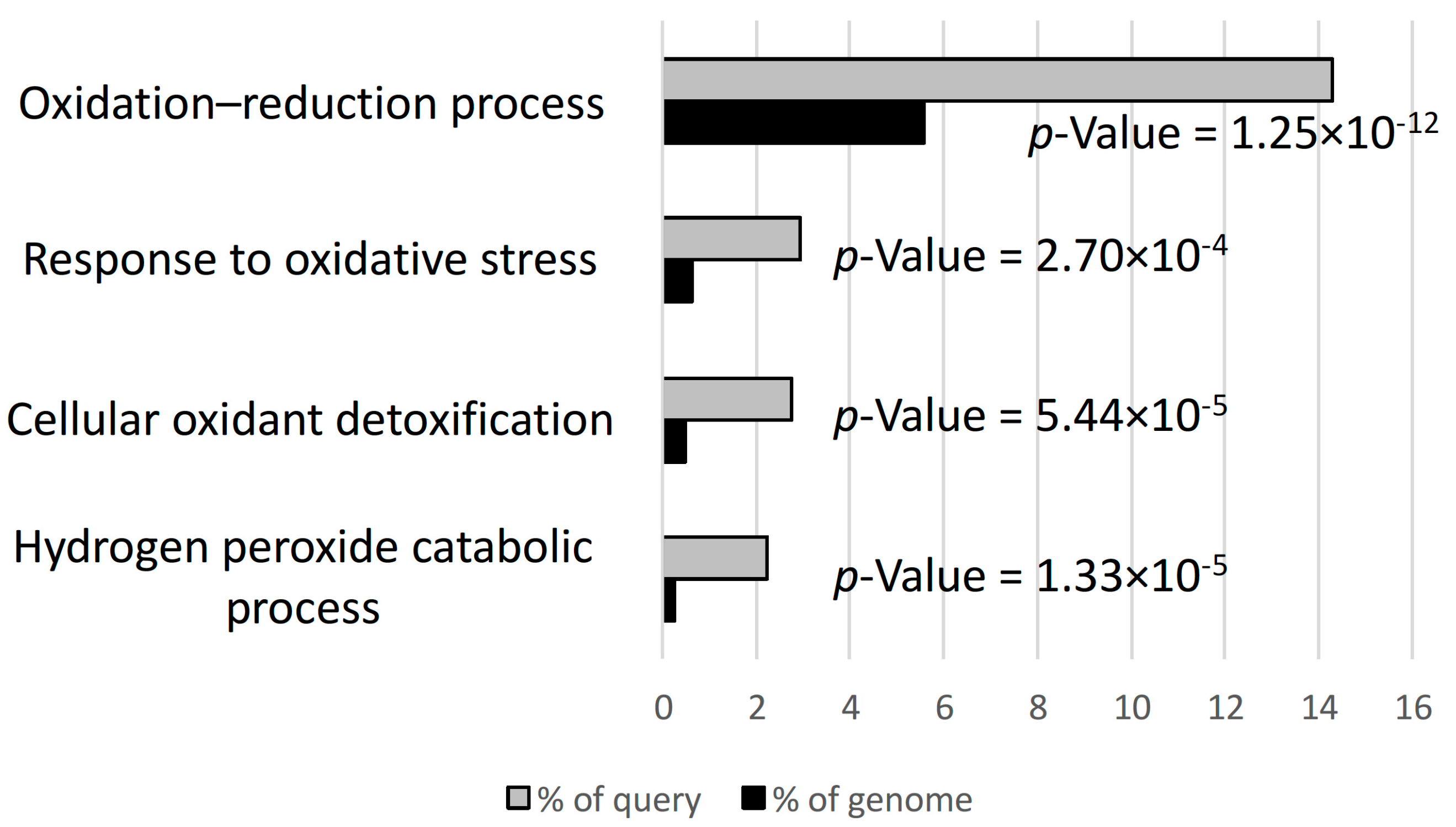
3.2. Hydrogen Peroxide and Oxidative Stress Responses were Differentially Regulated in D3
3.3. Implications of Expression-Level Dominance for Nodulation
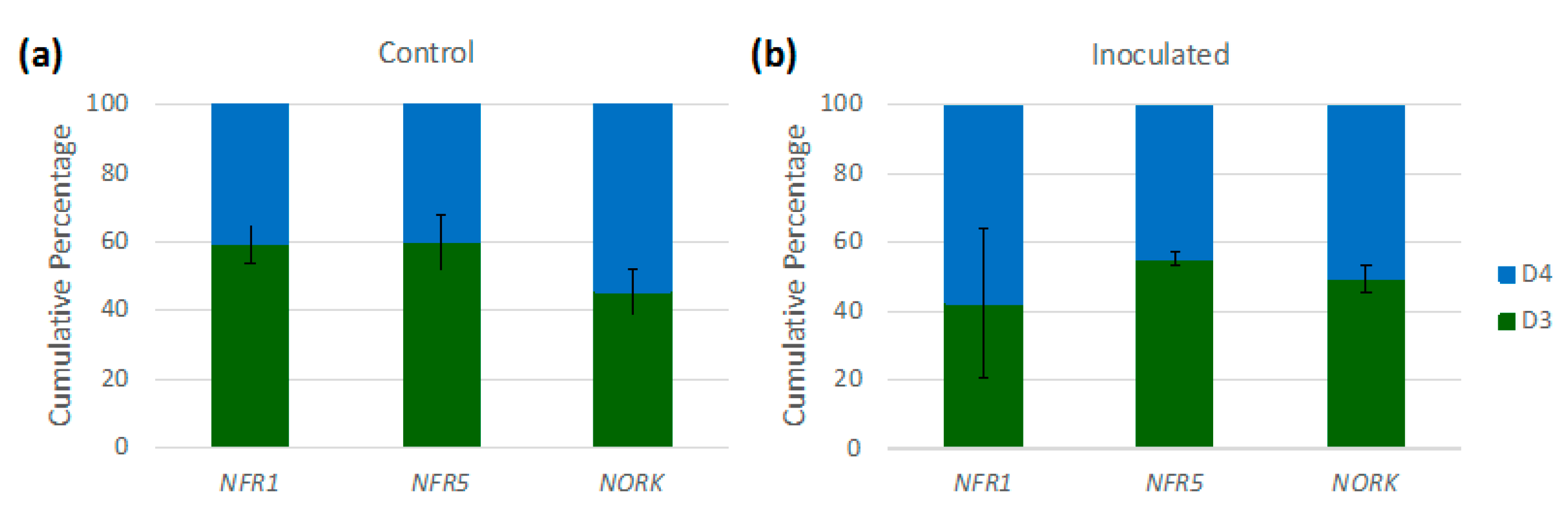
3.4. Implications of Homoeologue Usage for Nodulation
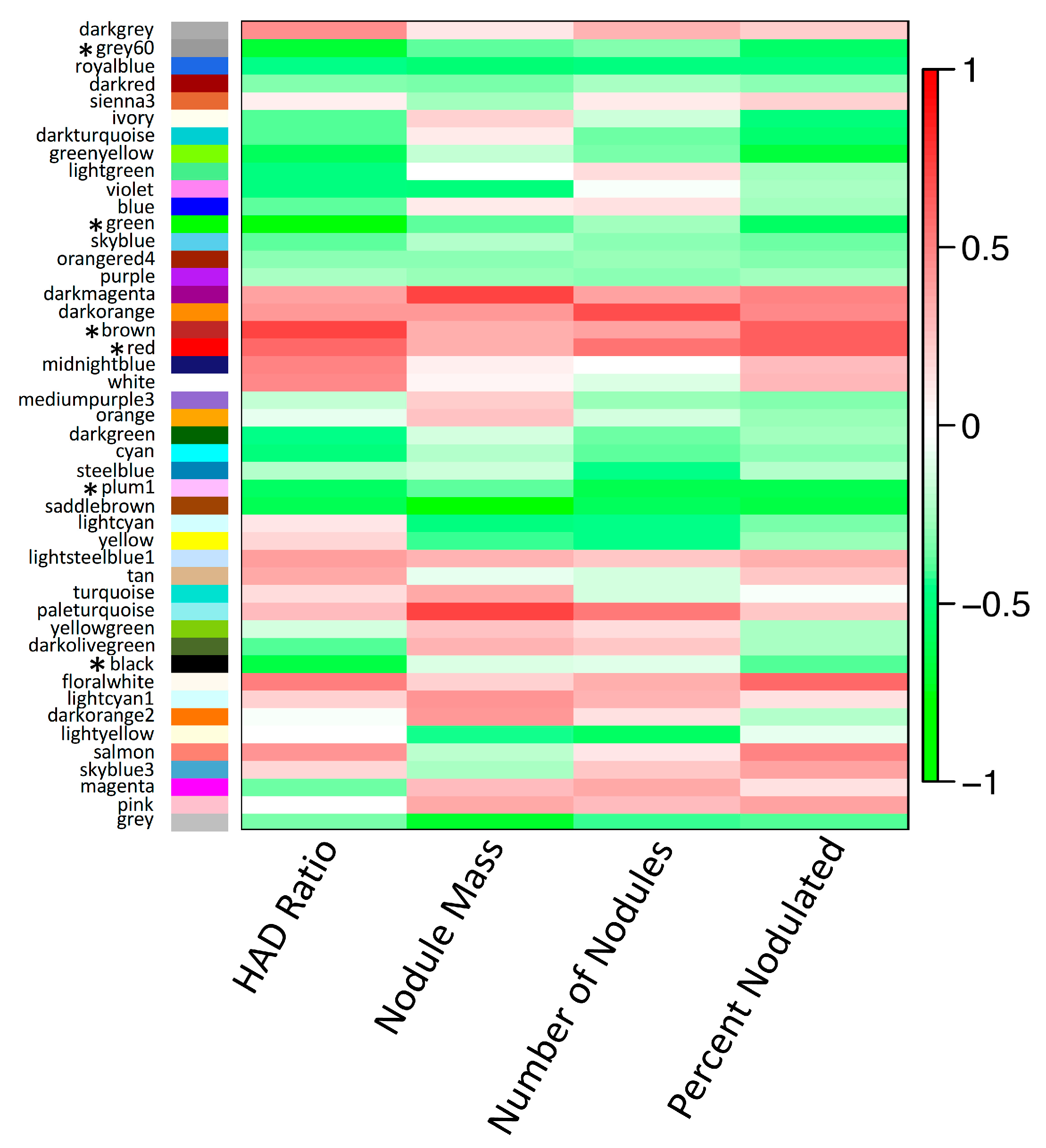
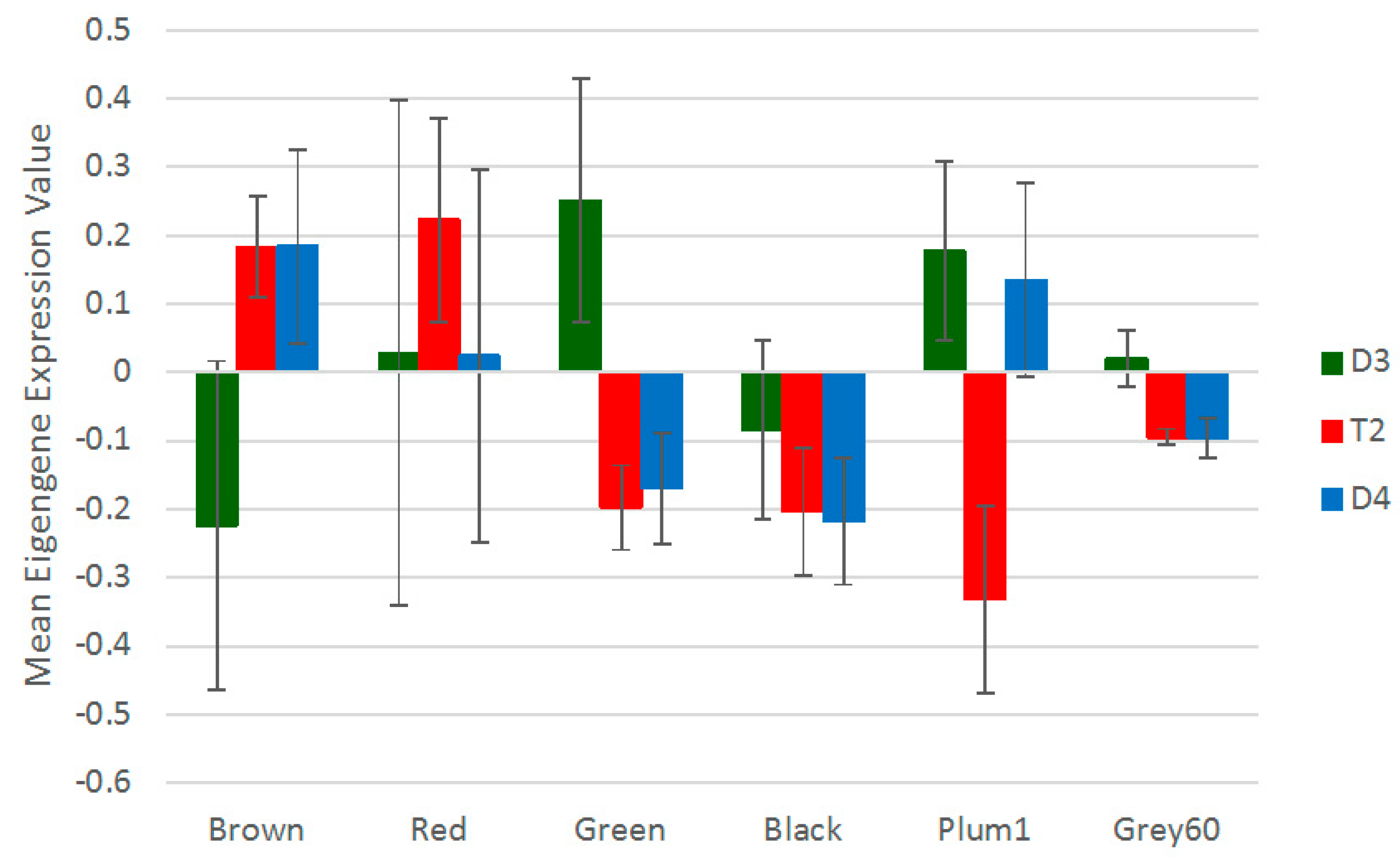
3.5. Non-Additive Expression Patterns of Nodulation-Related Modules and Hub Genes Identified by Network Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Smil, V. Nitrogen in crop production: An account of global flows. Glob. Biogeochem. Cycles 1999, 13, 647–662. [Google Scholar] [CrossRef]
- Graham, P.H.; Vance, C.P. Legumes: Importance and constraints to greater use. Plant Physiol. 2003, 131, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Sprent, J. Legume Nodulation: A Global Perspective; Wiley-Blackwell: Oxford, UK, 2009. [Google Scholar]
- Oldroyd, G.E. Speak, friend, and enter: Signalling systems that promote beneficial symbiotic associations in plants. Nat. Rev. Microbiol. 2013, 11, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Radutoiu, S.; Madsen, L.H.; Madsen, E.B.; Jurkiewicz, A.; Fukai, E.; Quistgaard, E.M.; Albrektsen, A.S.; James, E.K.; Thirup, S.; Stougaard, J. LysM domains mediate lipochitin–oligosaccharide recognition and Nfr genes extend the symbiotic host range. EMBO J. 2007, 26, 3923–3935. [Google Scholar] [CrossRef] [PubMed]
- Libault, M.; Farmer, A.; Brechenmacher, L.; Drnevich, J.; Langley, R.J.; Bilgin, D.D.; Radwan, O.; Neece, D.J.; Clough, S.J.; May, G.D.; et al. Complete transcriptome of the soybean root hair cell, a single-cell model, and its alteration in response to Bradyrhizobium japonicum infection. Plant Physiol. 2010, 152, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, S.; Reid, D.E.; Lorenc, M.T.; Stiller, J.; Edwards, D.; Gresshoff, P.M.; Ferguson, B.J. Transient Nod factor-dependent gene expression in the nodulation-competent zone of soybean (Glycine max [L.] Merr.) roots. Plant Biotechnol. J. 2012, 10, 995–1010. [Google Scholar] [CrossRef] [PubMed]
- Barros de Carvalho, G.A.; Batista, J.S.S.; Marcelino-Guimarães, F.C.; do Nascimento, L.C.; Hungria, M. Transcriptional analysis of genes involved in nodulation in soybean roots inoculated with Bradyrhizobium japonicum strain CPAC 15. BMC Genom. 2013, 14, 153. [Google Scholar] [CrossRef] [PubMed]
- Jardinaud, M.-F.; Boivin, S.; Rodde, N.; Catrice, O.; Kisiala, A.; Lepage, A.; Moreau, S.; Roux, B.; Cottret, L.; Sallet, E.; et al. A laser dissection-RNAseq analysis highlights the activation of cytokinin pathways by Nod factors in the Medicago truncatula root epidermis. Plant Physiol. 2016, 171, 2256–2276. [Google Scholar] [CrossRef] [PubMed]
- Roux, B.; Rodde, N.; Jardinaud, M.F.; Timmers, T.; Sauviac, L.; Cottret, L.; Carrère, S.; Sallet, E.; Courcelle, E.; Moreau, S.; et al. An integrated analysis of plant and bacterial gene expression in symbiotic root nodules using laser-capture microdissection coupled to RNA sequencing. Plant J. 2014, 77, 817–837. [Google Scholar] [CrossRef] [PubMed]
- Young, N.D.; Debellé, F.; Oldroyd, G.E.; Geurts, R.; Cannon, S.B.; Udvardi, M.K.; Benedito, V.A.; Mayer, K.F.; Gouzy, J.; Schoof, H. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 2011, 480, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.-G.; Zhang, L.; Li, C.; Dunwell, J.M.; Zhang, Y.-M. Comparative genomics suggests that an ancestral polyploidy event leads to enhanced root nodule symbiosis in the papilionoideae. Mol. Biol. Evol. 2013, 30, 2602–2611. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, P.; Li, Y.; Liu, Y.; Yu, J.; Ma, X.; Sun, S.; Yang, N.; Xia, R.; Lei, T.; et al. Hierarchically aligning 10 legume genomes establishes a family-level genomics platform. Plant Physiol. 2017, 174, 284–300. [Google Scholar] [CrossRef] [PubMed]
- Te Beest, M.; Le Roux, J.J.; Richardson, D.M.; Brysting, A.K.; Suda, J.; Kubešová, M.; Pyšek, P. The more the better? The role of polyploidy in facilitating plant invasions. Ann. Bot. 2012, 109, 19–45. [Google Scholar] [CrossRef] [PubMed]
- Madlung, A. Polyploidy and its effect on evolutionary success: Old questions revisited with new tools. Heredity 2013, 110, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Segraves, K.A. The effects of genome duplications in a community context. New Phytol. 2017, 215, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Yoo, M.-J.; Liu, X.; Pires, J.C.; Soltis, P.S.; Soltis, D.E. Nonadditive gene expression in polyploids. Annu. Rev. Genet. 2014, 48, 485–517. [Google Scholar] [CrossRef] [PubMed]
- Yoo, M.; Szadkowski, E.; Wendel, J. Homoeolog expression bias and expression level dominance in allopolyploid cotton. Heredity 2013, 110, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Buggs, R.J.; Wendel, J.F.; Doyle, J.J.; Soltis, D.E.; Soltis, P.S.; Coate, J.E. The legacy of diploid progenitors in allopolyploid gene expression patterns. Philos. Trans. R. Soc. B 2014, 369. [Google Scholar] [CrossRef] [PubMed]
- Hegarty, M.; Coate, J.; Sherman-Broyles, S.; Abbott, R.; Hiscock, S.; Doyle, J.J. Lessons from natural and artificial polyploids in higher plants. Cytogenet. Genome Res. 2013, 140, 204–225. [Google Scholar] [CrossRef] [PubMed]
- Bombarely, A.; Coate, J.E.; Doyle, J.J. Mining transcriptomic data to study the origins and evolution of a plant allopolyploid complex. PeerJ 2014, 2, e391. [Google Scholar] [CrossRef] [PubMed]
- Sherman-Broyles, S.; Bombarely, A.; Powell, A.F.; Doyle, J.L.; Egan, A.N.; Coate, J.E.; Doyle, J.J. The wild side of a major crop: Soybean’s perennial cousins from down under. Am. J. Bot. 2014, 101, 1651–1665. [Google Scholar] [CrossRef] [PubMed]
- Pueppke, S. Nodulating associations among rhizobia and legumes of the genus Glycine subgenus Glycine. Plant Soil 1988, 109, 189–193. [Google Scholar] [CrossRef]
- Pueppke, S.G.; Broughton, W.J. Rhizobium sp. strain NGR234 and R. fredii USDA257 share exceptionally broad, nested host ranges. Mol. Plant-Microbe Interact. 1999, 12, 293–318. [Google Scholar] [CrossRef] [PubMed]
- Brockwell, J.; McIlroy, R.; Hebb, D.M. The Australian Collection of Rhizobium Strains for Temperate Legumes. Catalogue 1998; CSIRO (Australia), Plant Industry, Divisional Report No. 98/1; Commonwealth Scientific and Industrial Research Organisation: Canberra, Australia, 1998. [Google Scholar]
- Powell, A.F.; Doyle, J.J. Enhanced rhizobial symbiotic capacity in an allopolyploid species of Glycine (Leguminosae). Am. J. Bot. 2016, 103, 1771–1782. [Google Scholar] [CrossRef] [PubMed]
- Coate, J.; Powell, A.; Owens, T.; Doyle, J.J. Transgressive physiological and transcriptomic responses to light stress in allopolyploid Glycine dolichocarpa (Leguminosae). Heredity 2013, 110, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Coate, J.E.; Doyle, J.J. Quantifying whole transcriptome size, a prerequisite for understanding transcriptome evolution across species: An example from a plant allopolyploid. Genome Biol. Evol. 2010, 2, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Ilut, D.C.; Coate, J.E.; Luciano, A.K.; Owens, T.G.; May, G.D.; Farmer, A.; Doyle, J.J. A comparative transcriptomic study of an allotetraploid and its diploid progenitors illustrates the unique advantages and challenges of RNA-seq in plant species. Am. J. Bot. 2012, 99, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Coate, J.E.; Luciano, A.K.; Seralathan, V.; Minchew, K.J.; Owens, T.G.; Doyle, J.J. Anatomical, biochemical, and photosynthetic responses to recent allopolyploidy in Glycine dolichocarpa (Fabaceae). Am. J. Bot. 2012, 99, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Coate, J.E.; Bar, H.; Doyle, J.J. Extensive translational regulation of gene expression in an allopolyploid (Glycine dolichocarpa). Plant Cell 2014, 26, 136–150. [Google Scholar] [CrossRef] [PubMed]
- Somasegaran, P.; Hoben, H.J. Handbook for Rhizobia: Methods in Legume-Rhizobium Technology; Springer: New York, NY, USA, 1994. [Google Scholar]
- Howieson, J.; Dilworth, M. Working with Rhizobia; Australian Centre for International Agricultural Research: Canberra, Australia, 2016.
- Aronesty, E. Comparison of sequencing utility programs. Open Bioinform. J. 2013, 7, 1–8. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; McCarthy, D.J.; Chen, Y.; Okoniewski, M.; Smyth, G.K.; Huber, W.; Robinson, M.D. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat. Protoc. 2013, 8, 1765–1786. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar]
- Rapp, R.A.; Udall, J.A.; Wendel, J.F. Genomic expression dominance in allopolyploids. BMC Biol. 2009, 7, 18. [Google Scholar] [CrossRef] [PubMed]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. Fast R functions for robust correlations and hierarchical clustering. J. Stat. Softw. 2012, 46, i11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 1128. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.; Tenhaken, R.; Dixon, R.; Lamb, C. H2O2 from the oxidative burst orchestrates the plant hypersensitive disease resistance response. Cell 1994, 79, 583–593. [Google Scholar] [CrossRef]
- Petrov, V.D.; Van Breusegem, F. Hydrogen peroxide—A central hub for information flow in plant cells. AoB Plants 2012, 2012, pls014. [Google Scholar] [CrossRef] [PubMed]
- Powell, A.F.; Doyle, J.J. Comparative analysis of defense responses in Glycine dolichocarpa and its diploid progenitors. In preparation.
- Lohar, D.P.; Haridas, S.; Gantt, J.S.; VandenBosch, K.A. A transient decrease in reactive oxygen species in roots leads to root hair deformation in the legume–rhizobia symbiosis. New Phytol. 2007, 173, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Kouchi, H.; Shimomura, K.; Hata, S.; Hirota, A.; Wu, G.-J.; Kumagai, H.; Tajima, S.; Suganuma, N.; Suzuki, A.; Aoki, T. Large-scale analysis of gene expression profiles during early stages of root nodule formation in a model legume, Lotus japonicus. DNA Res. 2004, 11, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Shaw, S.L.; Long, S.R. Nod factor elicits two separable calcium responses in Medicago truncatula root hair cells. Plant Physiol. 2003, 131, 976–984. [Google Scholar] [CrossRef] [PubMed]
- Bueno, P.; Soto, M.J.; Rodríguez-Rosales, M.P.; Sanjuan, J.; Olivares, J.; Donaire, J.P. Time-course of lipoxygenase, antioxidant enzyme activities and H2O2 accumulation during the early stages of rhizobium–legume symbiosis. New Phytol. 2001, 152, 91–96. [Google Scholar] [CrossRef]
- Jamet, A.; Mandon, K.; Puppo, A.; Hérouart, D. H2O2 is required for optimal establishment of the Medicago sativa/Sinorhizobium meliloti symbiosis. J. Bacteriol. 2007, 189, 8741–8745. [Google Scholar] [CrossRef] [PubMed]
- Andrio, E.; Marino, D.; Marmeys, A.; Segonzac, M.D.; Damiani, I.; Genre, A.; Huguet, S.; Frendo, P.; Puppo, A.; Pauly, N. Hydrogen peroxide-regulated genes in the Medicago truncatula–Sinorhizobium meliloti symbiosis. New Phytol. 2013, 198, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Marie, C.; Deakin, W.J.; Viprey, V.; Kopciñska, J.; Golinowski, W.; Krishnan, H.B.; Perret, X.; Broughton, W.J. Characterization of Nops, nodulation outer proteins, secreted via the type III secretion system of NGR234. Mol. Plant-Microbe Interact. 2003, 16, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.-J.; Zeng, Y.; Xie, Z.-P.; Staehelin, C. Symbiosis-promoting and deleterious effects of NopT, a novel type 3 effector of Rhizobium sp. strain NGR234. J. Bacteriol. 2008, 190, 5101–5110. [Google Scholar] [CrossRef] [PubMed]
- Schauser, L.; Roussis, A.; Stiller, J.; Stougaard, J. A plant regulator controlling development of symbiotic root nodules. Nature 1999, 402, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Grover, C.; Gallagher, J.; Szadkowski, E.; Yoo, M.; Flagel, L.; Wendel, J. Homoeolog expression bias and expression level dominance in allopolyploids. New Phytol. 2012, 196, 966–971. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, S.; Kim, J.; Muñoz, A.; Heckmann, A.B.; Downie, J.A.; Oldroyd, G.E. GRAS proteins form a DNA binding complex to induce gene expression during nodulation signaling in Medicago truncatula. Plant Cell 2009, 21, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Kaló, P.; Gleason, C.; Edwards, A.; Marsh, J.; Mitra, R.M.; Hirsch, S.; Jakab, J.; Sims, S.; Long, S.R.; Rogers, J. Nodulation signaling in legumes requires NSP2, a member of the GRAS family of transcriptional regulators. Science 2005, 308, 1786–1789. [Google Scholar] [CrossRef] [PubMed]
- Powell, A.F.; Doyle, J.J. The implications of polyploidy for the evolution of signalling in rhizobial nodulation symbiosis. Adv. Bot. Res. 2015, 75, 149–190. [Google Scholar]
- Madsen, E.B.; Antolín-Llovera, M.; Grossmann, C.; Ye, J.; Vieweg, S.; Broghammer, A.; Krusell, L.; Radutoiu, S.; Jensen, O.N.; Stougaard, J. Autophosphorylation is essential for the in vivo function of the Lotus japonicus Nod Factor Receptor 1 and receptor-mediated signalling in cooperation with Nod Factor Receptor 5. Plant J. 2011, 65, 404–417. [Google Scholar] [CrossRef] [PubMed]
- Ried, M.K.; Antolín-Llovera, M.; Parniske, M. Spontaneous symbiotic reprogramming of plant roots triggered by receptor-like kinases. eLife 2014, 3, e03891. [Google Scholar] [CrossRef] [PubMed]
- Indrasumunar, A.; Searle, I.; Lin, M.H.; Kereszt, A.; Men, A.; Carroll, B.J.; Gresshoff, P.M. Nodulation factor receptor kinase 1α controls nodule organ number in soybean (Glycine max L. Merr). Plant J. 2011, 65, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Indrasumunar, A.; Wilde, J.; Hayashi, S.; Li, D.; Gresshoff, P.M. Functional analysis of duplicated Symbiosis Receptor Kinase (SymRK) genes during nodulation and mycorrhizal infection in soybean (Glycine max). J. Plant Physiol. 2015, 176, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Hovav, R.; Grover, C.E.; Faigenboim-Doron, A.; Kadmon, N.; Page, J.T.; Udall, J.A.; Wendel, J.F. Evolutionary conservation and divergence of gene coexpression networks in Gossypium (cotton) seeds. Genome Biol. Evol. 2017, 8, 3765–3783. [Google Scholar]
- Gallagher, J.P.; Grover, C.E.; Hu, G.; Wendel, J.F. Insights into the ecology and evolution of polyploid plants through network analysis. Mol. Ecol. 2016, 25, 2644–2660. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, T.; Geldner, N.; Grebe, M.; Mangold, S.; Jackson, C.L.; Paris, S.; Gälweiler, L.; Palme, K.; Jürgens, G. Coordinated polar localization of auxin efflux carrier PIN1 by GNOM ARF GEF. Science 1999, 286, 316–318. [Google Scholar] [CrossRef] [PubMed]
- Geldner, N.; Anders, N.; Wolters, H.; Keicher, J.; Kornberger, W.; Muller, P.; Delbarre, A.; Ueda, T.; Nakano, A.; Jürgens, G. The Arabidopsis GNOM ARF-GEF mediates endosomal recycling, auxin transport, and auxin-dependent plant growth. Cell 2003, 112, 219–230. [Google Scholar] [CrossRef]
- Mathesius, U. Auxin: At the root of nodule development? Funct. Plant Biol. 2008, 35, 651–668. [Google Scholar] [CrossRef]
- Potuschak, T.; Lechner, E.; Parmentier, Y.; Yanagisawa, S.; Grava, S.; Koncz, C.; Genschik, P. EIN3-dependent regulation of plant ethylene hormone signaling by two Arabidopsis F box proteins: EBF1 and EBF2. Cell 2003, 115, 679–689. [Google Scholar] [CrossRef]
- Penmetsa, R.V.; Cook, D.R. A legume ethylene-insensitive mutant hyperinfected by its rhizobial symbiont. Science 1997, 275, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, B.J.; Mathesius, U. Phytohormone regulation of legume-rhizobia interactions. J. Chem. Ecol. 2014, 40, 770–790. [Google Scholar] [CrossRef] [PubMed]
- Miyata, K.; Kawaguchi, M.; Nakagawa, T. Two distinct EIN2 genes cooperatively regulate ethylene signaling in Lotus japonicus. Plant Cell Physiol. 2013, 54, 1469–1477. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.K.; Biswas, B.; Gresshoff, P.M. Classical ethylene insensitive mutants of the Arabidopsis EIN2 orthologue lack the expected ‘hypernodulation’ response in Lotus japonicus. J. Integr. Plant Biol. 2013, 55, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.S.; Harper, J.E.; Hoffman, T.K.; Bent, A.F. Regulation of soybean nodulation independent of ethylene signaling. Plant Physiol. 1999, 119, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Prayitno, J.; Rolfe, B.G.; Mathesius, U. The ethylene-insensitive sickle mutant of Medicago truncatula shows altered auxin transport regulation during nodulation. Plant Physiol. 2006, 142, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Powell, A.F.; Doyle, J.J. Effects of allopolyploidy and rhizobial inoculation on root metabolite profiles, diversity, exudation and biosynthetic gene expression in Glycine subgenus Glycine. In preparation.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Powell, A.F.; Doyle, J.J. Non-Additive Transcriptomic Responses to Inoculation with Rhizobia in a Young Allopolyploid Compared with Its Diploid Progenitors. Genes 2017, 8, 357. https://doi.org/10.3390/genes8120357
Powell AF, Doyle JJ. Non-Additive Transcriptomic Responses to Inoculation with Rhizobia in a Young Allopolyploid Compared with Its Diploid Progenitors. Genes. 2017; 8(12):357. https://doi.org/10.3390/genes8120357
Chicago/Turabian StylePowell, Adrian F., and Jeff J. Doyle. 2017. "Non-Additive Transcriptomic Responses to Inoculation with Rhizobia in a Young Allopolyploid Compared with Its Diploid Progenitors" Genes 8, no. 12: 357. https://doi.org/10.3390/genes8120357
APA StylePowell, A. F., & Doyle, J. J. (2017). Non-Additive Transcriptomic Responses to Inoculation with Rhizobia in a Young Allopolyploid Compared with Its Diploid Progenitors. Genes, 8(12), 357. https://doi.org/10.3390/genes8120357